

Mirk/Dyrk1B Kinase Inhibitors in Targeted Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
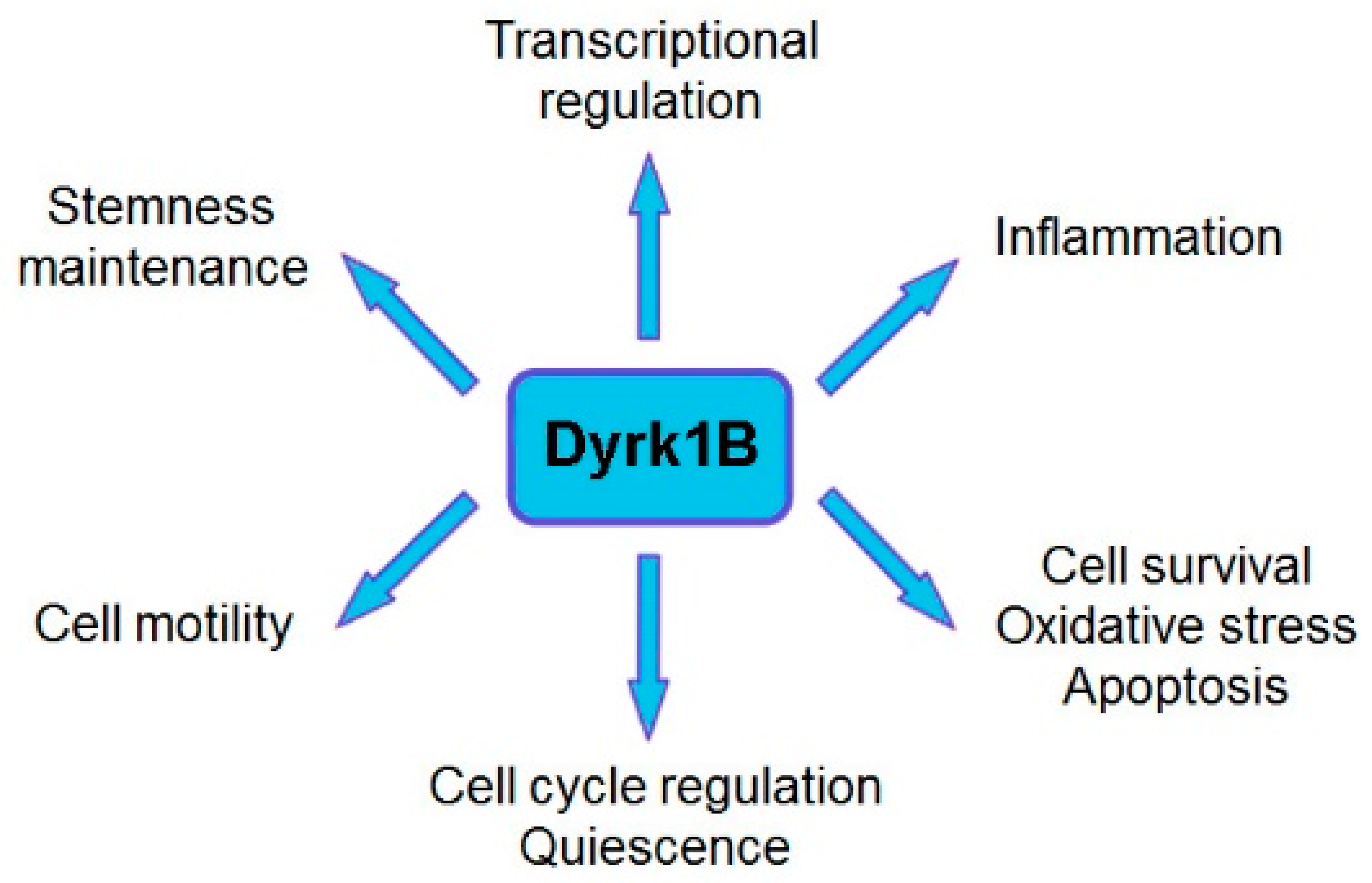
2. Mirk/Dyrk1B Kinase Is an Emerging Therapeutic Target in Cancer
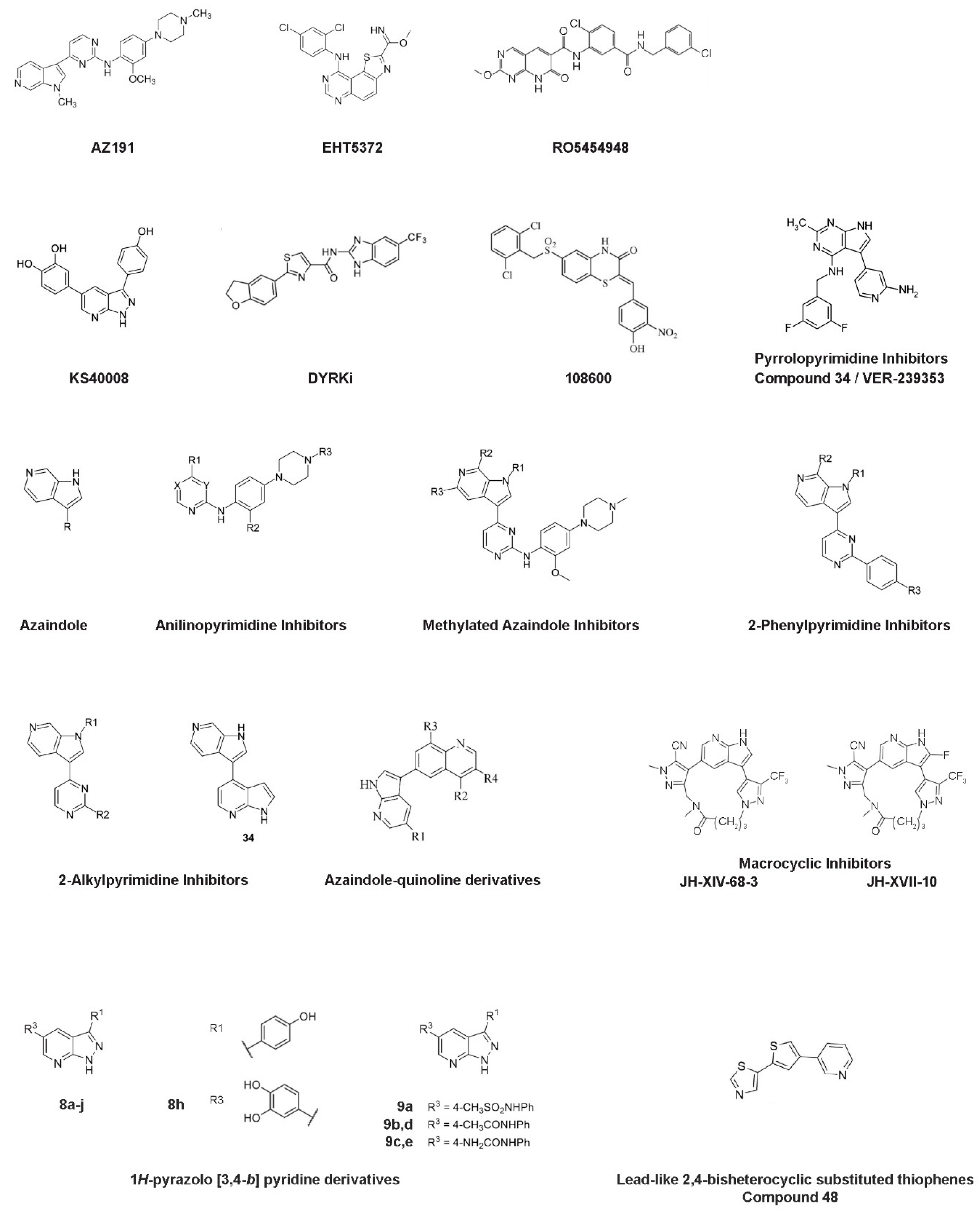
3. Mirk/Dyrk1B Kinase Inhibitors
3.1. AΖ191 Inhibitor
3.2. EHT5372 Inhibitor
3.3. RO5454948 Inhibitor
3.4. KS40008 Inhibitor
3.5. DYRKi Inhibitor
3.6. 108600 Inhibitor
3.7. Pyrrolopyrimidine Inhibitors—VER-239353 Inhibitor (Compound 34)
3.8. Anilinopyrimidine, Methylated Azaindole, 2-Phenylpyrimidine, and 2-Alkylpyrimidine Inhibitors
3.9. Azaindole-Quinoline-Based Inhibitors
3.10. Macrocyclic Inhibitors—JH-XVII-10 Inhibitor
3.11. 1H-pyrazolo [3,4-b] Pyridine Inhibitors—Compound 8h
3.12. Lead-Like 2,4-Bisheterocyclic Substituted Thiophenes Inhibitors—Compound 48
3.13. In Silico Identification of Potent and Selective Dyrk1B Inhibitors
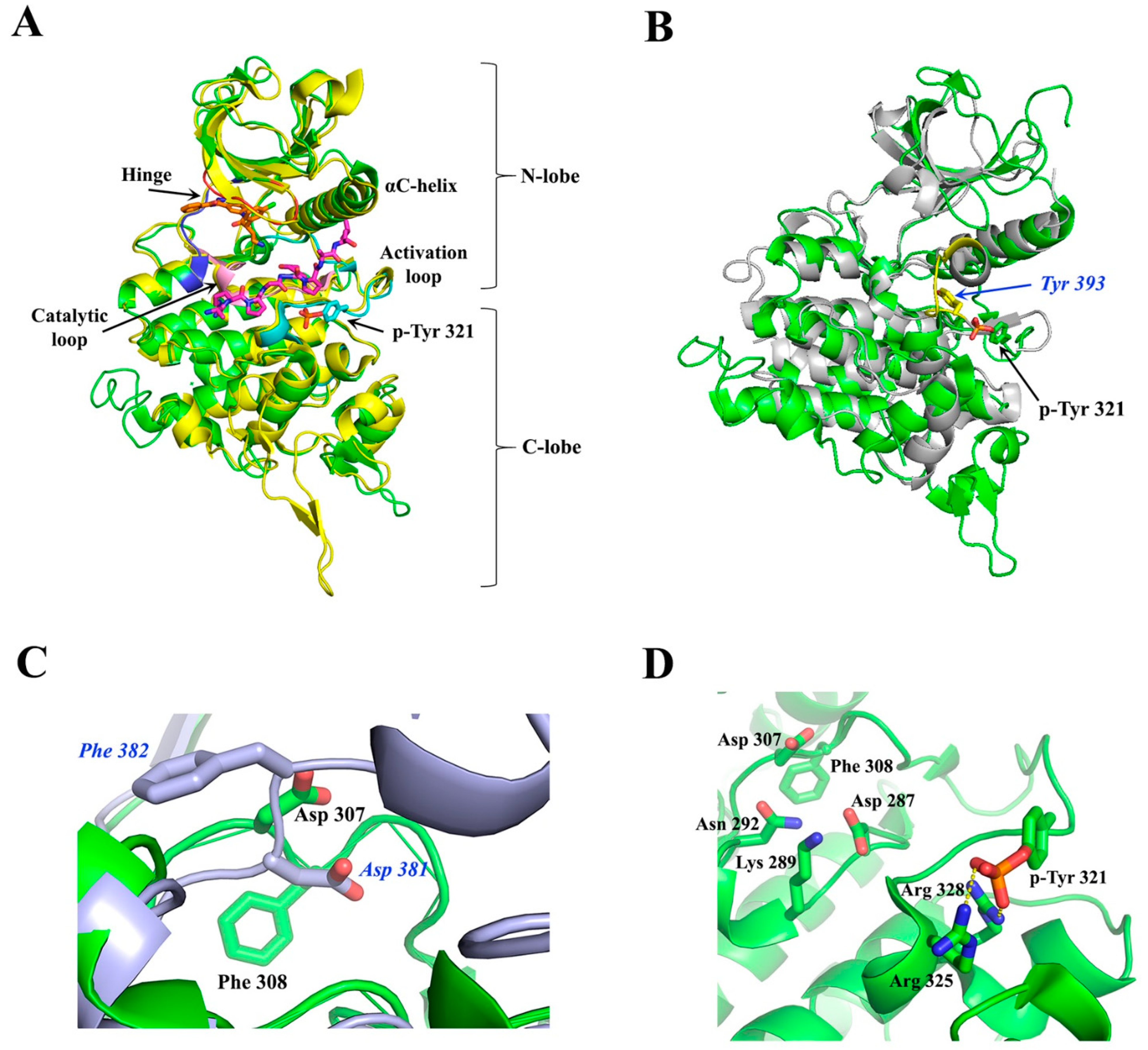
4. DYRK Structure, Crystallography Studies, and Inhibitor Selectivity
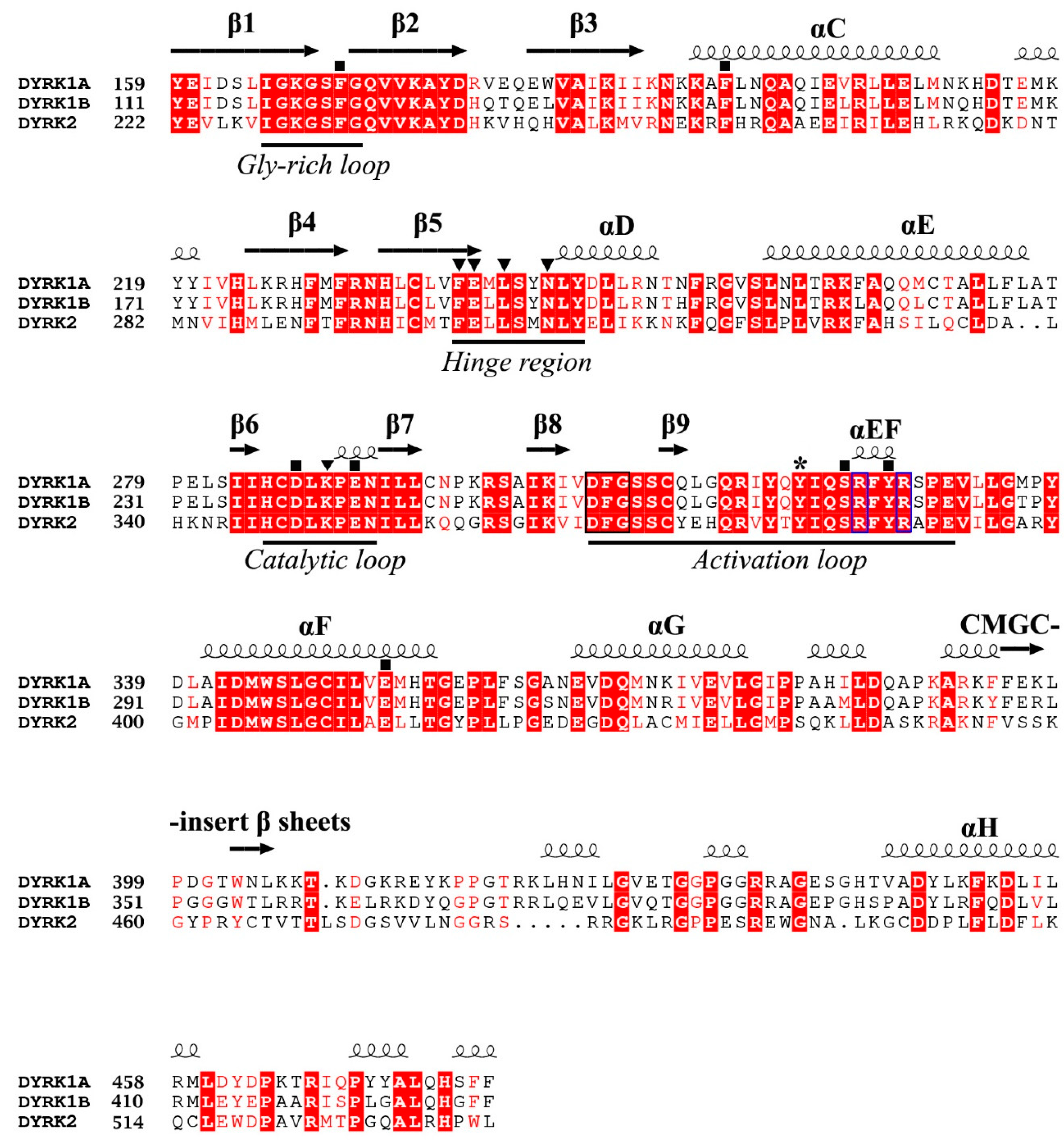
4.1. Overall Structure of the Kinase Domain of DYRKs and Activation Mechanism
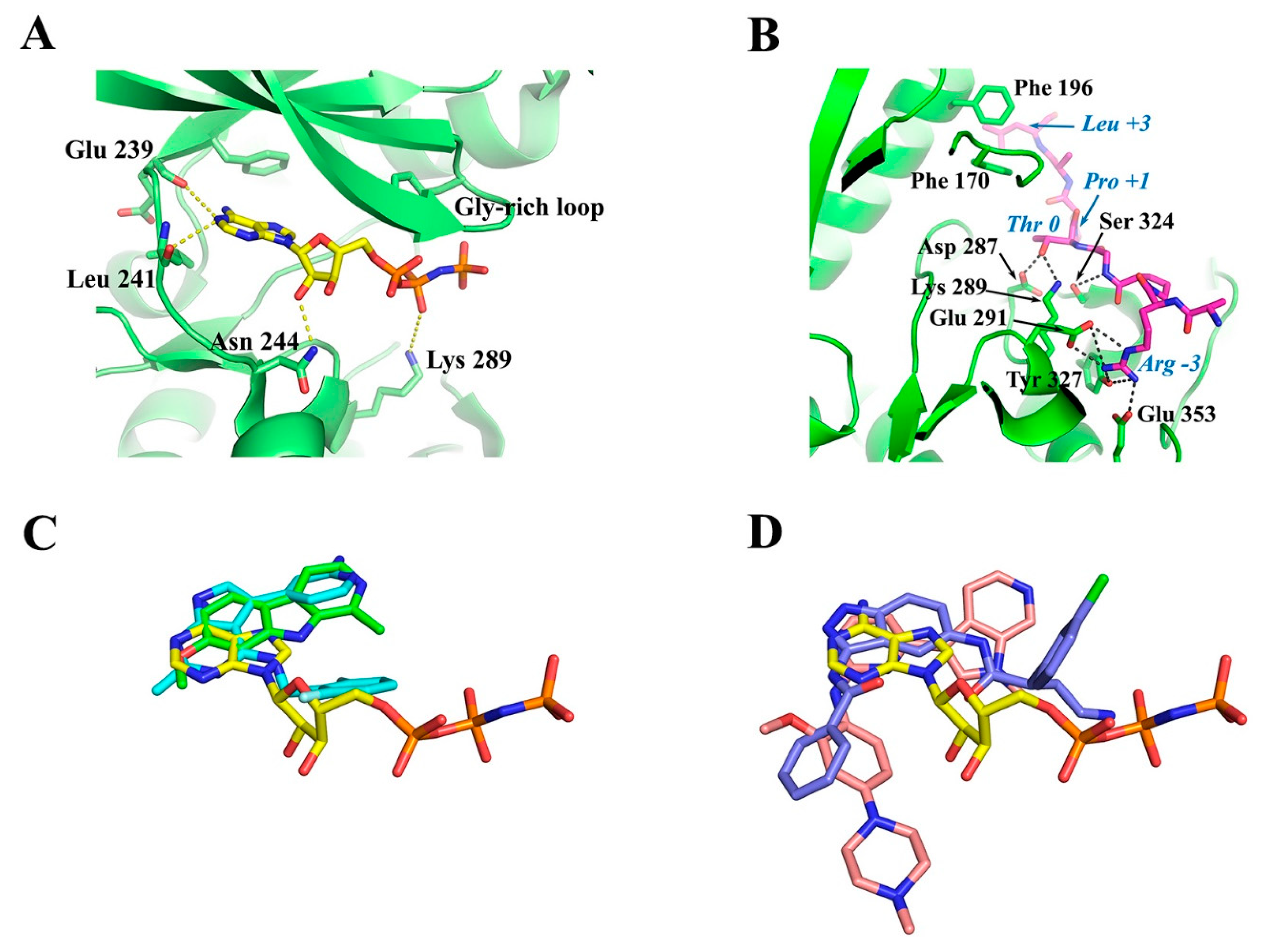
4.2. ATP-Binding Site of DYRKs
4.3. Substrate-Binding Site of DYRKs
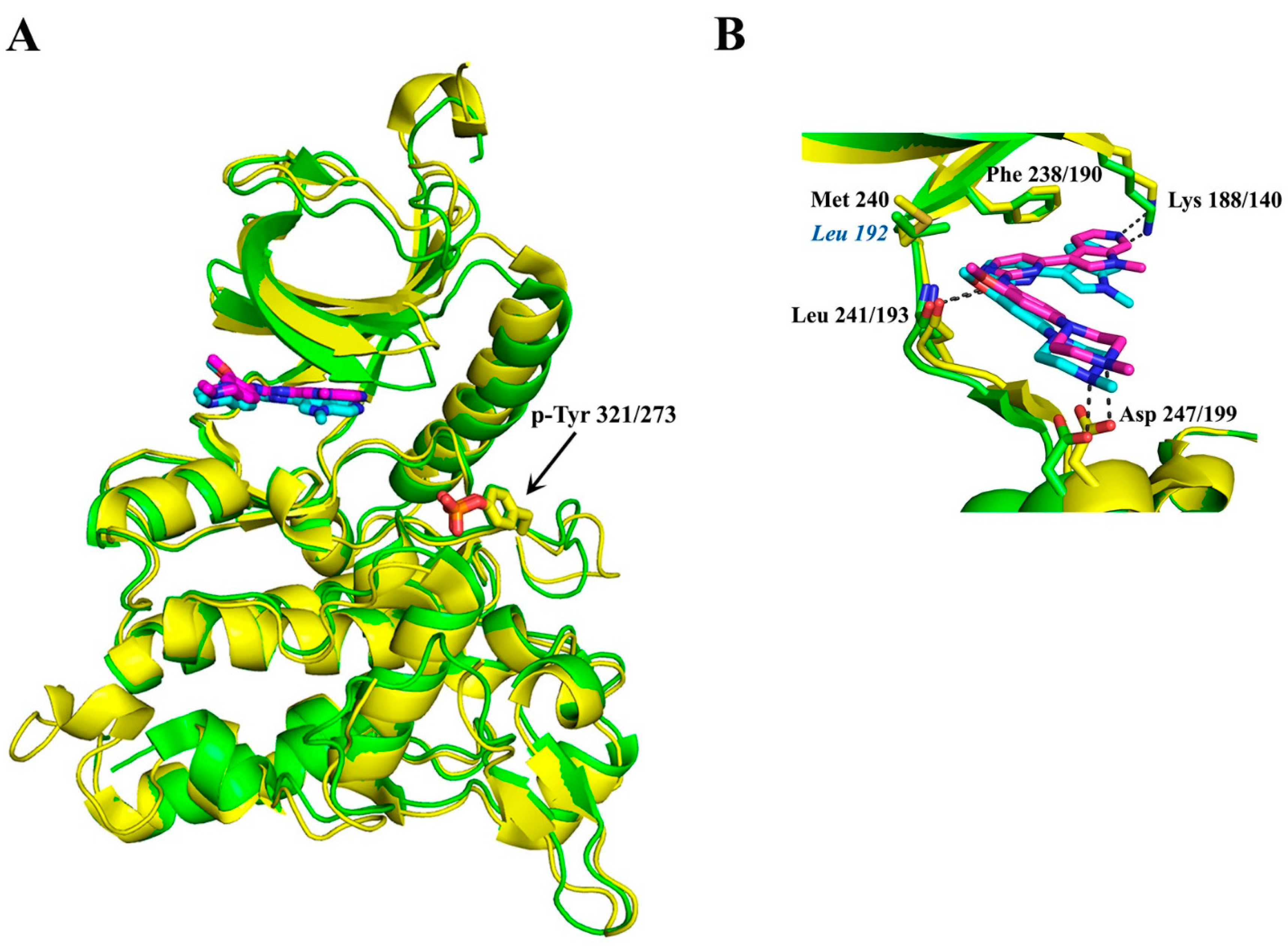
4.4. Development of Dyrk1A/B Inhibitors
4.5. Dyrk1B Crystal Structure
4.6. Current Limitations and Future Perspectives in the Development of Dyrk1B Inhibitors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dancey, J.; Sausville, E.A. Issues and Progress with Protein Kinase Inhibitors for Cancer Treatment. Nat. Rev. Drug Discov. 2003, 2, 296–313. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Scholar, E.M. Role of Tyrosine Kinase Inhibitors in Cancer Therapy. J. Pharmacol. Exp. Ther. 2005, 315, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.K. Therapeutic Protein Kinase Inhibitors. Cell. Mol. Life Sci. 2009, 66, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase Drug Discovery 20 Years after Imatinib. Nat. Rev. Drug Discov. 2022. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, L.G.; Ferreira, L.L.G.; Andricopulo, A.D. Recent Advances and Perspectives in Cancer Drug Design. An. Acad. Bras. Ciências 2018, 90, 1233–1250. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2023 Update. Pharmacol. Res. 2023, 187, 106552. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2024 Update. Pharmacol. Res. 2024, 200, 107059. [Google Scholar] [CrossRef] [PubMed]
- Valent, P. Imatinib-Resistant Chronic Myeloid Leukemia (CML): Current Concepts on Pathogenesis and New Emerging Pharmacologic Approaches. Biologics 2007, 1, 433. [Google Scholar]
- Aranda, S.; Laguna, A.; Luna, S. de la DYRK Family of Protein Kinases: Evolutionary Relationships, Biochemical Properties, and Functional Roles. FASEB J. 2011, 25, 449–462. [Google Scholar] [CrossRef]
- Soppa, U.; Becker, W. DYRK Protein Kinases. Curr. Biol. 2015, 25, R488–R489. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. Emerging Role of DYRK Family Protein Kinases as Regulators of Protein Stability in Cell Cycle Control. Cell Cycle 2012, 11, 3389–3394. [Google Scholar] [CrossRef] [PubMed]
- Mercer, S.E.; Friedman, E. Mirk/Dyrk1B: A Multifunctional Dual-Specificity Kinase Involved in Growth Arrest, Differentiation, and Cell Survival. Cell Biochem. Biophys. 2006, 45, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Arbones, M.L.; Thomazeau, A.; Nakano-Kobayashi, A.; Hagiwara, M.; Delabar, J.M. DYRK1A and Cognition: A Lifelong Relationship. Pharmacol. Ther. 2019, 194, 199–221. [Google Scholar] [CrossRef]
- Friedman, E. Mirk/Dyrk1B in Cancer. J. Cell. Biochem. 2007, 102, 274–279. [Google Scholar] [CrossRef]
- Becker, W. A Wake-up Call to Quiescent Cancer Cells—Potential Use of DYRK1B Inhibitors in Cancer Therapy. FEBS J. 2018, 285, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Song, W.J.; Chung, K.C. Function and Regulation of Dyrk1A: Towards Understanding Down Syndrome. Cell. Mol. Life Sci. 2009, 66, 3235–3240. [Google Scholar] [CrossRef]
- Friedman, E. Mirk/Dyrk1B Kinase in Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 5560–5575. [Google Scholar] [CrossRef]
- Deng, X.; Ewton, D.Z.; Pawlikowski, B.; Maimone, M.; Friedman, E. Mirk/Dyrk1B Is a Rho-Induced Kinase Active in Skeletal Muscle Differentiation. J. Biol. Chem. 2003, 278, 41347–41354. [Google Scholar] [CrossRef]
- Mercer, S.E.; Ewton, D.Z.; Deng, X.; Lim, S.; Mazur, T.R.; Friedman, E. Mirk/Dyrk1B Mediates Survival during the Differentiation of C2C12 Myoblasts. J. Biol. Chem. 2005, 280, 25788–25801. [Google Scholar] [CrossRef]
- Deng, X.; Ewton, D.Z.; Mercer, S.E.; Friedman, E. Mirk/Dyrk1B Decreases the Nuclear Accumulation of Class II Histone Deacetylases during Skeletal Muscle Differentiation. J. Biol. Chem. 2005, 280, 4894–4905. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Itoh, K.; Higashitsuji, H.; Higashitsuji, H.; Nakazawa, N.; Sakurai, T.; Liu, Y.; Tokuchi, H.; Fujita, T.; Zhao, Y.; et al. Cold-Inducible RNA-Binding Protein (Cirp) Interacts with Dyrk1b/Mirk and Promotes Proliferation of Immature Male Germ Cells in Mice. Proc. Natl. Acad. Sci. USA 2012, 109, 10885–10890. [Google Scholar] [CrossRef] [PubMed]
- Keramati, A.R.; Fathzadeh, M.; Go, G.W.; Singh, R.; Choi, M.; Faramarzi, S.; Mane, S.; Kasaei, M.; Sarajzadeh-Fard, K.; Hwa, J.; et al. A Form of the Metabolic Syndrome Associated with Mutations in DYRK1B. N. Engl. J. Med. 2014, 370, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Hickmott, J. DYRK1B Variant Linked to Autosomal Dominant Metabolic Syndrome. Clin. Genet. 2015, 87, 30–31. [Google Scholar] [CrossRef] [PubMed]
- Abu Jhaisha, S.; Widowati, E.W.; Kii, I.; Sonamoto, R.; Knapp, S.; Papadopoulos, C.; Becker, W. DYRK1B Mutations Associated with Metabolic Syndrome Impair the Chaperone-Dependent Maturation of the Kinase Domain. Sci. Rep. 2017, 7, 6420. [Google Scholar] [CrossRef] [PubMed]
- Armanmehr, A.; Jafari Khamirani, H.; Zoghi, S.; Dianatpour, M. Analysis of DYRK1B, PPARG, and CEBPB Expression Patterns in Adipose-Derived Stem Cells from Patients Carrying DYRK1B R102C and Healthy Individuals During Adipogenesis. Metab. Syndr. Relat. Disord. 2022, 20, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Tsioras, K.; Papastefanaki, F.; Politis, P.K.; Matsas, R.; Gaitanou, M. Functional Interactions between BM88/Cend1, Ran-Binding Protein M and Dyrk1B Kinase Affect Cyclin D1 Levels and Cell Cycle Progression/Exit in Mouse. PLoS ONE 2013, 8, e82172. [Google Scholar] [CrossRef] [PubMed]
- Gaitanou, M.; Segklia, K.; Matsas, R. CEND1, a Story with Many Tales: From Regulation of Cell Cycle Progression/Exit of Neural Stem Cells to Brain Structure and Function. Stem Cells Int. 2019, 2019, 2054783. [Google Scholar] [CrossRef] [PubMed]
- Kokkorakis, N.; Gaitanou, M. Minibrain-Related Kinase/Dual-Specificity Tyrosine-Regulated Kinase 1B Implication in Stem/Cancer Stem Cells Biology. World J. Stem Cells 2020, 12, 1553. [Google Scholar] [CrossRef]
- Kokkorakis, N.; Douka, K.; Nalmpanti, A.; Politis, P.K.; Zagoraiou, L.; Matsas, R.; Gaitanou, M. Mirk/Dyrk1B Controls Ventral Spinal Cord Development via Shh Pathway. Cell. Mol. Life Sci. 2024, 81. [Google Scholar] [CrossRef]
- Lee, K.; Deng, X.; Friedman, E. Mirk Protein Kinase Is a Mitogen-Activated Protein Kinase Substrate That Mediates Survival of Colon Cancer Cells. Cancer Res. 2000, 60, 3631–3637. [Google Scholar]
- Jin, K.; Park, S.; Ewton, D.Z.; Friedman, E. The Survival Kinase Mirk/Dyrk1B Is a Downstream Effector of Oncogenic K-Ras in Pancreatic Cancer. Cancer Res. 2007, 67, 7247–7255. [Google Scholar] [CrossRef]
- Hu, J.; Nakhla, H.; Friedman, E. Transient Arrest in a Quiescent State Allows Ovarian Cancer Cells to Survive Suboptimal Growth Conditions and Is Mediated by Both Mirk/Dyrk1b and P130/RB2. Int. J. Cancer 2011, 129, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yang, X.; Yin, P.; Hu, W.; Liao, H.; Miao, Z.; Pan, C.; Li, N. The Involvement of FoxO in Cell Survival and Chemosensitivity Mediated by Mirk/Dyrk1B in Ovarian Cancer. Int. J. Oncol. 2012, 40, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.J.; Sheppard, K.E.; Pearson, R.B.; Campbell, I.G.; Gorringe, K.L.; Simpson, K.J. Functional Analysis of Genes in Regions Commonly Amplified in High-Grade Serous and Endometrioid Ovarian Cancer. Clin. Cancer Res. 2013, 19, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Deng, H.; Friedman, E.A. Ovarian Cancer Cells, Not Normal Cells, Are Damaged by Mirk/Dyrk1B Kinase Inhibition. Int. J. Cancer 2013, 132, 2258–2269. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, S.; He, Z.; Sun, F.; Huang, Y.; Ni, Q.; Wang, H.; Wang, Y.; Cheng, C. Dyrk1B Overexpression Is Associated with Breast Cancer Growth and a Poor Prognosis. Hum. Pathol. 2017, 66, 48–58. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Gu, J.; Zhu, J.; Wang, X.; Wang, C.; Duan, C.; Ni, Y.; Lu, X.; Li, J. Up-Regulation of Dyrk1b Promote Astrocyte Activation Following Lipopolysaccharide-Induced Neuroinflammation. Neuropeptides 2018, 69, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Blom, K.; Rubin, J.; Berglund, M.; Jarvius, M.; Lenhammar, L.; Parrow, V.; Andersson, C.; Loskog, A.; Fryknäs, M.; Nygren, P.; et al. Mebendazole-Induced M1 Polarisation of THP-1 Macrophages May Involve DYRK1B Inhibition. BMC Res. Notes 2019, 12, 234. [Google Scholar] [CrossRef]
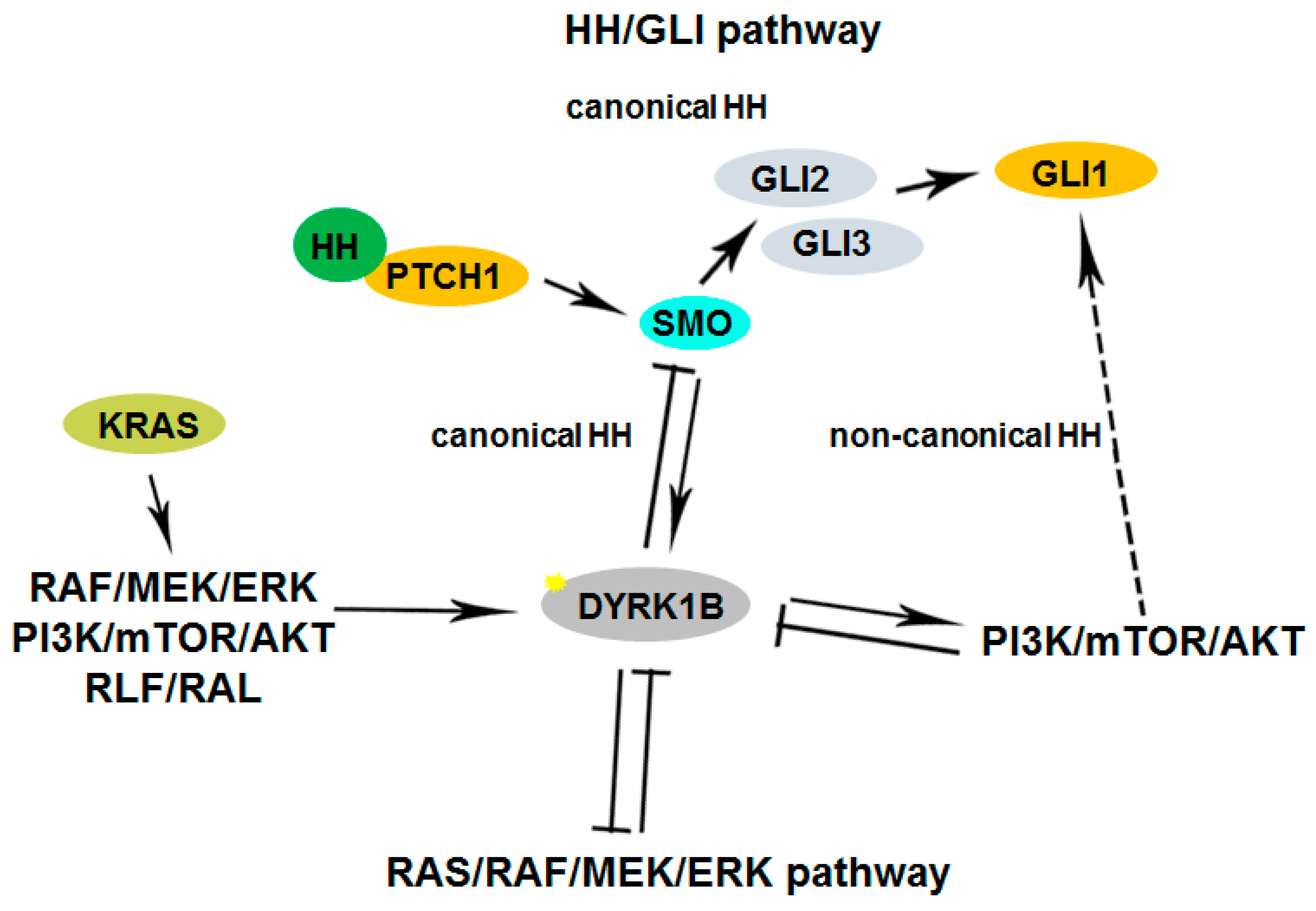
- Lauth, M.; Bergström, Å.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgård, R. DYRK1B-Dependent Autocrine-to-Paracrine Shift of Hedgehog Signaling by Mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar] [CrossRef]
- Gruber, W.; Hutzinger, M.; Elmer, D.P.; Parigger, T.; Sternberg, C.; Cegielkowski, L.; Zaja, M.; Leban, J.; Michel, S.; Hamm, S.; et al. DYRK1B as Therapeutic Target in Hedgehog/GLI-Dependent Cancer Cells with Smoothened Inhibitor Resistance. Oncotarget 2016, 7, 7134–7148. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B Blocks Canonical and Promotes Non-Canonical Hedgehog Signaling through Activation of the MTOR/AKT Pathway. Oncotarget 2017, 8, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Holz, P.S.; Roth, K.; Hupfer, A.; Meissner, W.; Müller, R.; Buchholz, M.; Gress, T.M.; Elsässer, H.P.; Jacob, R.; et al. DYRK1B Regulates Hedgehog-Induced Microtubule Acetylation. Cell. Mol. Life Sci. 2019, 76, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Hu, J.; Ewton, D.Z.; Friedman, E. Mirk/Dyrk1B Kinase Is Upregulated Following Inhibition of MTOR. Carcinogenesis 2014, 35, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhao, Y.; Lv, Y.; Chen, Y.; Wei, B.; Tian, J.; Yang, Z.; Kong, F.; Pang, J.; Liu, J.; et al. Mirk/Dyrk1B Mediates G0/G1 to S Phase Cell Cycle Progression and Cell Survival Involving MAPK/ERK Signaling in Human Cancer Cells. Cancer Cell Int. 2013, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Ashford, A.L.; Dunkley, T.P.J.; Cockerill, M.; Rowlinson, R.A.; Baak, L.M.; Gallo, R.; Balmanno, K.; Goodwin, L.M.; Ward, R.A.; Lochhead, P.A.; et al. Identification of DYRK1B as a Substrate of ERK1/2 and Characterisation of the Kinase Activity of DYRK1B Mutants from Cancer and Metabolic Syndrome. Cell. Mol. Life Sci. 2016, 73, 883–900. [Google Scholar] [CrossRef] [PubMed]
- Leder, S.; Czajkowska, H.; Maenz, B.; De Graaf, K.; Barthel, A.; Joost, H.G.; Becker, W. Alternative Splicing Variants of Dual Specificity Tyrosine Phosphorylated and Regulated Kinase 1B Exhibit Distinct Patterns of Expression and Functional Properties. Biochem. J. 2003, 372, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ewton, D.Z.; Deng, X.; Mercer, S.E.; Friedman, E. Mirk/Dyrk1B Kinase Destabilizes Cyclin D1 by Phosphorylation at Threonine 288. J. Biol. Chem. 2004, 279, 27790–27798. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Frattini, V.; Bansal, M.; Castano, A.M.; Sherman, D.; Hutchinson, K.; Bruce, J.N.; Califano, A.; Liu, G.; Cardozo, T.; et al. An ID2-Dependent Mechanism for VHL Inactivation in Cancer. Nature 2016, 529, 172–177. [Google Scholar] [CrossRef]
- Gao, J.; Zheng, Z.; Rawal, B.; Schell, M.J.; Bepler, G.; Haura, E.B. Mirk/Dyrk1B, a Novel Therapeutic Target, Mediates Cell Survival in Non-Small Cell Lung Cancer Cells. Cancer Biol. Ther. 2009, 8, 1671–1679. [Google Scholar] [CrossRef]
- Pérez-Sánchez, G.; Jiménez, A.; Quezada-Ramírez, M.A.; Estudillo, E.; Ayala-Sarmiento, A.E.; Mendoza-Hernández, G.; Hernández-Soto, J.; Hernández-Hernández, F.C.; Cázares-Raga, F.E.; Segovia, J. Annexin A1, Annexin A2, and Dyrk 1B Are Upregulated during GAS1-Induced Cell Cycle Arrest. J. Cell. Physiol. 2018, 233, 4166–4182. [Google Scholar] [CrossRef] [PubMed]
- MacKeigan, J.P.; Murphy, L.O.; Blenis, J. Sensitized RNAi Screen of Human Kinases and Phosphatases Identifies New Regulators of Apoptosis and Chemoresistance. Nat. Cell Biol. 2005, 7, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Ewton, D.Z.; Park, S.; Hu, J.; Friedman, E. Mirk Regulates the Exit of Colon Cancer Cells from Quiescence. J. Biol. Chem. 2009, 284, 22916–22925. [Google Scholar] [CrossRef] [PubMed]
- Song, L.N.; Silva, J.; Koller, A.; Rosenthal, A.; Chen, E.I.; Gelmann, E.P. The Tumor Suppressor NKX3.1 Is Targeted for Degradation by DYRK1B Kinase. Mol. Cancer Res. 2015, 13, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Bowen, C.; Ostrowski, M.C.; Leone, G.; Gelmann, E.P. Loss of PTEN Accelerates NKx3.1 Degradation to Promote Prostate Cancer Progression. Cancer Res. 2019, 79, 4124–4134. [Google Scholar] [CrossRef] [PubMed]
- Bowen, C.; Shibata, M.; Zhang, H.; Bergren, S.K.; Shen, M.M.; Gelmann, E.P. CRISPR/Cas9-Mediated Point Mutation in Nkx3.1 Prolongs Protein Half-Life and Reverses Effects Nkx3.1 Allelic Loss. Cancer Res. 2020, 80, 4805–4814. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Ewton, D.Z.; Li, S.; Naqvi, A.; Mercer, S.E.; Landas, S.; Friedman, E. The Kinase Mirk/Dyrk1B Mediates Cell Survival in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2006, 66, 4149–4158. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Ewton, D.Z.; Friedman, E. Mirk Dyrk1B Maintains the Viability of Quiescent Pancreatic Cancer Cells by Reducing Levels of Reactive Oxygen Species. Cancer Res. 2009, 69, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Ewton, D.Z.; Hu, J.; Vilenchik, M.; Deng, X.; Luk, K.C.; Polonskaia, A.; Hoffman, A.F.; Zipf, K.; Boylan, J.F.; Friedman, E.A. Inactivation of Mirk/Dyrk1b Kinase Targets Quiescent Pancreatic Cancer Cells. Mol. Cancer Ther. 2011, 10, 2104–2114. [Google Scholar] [CrossRef]
- Deng, X.; Friedman, E. Mirk Kinase Inhibition Blocks the in Vivo Growth of Pancreatic Cancer Cells. Genes Cancer 2014, 5, 337–347. [Google Scholar] [CrossRef]
- Mercer, S.E.; Ewton, D.Z.; Shah, S.; Naqvi, A.; Friedman, E. Mirk/Dyrk1b Mediates Cell Survival in Rhabdomyosarcomas. Cancer Res. 2006, 66, 5143–5150. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ji, D.; Weinstein, E.J.; Choy, E.; Hornicek, F.J.; Wood, K.B.; Liu, X.; Mankin, H.; Duan, Z. The Kinase Mirk Is a Potential Therapeutic Target in Osteosarcoma. Carcinogenesis 2010, 31, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shen, J.; Choy, E.; Hornicek, F.J.; Shan, A.; Duan, Z. Targeting DYRK1B Suppresses the Proliferation and Migration of Liposarcoma Cells. Oncotarget 2018, 9, 13154–13166. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wang, Y.; Strom, A.; Gustafsson, J.A.; Guan, X. Lapatinib Induces P27Kip1-Dependent G1 Arrest through Both Transcriptional and Post-Translational Mechanisms. Cell Cycle 2013, 12, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chiu, C.C.; Liu, P.F.; Wu, C.H.; Tseng, Y.C.; Lee, C.H.; Shu, C.W. Kinome-Wide SiRNA Screening Identifies DYRK1B as a Potential Therapeutic Target for Triple-Negative Breast Cancer Cells. Cancers 2021, 13, 5779. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Yuan, S.; Wang, R.; Zhang, W.; Chen, J.J. Role of Dual Specificity Tyrosine-Phosphorylation-Regulated Kinase 1B (Dyrk1B) in S-Phase Entry of HPV E7 Expressing Cells from Quiescence. Oncotarget 2015, 6, 30745–30761. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, K.; Zhu, X.; Lin, G.; Song, F.; Zhao, Y.; Piao, Y.; Liu, J.; Cheng, W.; Bi, X.; et al. Encorafenib (LGX818), a Potent BRAF Inhibitor, Induces Senescence Accompanied by Autophagy in BRAFV600E Melanoma Cells. Cancer Lett. 2016, 370, 332–344. [Google Scholar] [CrossRef]
- Ashford, A.L.; Oxley, D.; Kettle, J.; Hudson, K.; Guichard, S.; Cook, S.J.; Lochhead, P.A. A Novel DYRK1B Inhibitor AZ191 Demonstrates That DYRK1B Acts Independently of GSK3Β to Phosphorylate Cyclin D1 at Thr286, Not Thr 288. Biochem. J. 2014, 457, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Mercer, S.E.; Shah, S.; Ewton, D.Z.; Friedman, E. The Cyclin-Dependent Kinase Inhibitor P27Kip1 Is Stabilized in G0 by Mirk/Dyrk1B Kinase. J. Biol. Chem. 2004, 279, 22498–22504. [Google Scholar] [CrossRef]
- Litovchick, L.; Florens, L.A.; Swanson, S.K.; Washburn, M.P.; Decaprio, J.A. DYRK1A Protein Kinase Promotes Quiescence and Senescence through DREAM Complex Assembly. Genes Dev. 2011, 25, 801–813. [Google Scholar] [CrossRef]
- Hu, J.; Friedman, E. Depleting Mirk Kinase Increases Cisplatin Toxicity in Ovarian Cancer Cells. Genes Cancer 2010, 1, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zykova, T.A.; Peng, C.; Zhang, J.; Cho, Y.Y.; Zheng, D.; Yao, K.; Ma, W.Y.; Lau, A.T.Y.; Bode, A.M.; et al. Phosphorylation of H2AX at Ser139 and a New Phosphorylation Site Ser16 by RSK2 Decreases H2AX Ubiquitination and Inhibits Cell Transformation. Cancer Res. 2011, 71, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of Oxidative Stress as an Anticancer Strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E. The Kinase Mirk/Dyrk1B: A Possible Therapeutic Target in Pancreatic Cancer. Cancers 2010, 2, 1492–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wei, J.R.; Song, Y.; Fang, S.; Du, Y.; Li, Z.; Zeng, T.T.; Zhu, Y.H.; Li, Y.; Guan, X.Y. TROAP Switches DYRK1 Activity to Drive Hepatocellular Carcinoma Progression. Cell Death Dis. 2021, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, C.; Milbradt, J.; Hamilton, S.; Zaja, M.; Leban, J.; Henry, C.; Vitt, D.; Steingruber, M.; Sonntag, E.; Zeitträger, I.; et al. Inhibitors of Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases (DYRK) Exert a Strong Anti-Herpesviral Activity. Antiviral Res. 2017, 143, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Beckers, C.; Vasilikos, L.; Sanchez Fernandez, A.; Moor, L.; Pruschy, M. Targeting the Survival Kinase DYRK1B: A Novel Approach to Overcome Radiotherapy-Related Treatment Resistance. Radiother. Oncol. 2023, 190, 110039. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhang, P.; Pang, L.; Chen, J.; Sun, W.; Qi, W.; Lyu, Y.; Guan, H.; Gao, J. Expression and Clinical Significance of Dyrk1b in the Specimens and Cells of Cervical Lesions. Zhonghua Fu Chan Ke Za Zhi 2016, 51, 40–45. [Google Scholar] [CrossRef]
- Lee Walmsley, D.; Murray, J.B.; Dokurno, P.; Massey, A.J.; Benwell, K.; Fiumana, A.; Foloppe, N.; Ray, S.; Smith, J.; Surgenor, A.E.; et al. Fragment-Derived Selective Inhibitors of Dual-Specificity Kinases DYRK1A and DYRK1B. J. Med. Chem. 2021, 64, 8971–8991. [Google Scholar] [CrossRef]
- Kettle, J.G.; Ballard, P.; Bardelle, C.; Cockerill, M.; Colclough, N.; Critchlow, S.E.; Debreczeni, J.; Fairley, G.; Fillery, S.; Graham, M.A.; et al. Discovery and Optimization of a Novel Series of Dyrk1b Kinase Inhibitors to Explore a MEK Resistance Hypothesis. J. Med. Chem. 2015, 58, 2834–2844. [Google Scholar] [CrossRef]
- Saluja, T.S.; Kumar, V.; Agrawal, M.; Tripathi, A.; Meher, R.K.; Srivastava, K.; Gupta, A.; Singh, A.; Chaturvedi, A.; Singh, S.K. Mitochondrial Stress-Mediated Targeting of Quiescent Cancer Stem Cells in Oral Squamous Cell Carcinoma. Cancer Manag. Res. 2020, 12, 4519–4530. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Chu, C.; Chen, M.; Shi, Z.; Gan, S.; Qu, F.; Pan, X.; Yang, Q.; Tian, Y.; Wang, L.; et al. TROAP Regulates Prostate Cancer Progression via the WNT3/Survivin Signalling Pathways. Oncol. Rep. 2019, 41, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Mao, Q.; Ma, P. Decreased Expression of TROAP Suppresses Cellular Proliferation, Migration and Invasion in Gastric Cancer. Mol. Med. Rep. 2018, 18, 3020–3026. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Lv, H. MicroRNA-519d-3p Inhibits Cell Proliferation and Migration by Targeting TROAP in Colorectal Cancer. Biomed. Pharmacother. 2018, 105, 879–886. [Google Scholar] [CrossRef]
- Jiao, Y.; Li, Y.; Lu, Z.; Liu, Y. High Trophinin-Associated Protein Expression Is an Independent Predictor of Poor Survival in Liver Cancer. Dig. Dis. Sci. 2019, 64, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Xu, L.; Chen, Y.; Luo, S.J.; Wu, Y.; Xu, S.H.; Liu, M.T.; Lin, F.; Mei, Y.; Yang, Q.; et al. The Upregulation of Trophinin-Associated Protein (TROAP) Predicts a Poor Prognosis in Hepatocellular Carcinoma. J. Cancer 2019, 10, 957. [Google Scholar] [CrossRef]
- Hwang, J.; Park, A.; Kim, C.; Yu, D.; Byun, H.; Ku, M.; Yang, J.; Kim, T.I.; Jeong, K.S.; Kim, K.Y.; et al. Suppression of DYRK1A/B Drives Endoplasmic Reticulum Stress-Mediated Autophagic Cell Death through Metabolic Reprogramming in Colorectal Cancer Cells. Anticancer Res. 2022, 42, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.N.; Dhanyamraju, P.K. Role of Aberrant Sonic Hedgehog Signaling Pathway in Cancers and Developmental Anomalies. J. Biomed. Res. 2022, 36, 1. [Google Scholar] [CrossRef]
- Sato, K.; Padgaonkar, A.A.; Baker, S.J.; Cosenza, S.C.; Rechkoblit, O.; Subbaiah, D.R.C.V.; Domingo-Domenech, J.; Bartkowski, A.; Port, E.R.; Aggarwal, A.K.; et al. Simultaneous CK2/TNIK/DYRK1 Inhibition by 108600 Suppresses Triple Negative Breast Cancer Stem Cells and Chemotherapy-Resistant Disease. Nat. Commun. 2021, 12, 4671. [Google Scholar] [CrossRef]
- Massey, A.J.; Benwell, K.; Burbridge, M.; Kotschy, A.; Walmsley, D.L. Targeting DYRK1A/B Kinases to Modulate P21-Cyclin D1-P27 Signalling and Induce Anti-Tumour Activity in a Model of Human Glioblastoma. J. Cell. Mol. Med. 2021, 25, 10650–10662. [Google Scholar] [CrossRef] [PubMed]
- Szamborska-Gbur, A.; Rutkowska, E.; Dreas, A.; Frid, M.; Vilenchik, M.; Milik, M.; Brzózka, K.; Król, M. How to Design Potent and Selective DYRK1B Inhibitors? Molecular Modeling Study. J. Mol. Model. 2019, 25, 41. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.E.; Hatcher, J.M.; Jiang, J.; Vatsan, P.S.; Che, J.; Gray, N.S. Selective Macrocyclic Inhibitors of DYRK1A/B. ACS Med. Chem. Lett. 2022, 13, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Hwang, J.; Lee, J.Y.; Heo, E.J.; Na, Y.J.; Kang, S.; Jeong, K.S.; Kim, K.Y.; Shin, S.J.; Lee, H. Synthesis of Novel 1H-Pyrazolo[3,4-b] Pyridine Derivatives as DYRK 1A/1B Inhibitors. Bioorg. Med. Chem. Lett. 2021, 47, 128226. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Kail, D.; Mariano, M.; Empting, M.; Weber, N.; Paul, T.; Hartmann, R.W.; Engel, M. Design and Synthesis of a Library of Lead-like 2,4-Bisheterocyclic Substituted Thiophenes as Selective Dyrk/Clk Inhibitors. PLoS ONE 2014, 9, 87851. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, V.; Vilenchik, M.; Kantidze, O.; Tsutskiridze, N.; Kharchilava, D.; Lhewa, P.; Shishkin, A.; Gankin, Y.; Kirpich, A. Novel Efficient Multistage Lead Optimization Pipeline Experimentally Validated for DYRK1B Selective Inhibitors. J. Med. Chem. 2022, 65, 13784–13792. [Google Scholar] [CrossRef]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H.; et al. Development of a Novel Selective Inhibitor of the Down Syndrome-Related Kinase Dyrk1A. Nat. Commun. 2010, 1, 86. [Google Scholar] [CrossRef]
- Soundararajan, M.; Roos, A.K.; Savitsky, P.; Filippakopoulos, P.; Kettenbach, A.N.; Olsen, J.V.; Gerber, S.A.; Eswaran, J.; Knapp, S.; Elkins, J.M. Structures of down Syndrome Kinases, DYRKs, Reveal Mechanisms of Kinase Activation and Substrate Recognition. Structure 2013, 21, 986–996. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Sibbet, G.; Morrice, N.; Cleghon, V. Activation-Loop Autophosphorylation Is Mediated by a Novel Transitional Intermediate Form of DYRKs. Cell 2005, 121, 925–936. [Google Scholar] [CrossRef]
- Cowan-Jacob, S.W.; Fendrich, G.; Floersheimer, A.; Furet, P.; Liebetanz, J.; Rummel, G.; Rheinberger, P.; Centeleghe, M.; Fabbro, D.; Manley, P.W. Structural Biology Contributions to the Discovery of Drugs to Treat Chronic Myelogenous Leukaemia. Acta Crystallogr. D. Biol. Crystallogr. 2007, 63, 80–93. [Google Scholar] [CrossRef]
- Grygier, P.; Pustelny, K.; Menezes, F.; Jemiola-Rzeminska, M.; Suder, P.; Dubin, G.; Czarna, A. Structural Perspective on the Design of Selective DYRK1B Inhibitors. bioRxiv 2024, preprint. [Google Scholar] [CrossRef]
- Zheng, J.; Trafny, E.A.; Knighton, D.R.; Xuong, N.; Taylor, S.S.; Ten Eyck, L.F.; Sowadski, J.M. 2.2 A Refined Crystal Structure of the Catalytic Subunit of CAMP-Dependent Protein Kinase Complexed with MnATP and a Peptide Inhibitor. Acta Crystallogr. D. Biol. Crystallogr. 1993, 49, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Modugno, M.; Casale, E.; Soncini, C.; Rosettani, P.; Colombo, R.; Lupi, R.; Rusconi, L.; Fancelli, D.; Carpinelli, P.; Cameron, A.D.; et al. Crystal Structure of the T315I Abl Mutant in Complex with the Aurora Kinases Inhibitor PHA-739358. Cancer Res. 2007, 67, 7987–7990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Adrián, F.J.; Jahnke, W.; Cowan-Jacob, S.W.; Li, A.G.; Iacob, R.E.; Sim, T.; Powers, J.; Dierks, C.; Sun, F.; et al. Targeting Bcr–Abl by Combining Allosteric with ATP-Binding-Site Inhibitors. Nature 2010, 463, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, K.; Kii, I. Druggable Transient Pockets in Protein Kinases. Molecules 2021, 26, 651. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, Y.; Wang, J.; Ren, C.; Chen, H.; Zhang, J. DYRK1A Inhibitors for Disease Therapy: Current Status and Perspectives. Eur. J. Med. Chem. 2022, 229, 114062. [Google Scholar] [CrossRef] [PubMed]
- Kii, I.; Sumida, Y.; Goto, T.; Sonamoto, R.; Okuno, Y.; Yoshida, S.; Kato-Sumida, T.; Koike, Y.; Abe, M.; Nonaka, Y.; et al. Selective Inhibition of the Kinase DYRK1A by Targeting Its Folding Process. Nat. Commun. 2016 71 2016, 7, 11391. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; Mclauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The Selectivity of Protein Kinase Inhibitors: A Further Update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Göckler, N.; Jofre, G.; Papadopoulos, C.; Soppa, U.; Tejedor, F.J.; Becker, W. Harmine Specifically Inhibits Protein Kinase DYRK1A and Interferes with Neurite Formation. FEBS J. 2009, 276, 6324–6337. [Google Scholar] [CrossRef]
- Lindberg, M.F.; Meijer, L. Dual-Specificity, Tyrosine Phosphorylation-Regulated Kinases (DYRKs) and Cdc2-Like Kinases (CLKs) in Human Disease, an Overview. Int. J. Mol. Sci. 2021, 22, 6047. [Google Scholar] [CrossRef]
- Rothweiler, U.; Stensen, W.; Brandsdal, B.O.; Isaksson, J.; Leeson, F.A.; Engh, R.A.; Svendsen, J.S.M. Probing the ATP-Binding Pocket of Protein Kinase DYRK1A with Benzothiazole Fragment Molecules. J. Med. Chem. 2016, 59, 9814–9824. [Google Scholar] [CrossRef] [PubMed]
- Czarna, A.; Wang, J.; Zelencova, D.; Liu, Y.; Deng, X.; Choi, H.G.; Zhang, T.; Zhou, W.; Chang, J.W.; Kildalsen, H.; et al. Novel Scaffolds for Dual Specificity Tyrosine-Phosphorylation-Regulated Kinase (DYRK1A) Inhibitors. J. Med. Chem. 2018, 61, 7560–7572. [Google Scholar] [CrossRef] [PubMed]
- Wurzlbauer, A.; Rüben, K.; Gürdal, E.; Chaikuad, A.; Knapp, S.; Sippl, W.; Becker, W.; Bracher, F. How to Separate Kinase Inhibition from Undesired Monoamine Oxidase A Inhibition-The Development of the DYRK1A Inhibitor AnnH75 from the Alkaloid Harmine. Molecules 2020, 25, 5962. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Sipos, M.; Paczal, A.; Balint, B.; Kun, V.; Foloppe, N.; Dokurno, P.; Massey, A.J.; Walmsley, D.L.; Hubbard, R.E.; et al. Structure-Guided Discovery of Potent and Selective DYRK1A Inhibitors. J. Med. Chem. 2021, 64, 6745–6764. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokkorakis, N.; Zouridakis, M.; Gaitanou, M. Mirk/Dyrk1B Kinase Inhibitors in Targeted Cancer Therapy. Pharmaceutics 2024, 16, 528. https://doi.org/10.3390/pharmaceutics16040528
Kokkorakis N, Zouridakis M, Gaitanou M. Mirk/Dyrk1B Kinase Inhibitors in Targeted Cancer Therapy. Pharmaceutics. 2024; 16(4):528. https://doi.org/10.3390/pharmaceutics16040528
Chicago/Turabian StyleKokkorakis, Nikolaos, Marios Zouridakis, and Maria Gaitanou. 2024. "Mirk/Dyrk1B Kinase Inhibitors in Targeted Cancer Therapy" Pharmaceutics 16, no. 4: 528. https://doi.org/10.3390/pharmaceutics16040528
APA StyleKokkorakis, N., Zouridakis, M., & Gaitanou, M. (2024). Mirk/Dyrk1B Kinase Inhibitors in Targeted Cancer Therapy. Pharmaceutics, 16(4), 528. https://doi.org/10.3390/pharmaceutics16040528