

Phloretamide Protects against Diabetic Kidney Damage and Dysfunction in Diabetic Rats by Attenuating Hyperglycemia and Hyperlipidemia, Suppressing NF-κβ, and Upregulating Nrf2

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction of STZ-Mediated T1DM
2.3. Experimental Design
2.4. Dose Selection
2.5. Urine and Blood Sample Collection and Analyses
2.6. Biochemical Analysis of the Plasma, Serum, and Urine
2.7. Kidney Collection and Processing
2.8. Biochemical Analysis of the Renal Homogenates
2.9. Biochemical Analysis of the Cytoplasmic and Nuclear Extracts
2.10. Real-Time PCR (qPCR)
2.11. Statistical Analysis
3. Results
3.1. Effect of PHLTM on Body Weight and Metabolic Parameters
3.2. Effect of PHLTM on Markers of Kidney Function
3.3. Effect of PHLTM on Renal Markers of Oxidative Stress and Inflammation
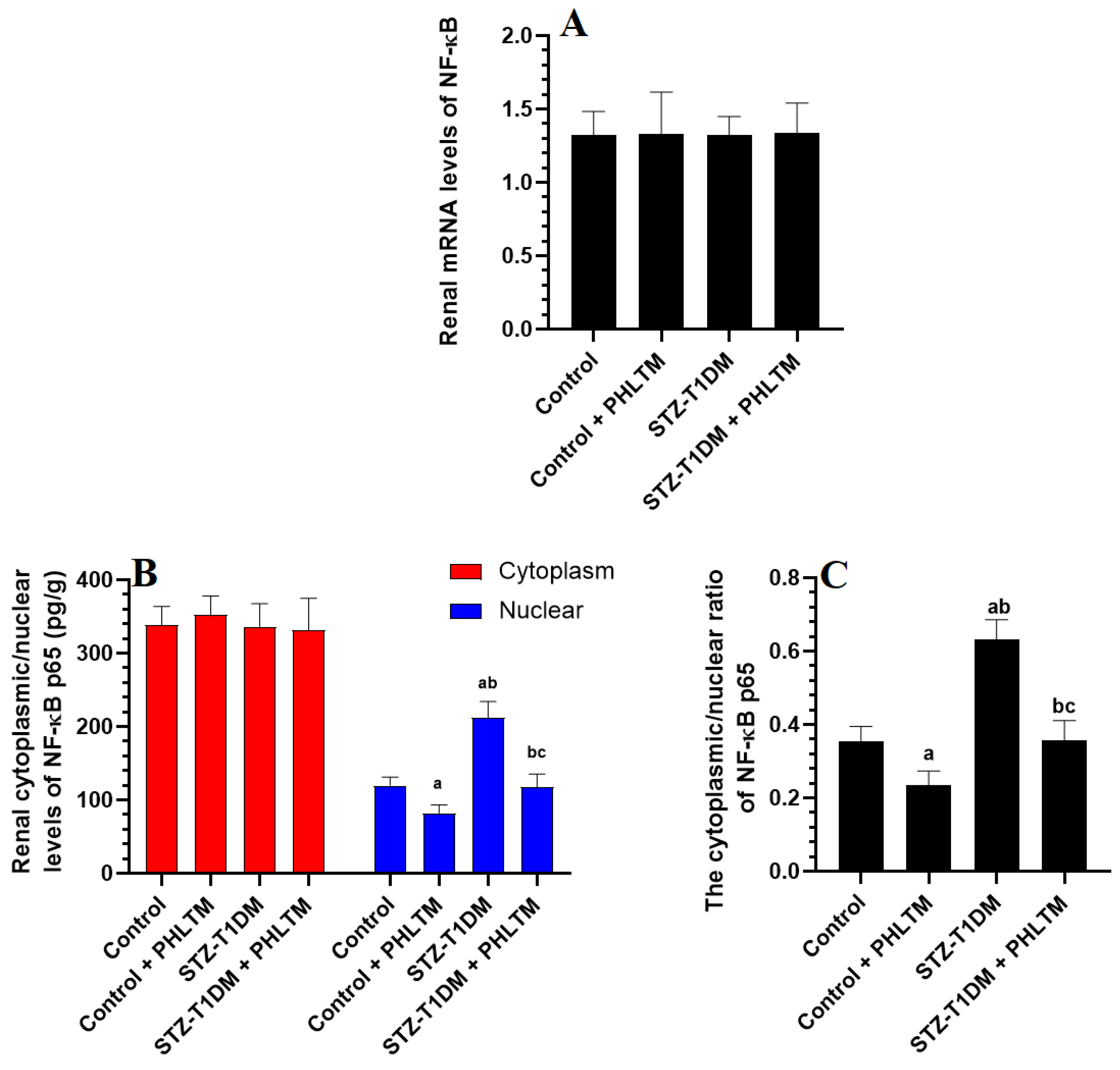
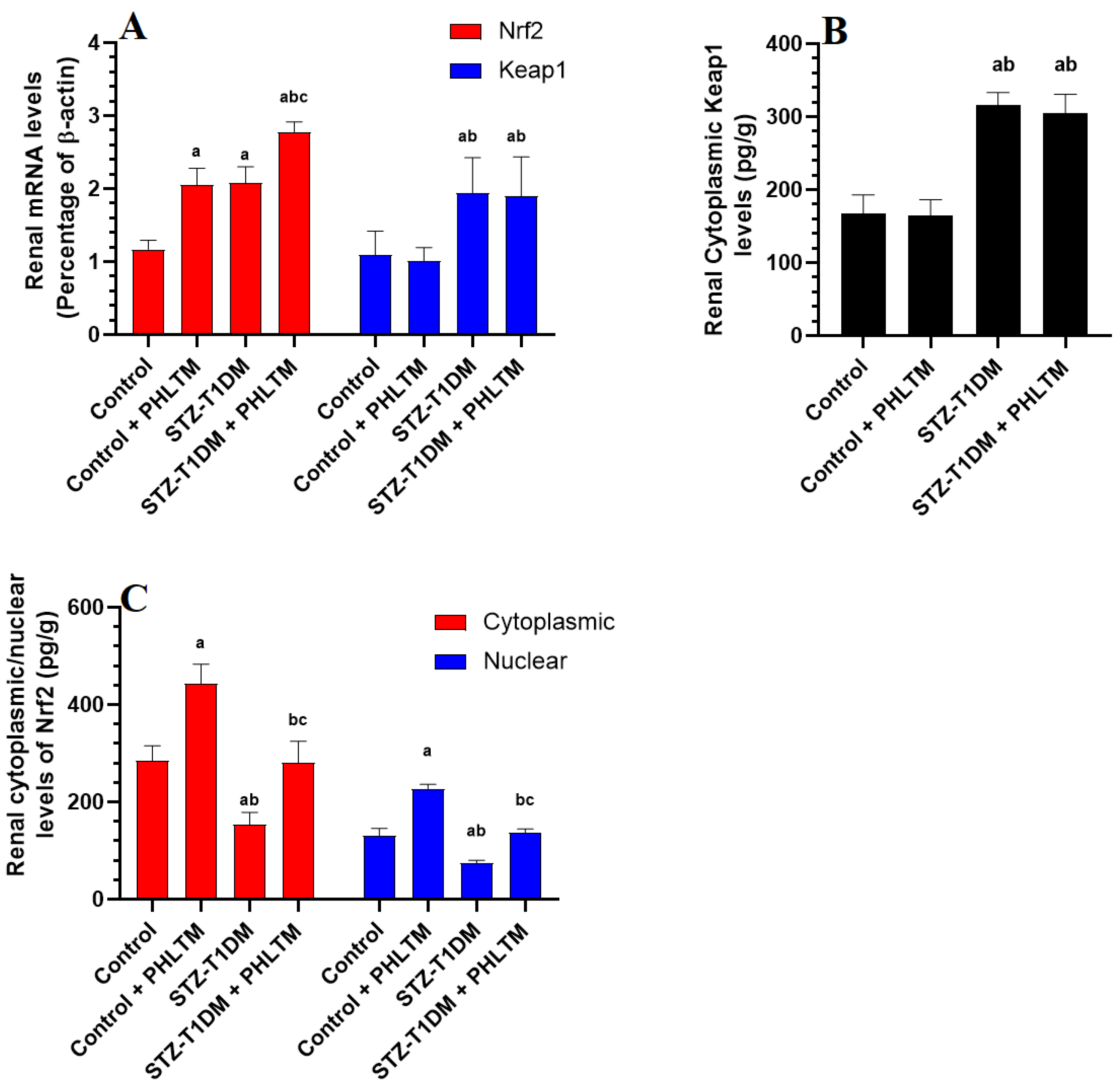
3.4. Changes in the Expression Levels of Nrf2 and NF-κβ
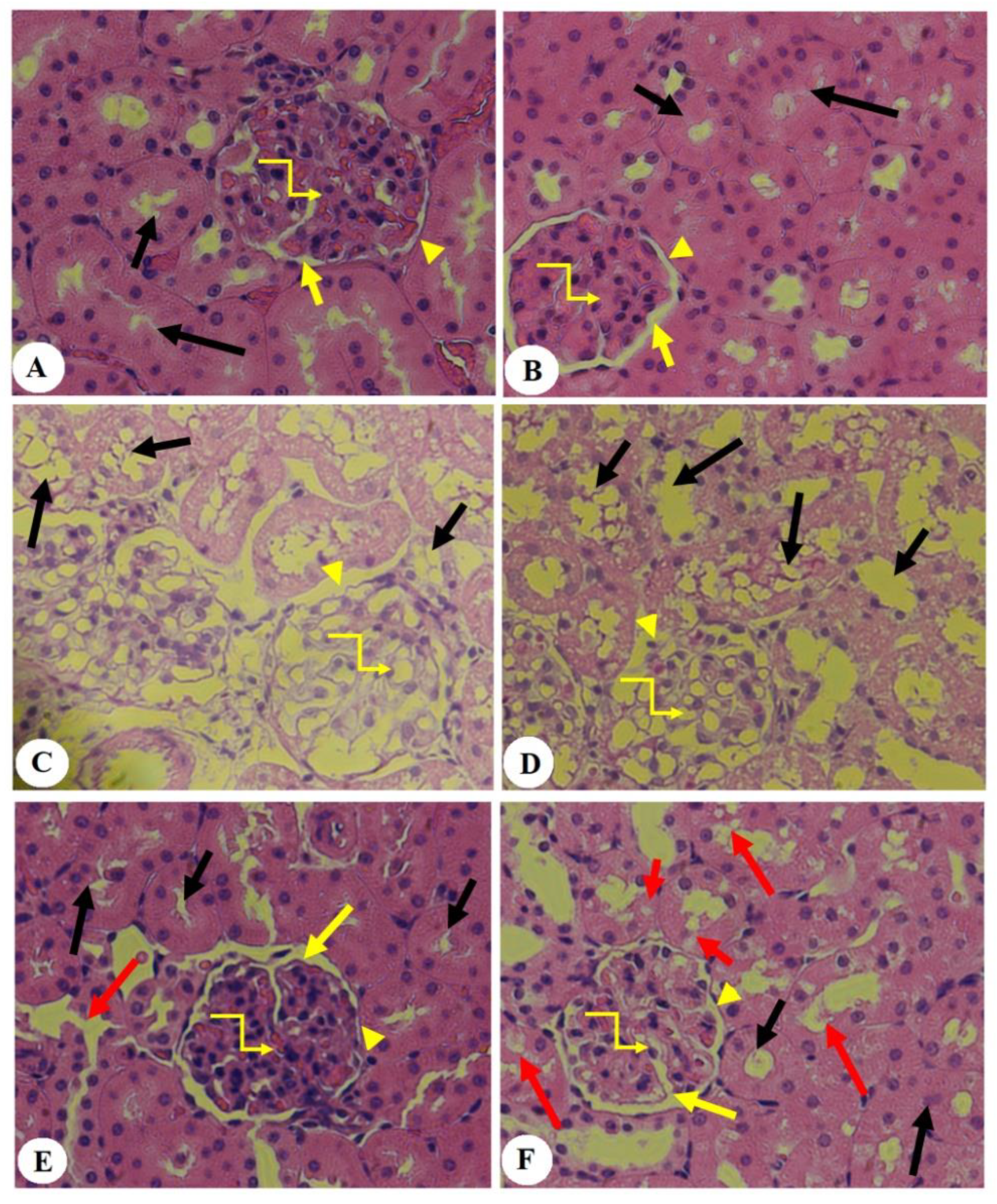
3.5. Effect of PHLTM on Kidney Morphology
4. Discussion and Conclusions
Study Limitation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. CJASN 2017, 12, 2032. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Agarwal, R.; Alpers, C.E.; Bakris, G.L.; Brosius, F.C.; Kolkhof, P.; Uribarri, J. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. 2022, 102, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Wang, X.; Xu, H.; Yang, Z.; Cheng, Y.; Zhou, B. Protective effect of ferulic acid on STZ-induced diabetic nephropathy in rats. Food Funct. 2020, 11, 3706–3718. [Google Scholar] [CrossRef] [PubMed]
- Naaman, S.C.; Bakris, G.L. Diabetic nephropathy: Update on pillars of therapy slowing progression. Diabetes Care 2023, 46, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Mima, A. Inflammation and oxidative stress in diabetic nephropathy: New insights on its inhibition as new therapeutic targets. J. Diabetes Res. 2013, 2013, 248563. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Sato, E.; Mishima, E.; Miyazaki, M.; Tanaka, T. What’s new in the molecular mechanisms of diabetic kidney disease: Recent advances. Int. J. Mol. Sci. 2022, 24, 570. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Liu, T.; Qiao, Y.; Liu, D.; Yang, L.; Mao, H.; Ma, F.; Wang, Y.; Peng, L.; Zhan, Y. Oxidative stress and inflammation in diabetic nephropathy: Role of polyphenols. Front. Immunol. 2023, 14, 1185317. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.B.; Sinha, K.; Sil, P.C. Mangiferin attenuates diabetic nephropathy by inhibiting oxidative stress mediated signaling cascade, TNFα related and mitochondrial dependent apoptotic pathways in streptozotocin-induced diabetic rats. PLoS ONE 2014, 9, e107220. [Google Scholar] [CrossRef]
- Sahakyan, G.; Vejux, A.; Sahakyan, N. The role of oxidative stress-mediated inflammation in the development of T2DM-induced diabetic nephropathy: Possible preventive action of tannins and other oligomeric polyphenols. Molecules 2022, 27, 9035. [Google Scholar] [CrossRef]
- Ma, X.; Ma, J.; Leng, T.; Yuan, Z.; Hu, T.; Liu, Q.; Shen, T. Advances in oxidative stress in pathogenesis of diabetic kidney disease and efficacy of TCM intervention. Ren. Fail. 2023, 45, 2146512. [Google Scholar] [CrossRef]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef]
- Gao, W.; Guo, L.; Yang, Y.; Wang, Y.; Xia, S.; Gong, H.; Zhang, B.-K.; Yan, M. Dissecting the crosstalk between Nrf2 and NF-κB response pathways in drug-induced toxicity. Front. Cell Dev. Biol. 2022, 9, 809952. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2: INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Sergio, M.; Claudio, A.; Alejandra, D.; Eugenia, B.M.; Leopoldo, A.; Claudio, F.; Herman, S.; Marta, R.-O.; Jesús, E. NF-κB activation and overexpression of regulated genes in human diabetic nephropathy. In Nephrology Dialysis Transplantation; Official Publication of the European Dialysis and Transplant Association; European Renal Association: Parma, Italy, 2004. [Google Scholar]
- Pérez-Morales, R.E.; Del Pino, M.D.; Valdivielso, J.M.; Ortiz, A.; Mora-Fernández, C.; Navarro-González, J.F. Inflammation in diabetic kidney disease. Nephron 2019, 143, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Yerra, V.G.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-κβ: A potential target in the management of vascular complications of diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef] [PubMed]
- Foresto-Neto, O.; Albino, A.H.; Arias, S.C.A.; Faustino, V.D.; Zambom, F.F.F.; Cenedeze, M.A.; Elias, R.M.; Malheiros, D.M.A.C.; Camara, N.O.S.; Fujihara, C.K. NF-κB system is chronically activated and promotes glomerular injury in experimental type 1 diabetic kidney disease. Front. Physiol. 2020, 11, 84. [Google Scholar] [CrossRef]
- Adelusi, T.I.; Du, L.; Hao, M.; Zhou, X.; Xuan, Q.; Apu, C.; Sun, Y.; Lu, Q.; Yin, X. Keap1/Nrf2/ARE signaling unfolds therapeutic targets for redox imbalanced-mediated diseases and diabetic nephropathy. Biomed. Pharmacother. 2020, 123, 109732. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Anton, M.I.; Floria, M.; Seritean Isac, P.N.; Hurjui, L.L.; Tarniceriu, C.C.; Costea, C.F.; Ciocoiu, M.; Rezus, C. Oxidative stress and NRF2/KEAP1/ARE pathway in diabetic kidney disease (DKD): New perspectives. Biomolecules 2022, 12, 1227. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mittal, R. Nrf2: A potential therapeutic target for diabetic neuropathy. Inflammopharmacology 2017, 25, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K. Diabetic nephropathy–complications and treatment. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 361–381. [Google Scholar] [CrossRef] [PubMed]
- Nithiya, T.; Udayakumar, R. In vitro antioxidant properties of phloretin—An important phytocompound. J. Biosci. Med. 2016, 4, 85. [Google Scholar]
- Krajka-Kuźniak, V.; Paluszczak, J.; Celewicz, L.; Barciszewski, J.; Baer-Dubowska, W. Phloretamide, an apple phenolic compound, activates the Nrf2/ARE pathway in human hepatocytes. Food Chem. Toxicol. 2013, 51, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussan, R.; Albadr, N.A.; Alshammari, G.M.; Almasri, S.A.; Yahya, M.A. Phloretamide Prevent Hepatic and Pancreatic Damage in Diabetic Male Rats by Modulating Nrf2 and NF-κB. Nutrients 2023, 15, 1456. [Google Scholar] [CrossRef] [PubMed]
- AlTamimi, J.Z.; AlFaris, N.A.; Alshammari, G.M.; Alagal, R.I.; Aljabryn, D.H.; Yahya, M.A. Protective effect of eriodictyol against hyperglycemia-induced diabetic nephropathy in rats entails antioxidant and anti-inflammatory effects mediated by activating Nrf2. Saudi Pharm. J. 2023, 31, 101817. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Koya, D. Rodent models of diabetic nephropathy: Their utility and limitations. Int. J. Nephrol. Renovasc. Dis. 2016, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Kamli-Salino, S.E.; Brown, P.A.; Haschler, T.N.; Liang, L.; Feliers, D.; Wilson, H.M.; Delibegovic, M. Induction of experimental diabetes and diabetic nephropathy using anomer-equilibrated streptozotocin in male C57Bl/6J mice. Biochem. Biophys. Res. Commun. 2023, 650, 109–116. [Google Scholar] [CrossRef]
- Kaur, N.; Kishore, L.; Singh, R. Dillenia indica L. attenuates diabetic nephropathy via inhibition of advanced glycation end products accumulation in STZ-nicotinamide induced diabetic rats. J. Tradit. Complement. Med. 2018, 8, 226–238. [Google Scholar] [CrossRef]
- Kumari, S.; Kamboj, A.; Wanjari, M.; Sharma, A.K. Nephroprotective effect of Vanillic acid in STZ-induced diabetic rats. J. Diabetes Metab. Disord. 2021, 20, 571–582. [Google Scholar] [CrossRef]
- Kaikini, A.A.; Dhodi, D.; Muke, S.; Peshattiwar, V.; Bagle, S.; Korde, A.; Sarnaik, J.; Kadwad, V.; Sachdev, S.; Sathaye, S. Standardization of type 1 and type 2 diabetic nephropathy models in rats: Assessment and characterization of metabolic features and renal injury. J. Pharm. Bioallied Sci. 2020, 12, 295. [Google Scholar]
- Han, W.K.; Bailly, V.; Abichandani, R.; Thadhani, R.; Bonventre, J.V. Kidney Injury Molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002, 62, 237–244. [Google Scholar] [CrossRef]
- Bonventre, J.V. Kidney Injury Molecule-1 (KIM-1): A specific and sensitive biomarker of kidney injury. Scand. J. Clin. Lab. Investig. 2008, 68, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Latoch, E.; Konończuk, K.; Muszyńska-Rosłan, K.; Taranta-Janusz, K.; Wasilewska, A.; Szymczak, E.; Trochim, J.; Krawczuk-Rybak, M. Urine NGAL and KIM-1—Tubular injury biomarkers in long-term survivors of childhood solid tumors: A Cross-Sectional Study. J. Clin. Med. 2021, 10, 399. [Google Scholar] [CrossRef]
- Kandasamy, Y.; Smith, R.; Lumbers, E.R.; Rudd, D. Nephrin–a biomarker of early glomerular injury. Biomark. Res. 2014, 2, 21. [Google Scholar] [CrossRef]
- Patari, A.; Forsblom, C.; Havana, M.; Taipale, H.; Groop, P.-H.; Holthofer, H.; Group, F.S. Nephrinuria in diabetic nephropathy of type 1 diabetes. Diabetes 2003, 52, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, J.A.M.; Tatsch, E.; Hausen, B.S.; Bollick, Y.S.; Moretto, M.B.; Duarte, T.; Duarte, M.M.; Londero, S.W.; Premaor, M.O.; Comim, F.V. Urinary kidney injury molecule-1 and neutrophil gelatinase-associated lipocalin as indicators of tubular damage in normoalbuminuric patients with type 2 diabetes. Clin. Biochem. 2016, 49, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Quang, T.H.; Nguyet, M.P.; Thao, D.P.; Thi, M.H.; Phuong Thi Dam, L.; Thi, H.H.; Van, A.P.; Luong, T.C.; Tuyet, M.N.T.; Duy, Q.D. Evaluation of urinary neutrophil gelatinase associated lipocalin and kidney injury molecule-1 as diagnostic markers for early nephropathy in patients with type 2 diabetes mellitus. Diabetes Metab. Syndr. Obes. 2020, 13, 2199–2207. [Google Scholar]
- Rico-Fontalvo, J.; Aroca-Martínez, G.; Daza-Arnedo, R.; Cabrales, J.; Rodríguez-Yanez, T.; Cardona-Blanco, M.; Montejo-Hernández, J.; Rodelo Barrios, D.; Patiño-Patiño, J.; Osorio Rodríguez, E. Novel Biomarkers of Diabetic Kidney Disease. Biomolecules 2023, 13, 633. [Google Scholar] [CrossRef]
- Cui, D.; Liu, S.; Tang, M.; Lu, Y.; Zhao, M.; Mao, R.; Wang, C.; Yuan, Y.; Li, L.; Chen, Y. Phloretin ameliorates hyperuricemia-induced chronic renal dysfunction through inhibiting NLRP3 inflammasome and uric acid reabsorption. Phytomedicine 2020, 66, 153111. [Google Scholar] [CrossRef] [PubMed]
- Un, H.; Ugan, R.A.; Gurbuz, M.A.; Bayir, Y.; Kahramanlar, A.; Kaya, G.; Cadirci, E.; Halici, Z. Phloretin and phloridzin guard against cisplatin-induced nephrotoxicity in mice through inhibiting oxidative stress and inflammation. Life Sci. 2021, 266, 118869. [Google Scholar] [CrossRef] [PubMed]
- Chhimwal, J.; Goel, A.; Sukapaka, M.; Patial, V.; Padwad, Y. Phloretin mitigates oxidative injury, inflammation, and fibrogenic responses via restoration of autophagic flux in in vitro and preclinical models of NAFLD. J. Nutr. Biochem. 2022, 107, 109062. [Google Scholar] [CrossRef] [PubMed]
- Shelke, V.; Kale, A.; Dagar, N.; Habshi, T.; Gaikwad, A.B. Concomitant inhibition of TLR-4 and SGLT2 by phloretin and empagliflozin prevents diabetes-associated ischemic acute kidney injury. Food Funct. 2023, 14, 5391–5403. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-c.; Tseng, C.-H. Dyslipidemia, kidney disease, and cardiovascular disease in diabetic patients. Rev. Diabet. Stud. RDS 2013, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodnia, L.; Aghadavod, E.; Beigrezaei, S.; Rafieian-Kopaei, M. An update on diabetic kidney disease, oxidative stress and antioxidant agents. J. Ren. Inj. Prev. 2017, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Satoh, N.; Kodera, R.; Hirashima, T.; Suzuki, N.; Aoki, E.; Oshima, T.; Hosoya, M.; Fujita, M.; Hayashi, T. Dyslipidemia in diabetic kidney disease classified by proteinuria and renal dysfunction: A cross-sectional study from a regional diabetes cohort. J. Diabetes Investig. 2022, 13, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Mottalib, A.; Kasetty, M.; Mar, J.Y.; Elseaidy, T.; Ashrafzadeh, S.; Hamdy, O. Weight management in patients with type 1 diabetes and obesity. Curr. Diab. Rep. 2017, 17, 92. [Google Scholar] [CrossRef]
- Yamagishi, S.-i.; Matsui, T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxidative Med. Cell. Longev. 2010, 3, 101–108. [Google Scholar] [CrossRef]
- Rivero, A.; Mora, C.; Muros, M.; García, J.; Herrera, H.; Navarro-González, J.F. Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clin. Sci. 2009, 116, 479–492. [Google Scholar] [CrossRef]
- Navarro-González, J.F.; Mora-Fernández, C.; De Fuentes, M.M.; García-Pérez, J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Opazo-Ríos, L.; Guerrero-Hue, M.; García-Caballero, C.; Vázquez-Carballo, C.; Mas, S.; Sanz, A.B.; Herencia, C.; Mezzano, S. Pathogenic pathways and therapeutic approaches targeting inflammation in diabetic nephropathy. Int. J. Mol. Sci. 2020, 21, 3798. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Liang, R.; Huang, B.; Hou, J.; Yin, J.; Zhao, T.; Zhou, L.; Wu, R.; Qian, Y.; Wang, F. Tumor necrosis factor-α blockade ameliorates diabetic nephropathy in rats. Clin. Kidney J. 2021, 14, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Kreiner, F.F.; Kraaijenhof, J.M.; von Herrath, M.; Hovingh, G.K.K.; von Scholten, B.J. Interleukin 6 in diabetes, chronic kidney disease, and cardiovascular disease: Mechanisms and therapeutic perspectives. Expert Rev. Clin. Immunol. 2022, 18, 377–389. [Google Scholar] [CrossRef]
- Wu, C.-C.; Sytwu, H.-K.; Lu, K.-C.; Lin, Y.-F. Role of T cells in type 2 diabetic nephropathy. J. Diabetes Res. 2011, 2011, 514738. [Google Scholar] [CrossRef] [PubMed]
- Fordham, J.B.; Raza Naqvi, A.; Nares, S. Leukocyte production of inflammatory mediators is inhibited by the antioxidants phloretin, silymarin, hesperetin, and resveratrol. Mediat. Inflamm. 2014, 2014, 938712. [Google Scholar] [CrossRef]
- Chang, W.-T.; Huang, W.-C.; Liou, C.-J. Evaluation of the anti-inflammatory effects of phloretin and phlorizin in lipopolysaccharide-stimulated mouse macrophages. Food Chem. 2012, 134, 972–979. [Google Scholar] [CrossRef] [PubMed]
- VS, A.; SK, K. Phloretin ameliorates acetic acid induced colitis through modulation of immune and inflammatory reactions in rats. Endocr. Metab. Immune Disord. Drug Targets (Former. Curr. Drug Targets-Immune Endocr. Metab. Disord.) 2021, 21, 163–172. [Google Scholar]
- Huang, W.-C.; Lai, C.-L.; Liang, Y.-T.; Hung, H.-C.; Liu, H.-C.; Liou, C.-J. Phloretin attenuates LPS-induced acute lung injury in mice via modulation of the NF-κB and MAPK pathways. Int. Immunopharmacol. 2016, 40, 98–105. [Google Scholar] [CrossRef]
- Jeon, D.; Jeong, M.-C.; Jnawali, H.N.; Kwak, C.; Ryoo, S.; Jung, I.D.; Kim, Y. Phloretin exerts anti-tuberculosis activity and suppresses lung inflammation. Molecules 2017, 22, 183. [Google Scholar] [CrossRef]
- Ahn, C.-B.; Jung, W.-K.; Park, S.-J.; Kim, Y.-T.; Kim, W.-S.; Je, J.-Y. Gallic acid-g-chitosan modulates inflammatory responses in LPS-stimulated RAW264. 7 cells via NF-κB, AP-1, and MAPK pathways. Inflammation 2016, 39, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Tang, Q.; Huang, H.; Hao, W.; Wei, X. Grape-seed proanthocyanidins inhibit the lipopolysaccharide-induced inflammatory mediator expression in RAW264. 7 macrophages by suppressing MAPK and NF-κb signal pathways. Environ. Toxicol. Pharmacol. 2016, 41, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M. Epigenetic regulation of redox signaling in diabetic retinopathy: Role of Nrf2. Free Radic. Biol. Med. 2017, 103, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Zhong, Q.; Kowluru, R.A. Epigenetic modifications of Nrf2-mediated glutamate–cysteine ligase: Implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic. Biol. Med. 2014, 75, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.P.; Sunilkumar, S.; Giordano, J.F.; Toro, A.L.; Barber, A.J.; Dennis, M.D. The stress response protein REDD1 promotes diabetes-induced oxidative stress in the retina by Keap1-independent Nrf2 degradation. J. Biol. Chem. 2020, 295, 7350–7361. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Guo, H.; Meng, S.; Zhu, B.; Fang, J.; Huang, J.; Chen, J.; Wang, Y.; Wang, L.; Yao, X. Klotho ameliorates diabetic nephropathy by activating Nrf2 signaling pathway in podocytes. Biochem. Biophys. Res. Commun. 2021, 534, 450–456. [Google Scholar] [CrossRef]
- Li, S.; Zheng, L.; Zhang, J.; Liu, X.; Wu, Z. Inhibition of ferroptosis by up-regulating Nrf2 delayed the progression of diabetic nephropathy. Free Radic. Biol. Med. 2021, 162, 435–449. [Google Scholar] [CrossRef]
- Song, D.; Liu, F.; Tao, W.; Wu, X.; Bi, H.; Li, X.; Shu, J.; Wang, D. Protective Effect of Phloretin against Hydrogen Peroxide-Induced Oxidative Damage by Enhancing Autophagic Flux in DF-1 Cells. Oxidative Med. Cell. Longev. 2022, 2022, 8359118. [Google Scholar] [CrossRef]
- Ying, Y.; Jin, J.; Ye, L.; Sun, P.; Wang, H.; Wang, X. Phloretin prevents diabetic cardiomyopathy by dissociating Keap1/Nrf2 complex and inhibiting oxidative stress. Front. Endocrinol. 2018, 9, 774. [Google Scholar] [CrossRef]
- Yang, Q.; Han, L.; Li, J.; Xu, H.; Liu, X.; Wang, X.; Pan, C.; Lei, C.; Chen, H.; Lan, X. Activation of Nrf2 by phloretin attenuates palmitic acid-induced endothelial cell oxidative stress via AMPK-dependent signaling. J. Agric. Food Chem. 2018, 67, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Shirgadwar, S.M.; Kumar, R.; Preeti, K.; Khatri, D.K.; Singh, S.B. Neuroprotective effect of phloretin in rotenone-induced mice model of Parkinson’s disease: Modulating mTOR-NRF2-p62 mediated autophagy-oxidative stress crosstalk. J. Alzheimer’s Dis. 2023, 94, S109–S124. [Google Scholar] [CrossRef] [PubMed]
- Nithiya, T.; Udayakumar, R. Hepato and renal protective effect of phloretin on streptozotocin induced diabetic rats. J. Biomed. Pharm. Sci. 2018, 1, 3–6. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Control | Control + PHLTM | STZ-T1DM | STZ-T1DM + PHLTM | ||
|---|---|---|---|---|---|
| Final body weight | 534.1 ± 53.2 | 547.6 ± 49.3 | 322.2 ± 36.5 ab | 449.4 ± 39.5 abc | |
| Plasma | Glucose (mg/dL) | 113.3 ± 10.3 | 92.5 ± 7.9 a | 344.2 ± 34.6 ab | 172.4 ± 25.4 abc |
| Insulin (ng/mL) | 4.3 ± 0.6 | 4.1 ± 0.5 | 2.1 ± 0.2 ab | 1.9 ± 0.2 abc | |
| Serum | TGs (mg/dL) | 83.4 ± 7.8 | 88.3 ± 8.2 | 169.3 ± 14.5 ab | 103.2 ± 15.9 abc |
| CHOL (mg/dL) | 81.9 ± 8.4 | 88.5 ± 9.1 | 212.2 ± 18.7 ab | 148.2 ± 20.1 abc | |
| LDL-c (mg/dL) | 48.5 ± 5.7 | 44.3 ± 4.9 | 110.2 ± 9.5 ab | 76.3 ± 10.5 abc | |
| HDL-c (mg/dL) | 38.4 ± 3.6 | 40.8 ± 5.1 | 22.2 ± 2.7 ab | 31.2 ± 3.9 abc | |
| FFAs (μmol/mg) | 367.6 ± 35.9 | 293.3 ± 24.5 a | 663.3 ± 58.9 ab | 447.4 ± 48.1 abc |
| Control | Control + PHLTM | STZ-T1DM | STZ-T1DM + PHLTM | |
|---|---|---|---|---|
| Serum | ||||
| Cr (μmol/L) | 58.8 ± 6.9 | 54.4 ± 5.7 | 134.5 ± 11.6 ab | 67.5 ± 7.7 abc |
| Albumin (g/dL) | 3.89 ± 0.4 | 4.11 ± 0.6 | 1.63 ± 0.2 ab | 3.11 ± 0.4 abc |
| Urine | ||||
| Volume (mL) | 13.6 ± 1.9 | 12.7 ± 1.7 | 7.4 ± 0.9 ab | 12.2 ± 1.2 c |
| Albumin (µg/dL) | 31.6 ± 2.5 | 34.5 ± 2.1 | 356.2 ± 23.2 ab | 46.3 ± 4.8 abc |
| Cr (µg/dL) | 101.7 ± 8.5 | 108.1 ± 9.8 | 54.3 ± 4.9 ab | 90.3 ± 8.6 abc |
| 8-OHdG (ng/mL) | 2.57 ± 0.4 | 2.73 ± 0.3 | 5.64 ± 0.6 ab | 3.22 ± 0.3 abc |
| KIM-1 (pg/mL) | 312.2 ± 27.5 | 324.4 ± 29.2 | 761.2 ± 66.5 ab | 415.9 ± 39.5 abc |
| Nephrin pg/mL) | 198.3 ± 20.4 | 212.4 ± 25.6 | 633.3 ± 54.6 ab | 274.4 ± 23.4 abc |
| NGAL (pg/mL) | 55.7 ± 6.7 | 51.2 ± 4.9 | 246.1 ± 22.8 ab | 88.9 ± 8.7 abc |
| Control | Control + PHLTM | STZ-T1DM | STZ-T1DM + PHLTM | |
|---|---|---|---|---|
| MDA (nmol/g) | 0.48 ± 0.05 | 0.51 ± 0.03 a | 1.66 ± 0.21 ab | 0.71 ± 0.06 abc |
| GSH (μg/g) | 46.7 ± 4.6 | 73.4 ± 5.8 a | 22.3 ± 3.1 ab | 48.5 ± 5.1 bc |
| SOD (U/g) | 14.6 ± 1.5 | 24.5 ± 2.1 a | 7.2 ± 0.8 ab | 15.2 ± 1.7 bc |
| HO-1 (pg/g) | 22.5 ± 2.5 | 36.8 ± 4.2 a | 10.9 ± 1.3 ab | 25.4 ± 2.1 bc |
| AGEs (ng/g) | 17.6 ± 1.1 | 15.4 ± 1.2 | 66.7 ± 4.9 ab | 27.4 ± 2.4 abc |
| TNF-α (pg/g) | 3.6 ± 0.42 | 4.2 ± 0.58 | 22.6 ± 1.9 ab | 8.8 ± 0.74 abc |
| IL-6 (pg/g) | 14.9 ± 1.3 | 16.4 ± 1.5 | 64.6 ± 3.7 ab | 22.6 ± 2.1 abc |
| Control | Control + PHLTM | STZ-T1DM | STZ-T1DM + PHLTM | |
|---|---|---|---|---|
| Bax (pg/g) | 8.3 ± 0.7 | 9.1 ± 0.7 | 33.5 ± 2.7 ab | 13.2 ± 1.1 abc |
| Bcl2 (nmol/g) | 14.5 ± 1.2 | 22.9 ± 1.9 a | 6.5 ± 0.5 ab | 12.4 ± 0.9 bc |
| Caspapse-3 (nmol/g) | 4.8 ± 0.5 | 5.1 ± 0.5 | 22.6 ± 1.8 ab | 7.6 ± 0.8 c |
| Bax/Bcl2 (%) | 0.58 ± 0.06 | 0.38 ± 0.05 a | 4.9 ± 0. 7 ab | 1.1 ± 0.1 abc |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Hussan, R.; Albadr, N.A.; Alshammari, G.M.; Almasri, S.A.; Alfayez, F.F.; Yahya, M.A. Phloretamide Protects against Diabetic Kidney Damage and Dysfunction in Diabetic Rats by Attenuating Hyperglycemia and Hyperlipidemia, Suppressing NF-κβ, and Upregulating Nrf2. Pharmaceutics 2024, 16, 505. https://doi.org/10.3390/pharmaceutics16040505
Al-Hussan R, Albadr NA, Alshammari GM, Almasri SA, Alfayez FF, Yahya MA. Phloretamide Protects against Diabetic Kidney Damage and Dysfunction in Diabetic Rats by Attenuating Hyperglycemia and Hyperlipidemia, Suppressing NF-κβ, and Upregulating Nrf2. Pharmaceutics. 2024; 16(4):505. https://doi.org/10.3390/pharmaceutics16040505
Chicago/Turabian StyleAl-Hussan, Rasha, Nawal A. Albadr, Ghedeir M. Alshammari, Soheir A. Almasri, Farah Fayez Alfayez, and Mohammed Abdo Yahya. 2024. "Phloretamide Protects against Diabetic Kidney Damage and Dysfunction in Diabetic Rats by Attenuating Hyperglycemia and Hyperlipidemia, Suppressing NF-κβ, and Upregulating Nrf2" Pharmaceutics 16, no. 4: 505. https://doi.org/10.3390/pharmaceutics16040505
APA StyleAl-Hussan, R., Albadr, N. A., Alshammari, G. M., Almasri, S. A., Alfayez, F. F., & Yahya, M. A. (2024). Phloretamide Protects against Diabetic Kidney Damage and Dysfunction in Diabetic Rats by Attenuating Hyperglycemia and Hyperlipidemia, Suppressing NF-κβ, and Upregulating Nrf2. Pharmaceutics, 16(4), 505. https://doi.org/10.3390/pharmaceutics16040505