Quantification of Process Induced Disorder in Milled Samples Using Different Analytical Techniques

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials

2.2. Preparative Methods
2.2.1. Sample preparation by ball milling (cold room)
2.2.2. Cryo milling to produce an amorphous standard

2.2.3. Melt quench cooling to produce an amorphous standard
2.2.4. Preparation of binary mixtures
2.3. Analytical methods
2.3.1. X-ray powder diffraction (XRPD)
2.3.2. Differential scanning calorimetry (DSC)
2.3.3. FT-Raman-spectroscopy
2.3.4. HPLC
2.3.5. Multivariate Analysis
2.3.6. Statistical Analysis
2.3.7. Cambridge Structural Database (CSD)
3. Results and Discussion
3.1. Chemical stability
3.2. Influence of the calibration standard on the quantification outcome for process induced disorder
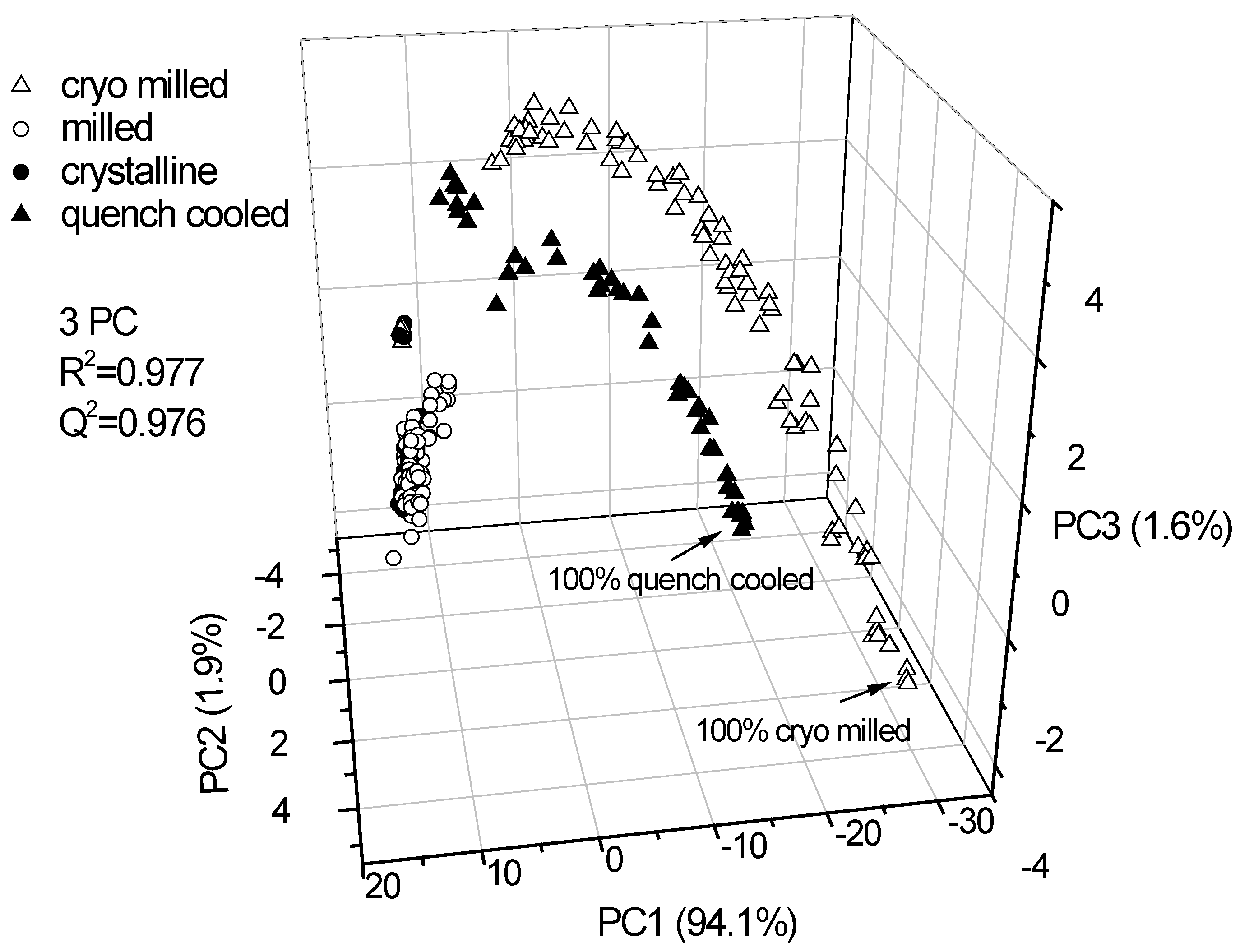
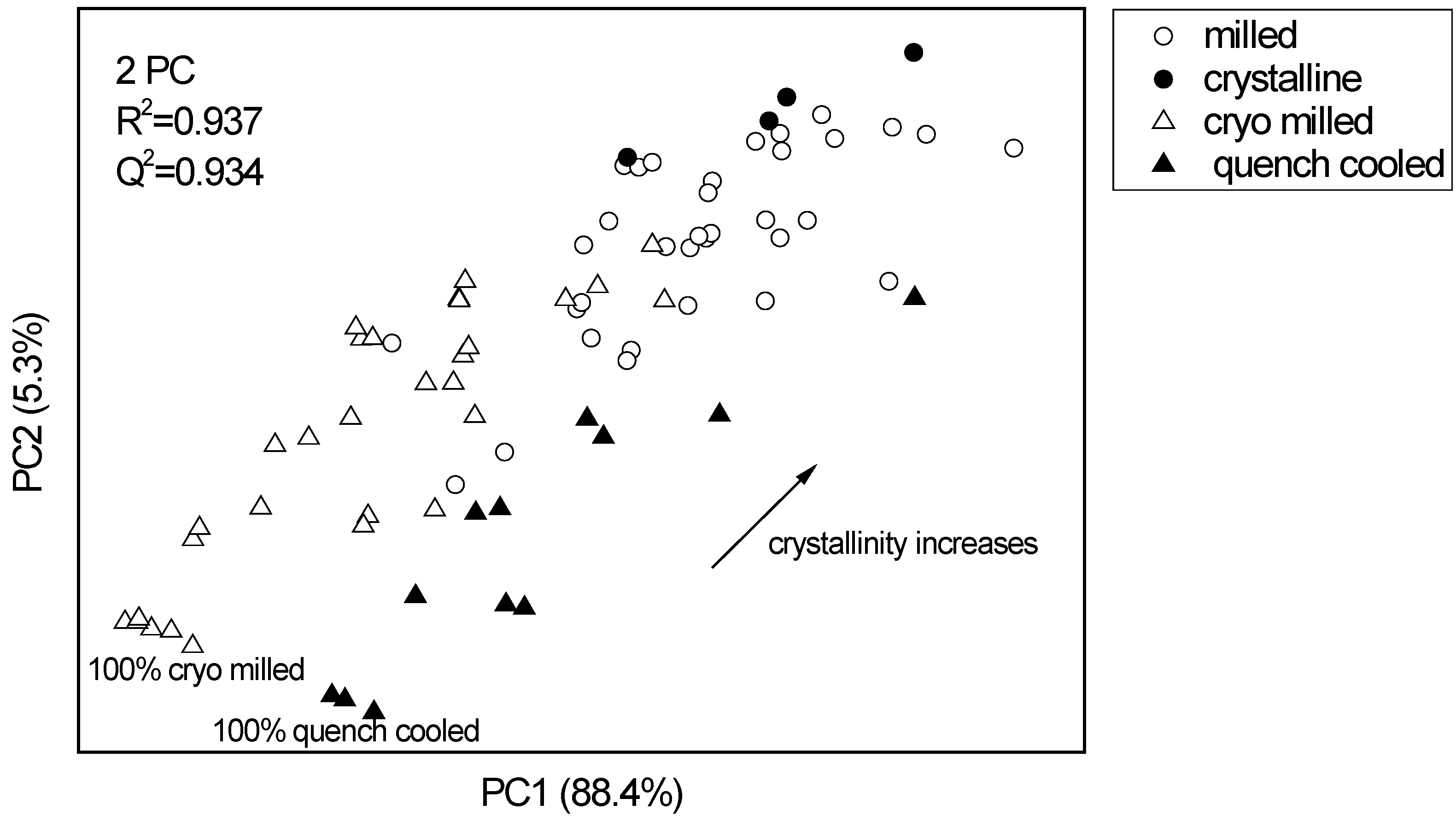
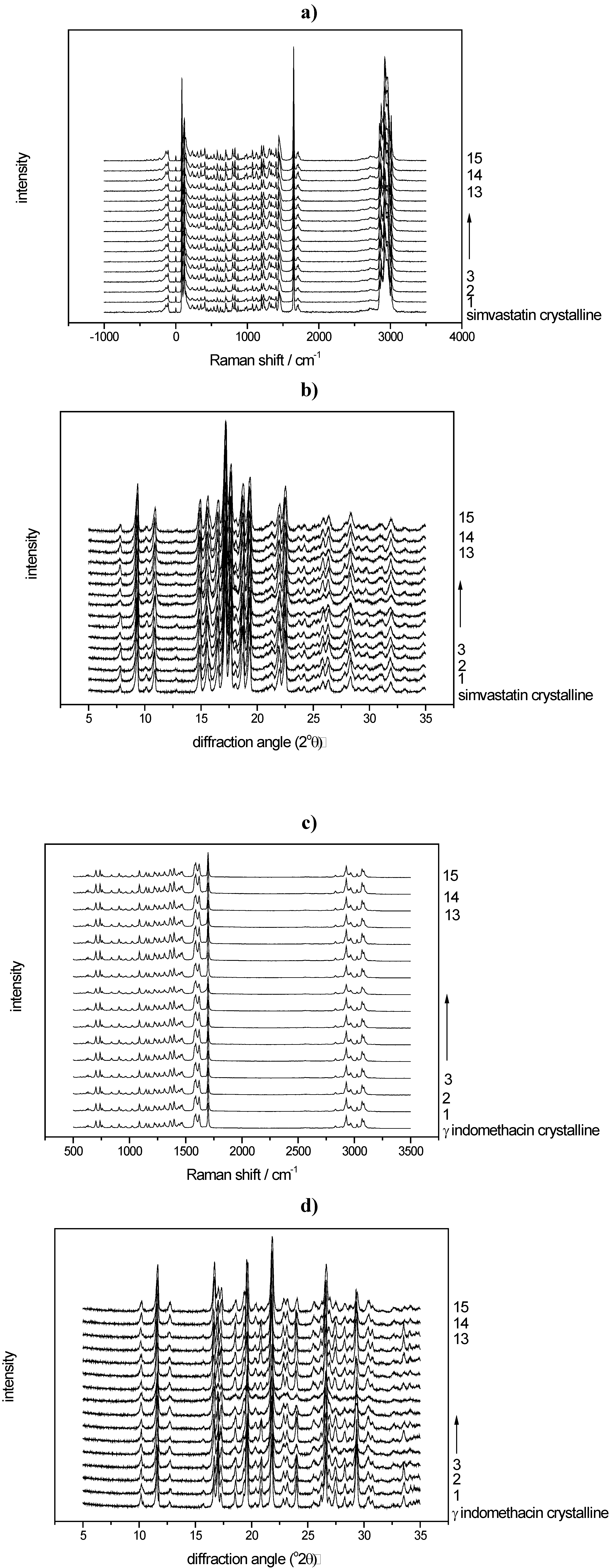
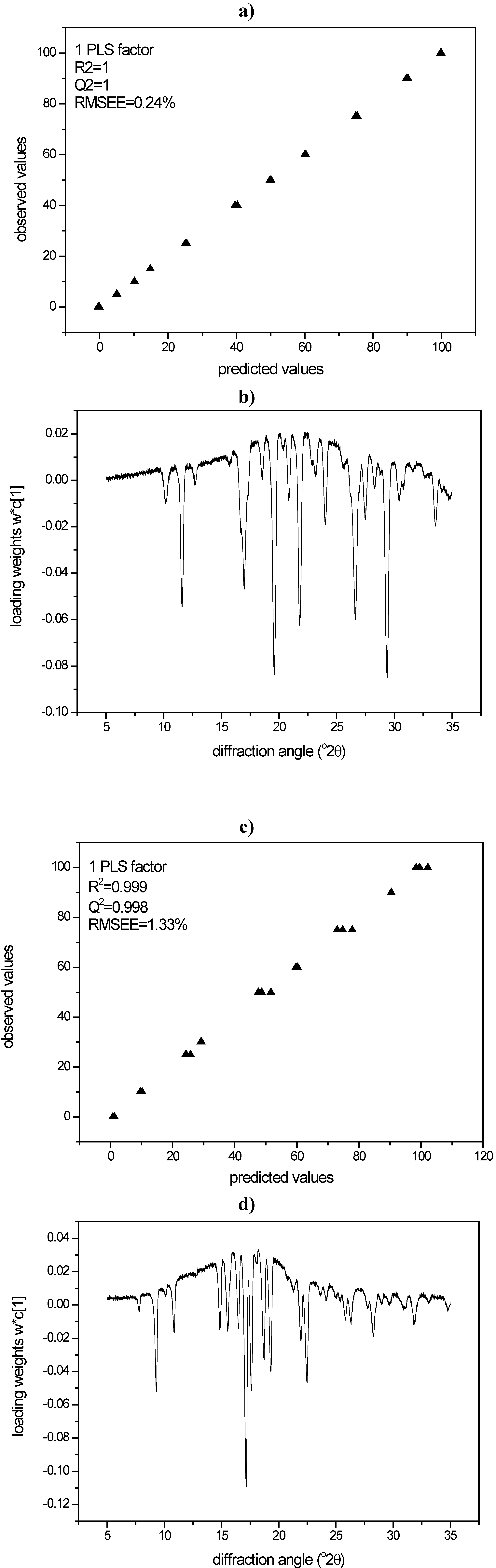
3.2.1. PCA for Raman spectra of the binary mixtures of indomethacin

3.2.2. PCA for XRPD data of the binary mixtures of indomethacin

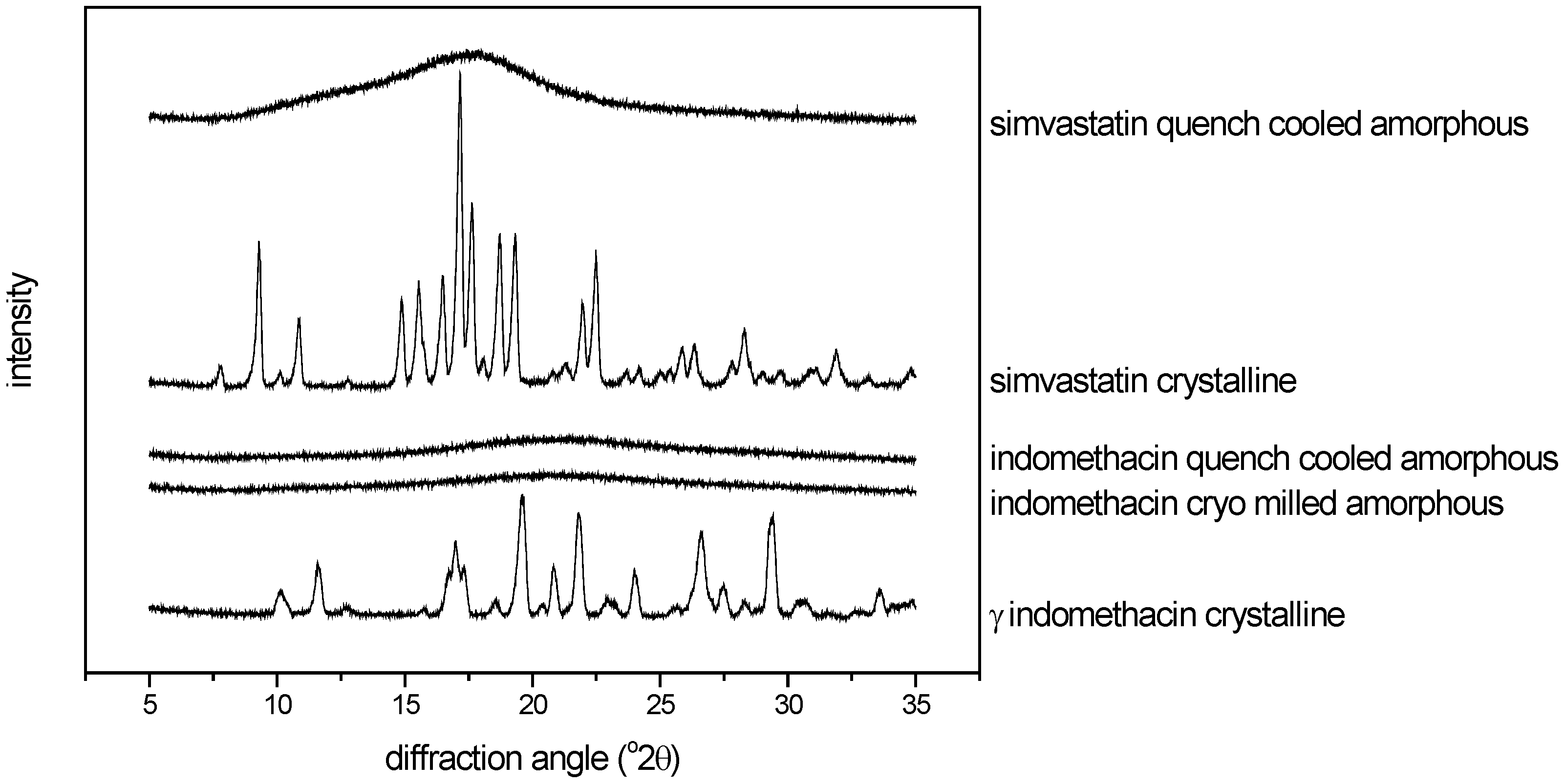
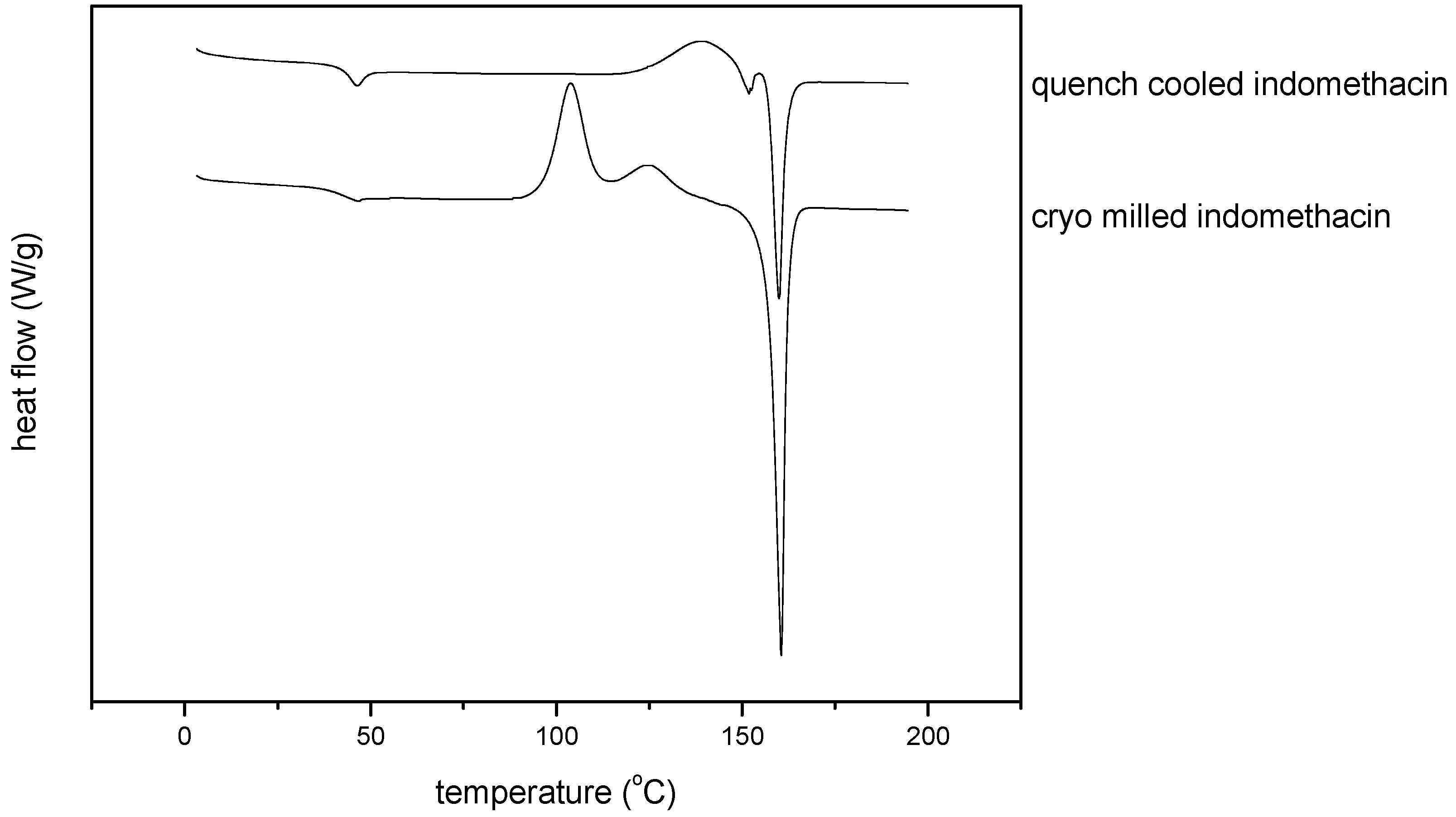
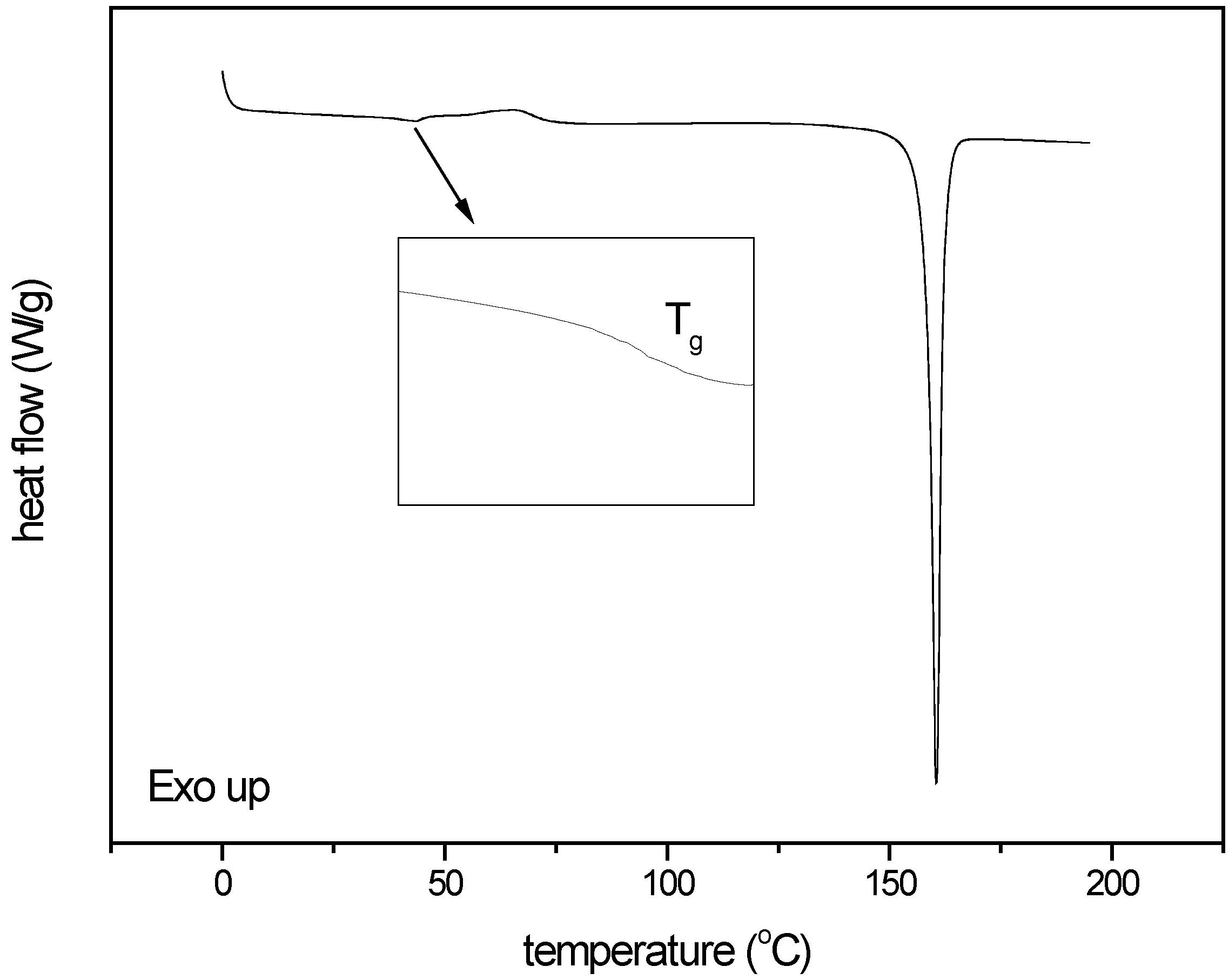
3.2.3. Thermal analysis of quench cooled amorphous and cryo milled amorphous indomethacin

3.3. Influence of the analytical method on the quantification outcome for process induced disorder
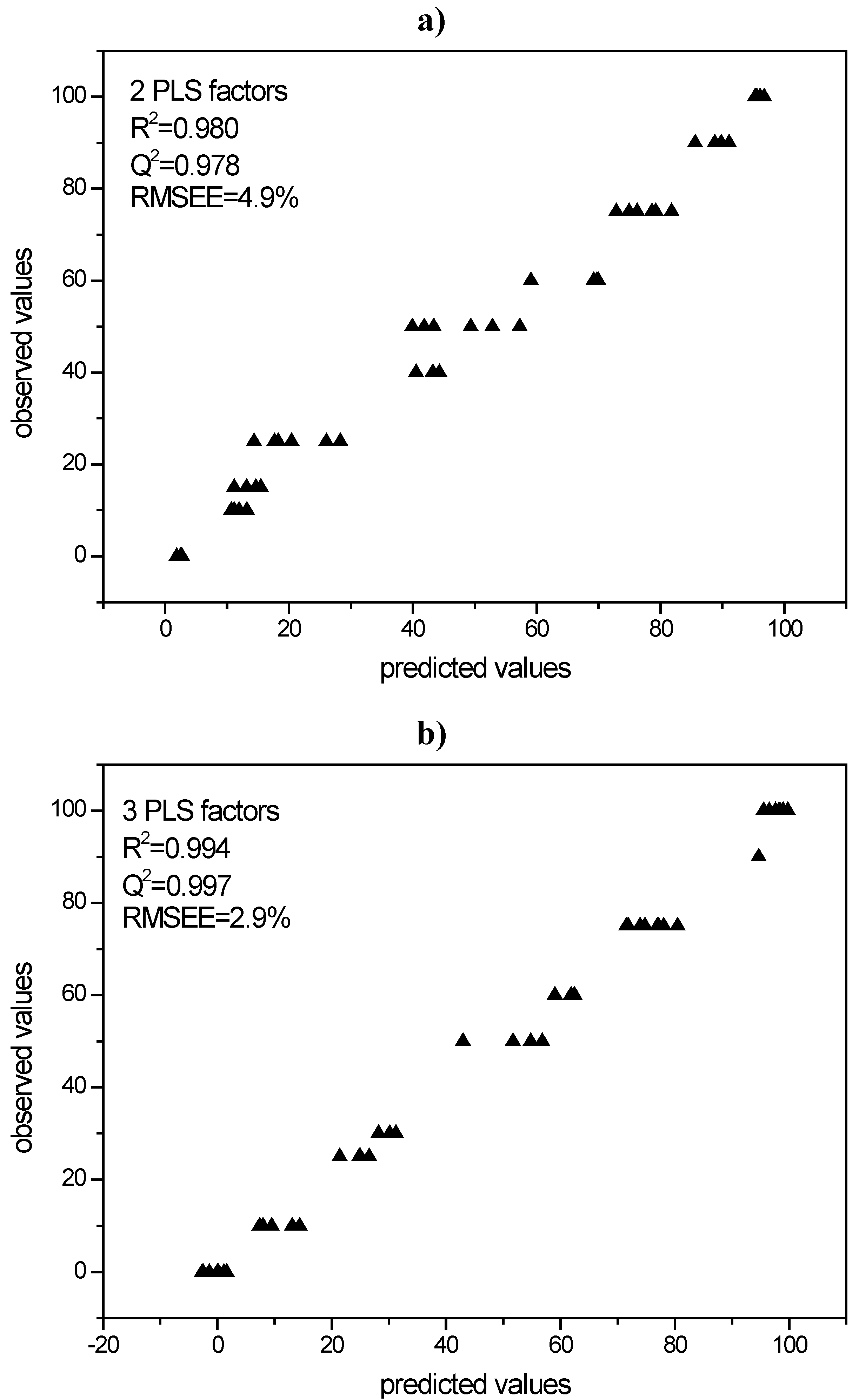
3.3.1. PLS modeling of Raman spectroscopic data of quench cooled amorphous/ crystalline binary mixtures of indomethacin and simvastatin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample No | Time Minutes | Frequency Hertz | No of Balls | Raman Simvastatin | XRPD Simvastatin | DSC Sim-vastatin | Raman Indo-methacin | XRPD Indo-methacin | DSC Indo-methacin |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 5 | 5 | 3 | 0.4 | 0.5 | n.d. | 1.8 | -1.7 | n.d. |
| 2 | 60 | 5 | 3 | 0.4 | 0.2 | n.d. | 1.5 | 3.9 | n.d. |
| 3 | 5 | 25 | 3 | 2.2 | -2.4 | n.d. | 1.6 | 8.1 | n.d. |
| 4 | 60 | 25 | 3 | 7.5 | 10.7 | n.d. | 2.8 | 21.6 | n.d. |
| 5 | 5 | 5 | 60 | -0.4 | 1.4 | n.d. | 1.9 | 0.6 | n.d. |
| 6 | 60 | 5 | 60 | 0.2 | 0.7 | n.d. | 1.7 | 6.6 | n.d. |
| 7 | 5 | 25 | 60 | 3.7 | 8.0 | n.d. | 1.9 | 16.0 | n.d. |
| 8 | 60 | 25 | 60 | 15.5 | 18.3 | n.d. | 10.8 | 44.0 | 17.8 |
| 9 | 5 | 15 | 31 | 1.4 | -1.1 | n.d. | 1.8 | 9.8 | n.d. |
| 10 | 60 | 15 | 31 | 3.2 | 2.1 | n.d. | 1.6 | 12.3 | n.d. |
| 11 | 32.5 | 5 | 31 | 0.2 | -0.6 | n.d. | 1.8 | 4.5 | n.d. |
| 12 | 32.5 | 25 | 31 | 7.3 | 7.9 | n.d. | 5.0 | 24.2 | n.d. |
| 13 | 32.5 | 15 | 3 | 0.0 | 0.5 | n.d. | 1.7 | 5.7 | n.d. |
| 14 | 32.5 | 15 | 60 | 1.9 | 2.2 | n.d. | 2.4 | 11.9 | n.d. |
| 15 | 32.5 | 15 | 31 | 1.7 | 1.7 | n.d. | 1.8 | 17.6 | n.d. |
3.3.2. PLS modeling of XRPD data of quench cooled amorphous/ crystalline binary mixtures of indomethacin and simvastatin

3.3.3. DSC


3.4. Influence of the drug itself on the quantification outcome for process induced disorder
4. Conclusions
Acknowledgements
References and Notes
- Fisher, E.S. Milling of Active Pharmaceutical Ingredients. In Encyclopedia of Pharmaceutical Technology, 3rd ed.; Swarbrick, J., Ed.; informa healthcare: New York, NY, USA; London, UK, 2006; Vol. 4, pp. 2339–2351. [Google Scholar]
- Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef]
- Date, A.A.; Patravale, V.B. Current strategies for engineering drug nanoparticles. Curr. Opin. Colloid Interface Sci. 2004, 9, 222–235. [Google Scholar] [CrossRef]
- Chieng, N.; Zujovic, Z.; Bowmaker, G.; Rades, T.; Saville, D. Effect of milling conditions on the solid-state conversion of ranitidine hydrochloride form 1. Int. J. Pharm. 2006, 327, 36–44. [Google Scholar] [CrossRef]
- Savolainen, M.; Heinz, A.; Strachan, C.; Gordon, K.C.; Yliruusi, J.; Rades, T.; Sandler, N. Screening for differences in the amorphous state of indomethacin using multivariate visualization. Eur. J. Pharm. Sci. 2007, 30, 113–123. [Google Scholar] [CrossRef]
- Feng, T.; Pinal, R.; Carvajal, M.T. Process induced disorder in crystalline materials: Differentiating defective crystals from the amorphous form of griseofulvin. J. Pharm. Sci. 2008, 97, 3207–3221. [Google Scholar] [CrossRef]
- Shah, B.; Kakumanu, V.K.; Bansal, A.K. Analytical techniques for quantification of amorphous/crystalline phases in pharmaceutical solids. J. Pharm. Sci. 2006, 95, 1641–1665. [Google Scholar] [CrossRef]
- Brittain, H.G. Overview of Physical Characterization Methodology. In Physical Characterisation of Pharmaceutical Solids; Brittain, H.G., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1995; pp. 1–35. [Google Scholar]
- Suryanarayanan, R. X-Ray Powder Diffractometry. In Physical Characterisation of Pharmaceutical Solids; Brittain, H.G., Ed.; Marcel Dekker, Inc: New York, NY, USA, 1995; pp. 187–221. [Google Scholar]
- Chieng, N.; Rehder, S.; Saville, D.; Rades, T.; Aaltonen, J. Quantitative solid-state analysis of three solid forms of ranitidine hydrochloride in ternary mixtures using Raman spectroscopy and X-ray powder diffraction. J. Pharm. Biomed. Anal. 2009, 49, 18–25. [Google Scholar] [CrossRef]
- Zhang, F.; Aaltonen, J.; Tian, F.; Saville, D.J.; Rades, T. Influence of particle size and preparation methods on the physical and chemical stability of amorphous simvastatin. Eur. J. Pharm. Biopharm. 2009, 71, 64–70. [Google Scholar]
- Graeser, K.A.; Strachan, C.J.; Patterson, J.E.; Gordon, K.C.; Rades, T. Physicochemical properties and stability of two differently prepared amorphous forms of simvastatin. Cryst. Growth Des. 2008, 8, 128–135. [Google Scholar] [CrossRef]
- Graeser, K.A.; Patterson, J.E.; Zeitler, J.A.; Gordon, K.C.; Rades, T. Correlating thermodynamic and kinetic parameters with amorphous stability. Eur. J. Pharm. Sci. 2009, 37, 492–498. [Google Scholar] [CrossRef]
- Willart, J.F.; Descamps, M. Solid State Amorphization of Pharmaceuticals. Mol. Pharmaceutics 2008, 5, 905–920. [Google Scholar]
- Descamps, M.; Willart, J.F.; Dudognon, E.; Caron, V. Transformation of pharmaceutical compounds upon milling and comilling: The role of T-g. J. Pharm. Sci. 2007, 96, 1398–1407. [Google Scholar] [CrossRef]
- Desprez, S.; Descamps, M. Transformations of glassy indomethacin induced by ball-milling. J. Non-Cryst. Solids 2006, 352, 4480–4485. [Google Scholar] [CrossRef]
- Crowley, K.J.; Zografi, G. Cryogenic grinding of indomethacin polymorphs and solvates: Assessment of amorphous phase formation and amorphous phase physical stability. J. Pharm. Sci. 2002, 91, 492–507. [Google Scholar] [CrossRef]
- Otsuka, M.; Matsumoto, T.; Kaneniwa, N. Effect of environmental-temperature on polymorphic solid-state transformation of indomethacin during grinding. Chem. Pharm. Bull. 1986, 34, 1784–1793. [Google Scholar] [CrossRef]
- Hess, S.; Teubert, U.; Ortwein, J.; Eger, K. Profiling indomethacin impurities using high-performance liquid chromatography and nuclear magnetic resonance. Eur. J. Pharm. Sci. 2001, 14, 301–311. [Google Scholar] [CrossRef]
- Meng, J.; Zheng, L.Y. Application of mixture experimental design to simvastatin apparent solubility predictions in the microemulsifion formed by self-microemulsifying. Drug Dev. Ind. Pharm. 2007, 33, 927–931. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr., Sect. B: Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Aaltonen, J.; Rantanen, J.; Siiria, S.; Karjalainen, M.; Jorgensen, A.; Laitinen, N.; Savolainen, M.; Seitavuopio, P.; Louhi-Kultanen, M.; Yliruusi, J. Polymorph screening using near-infrared spectroscopy. Anal. Chem. 2003, 75, 5267–5273. [Google Scholar]
- Qi, S.; Avalle, P.; Saklatvala, R.; Craig, D.Q.M. An investigation into the effects of thermal history on the crystallisation behaviour of amorphous paracetamol. Eur. J. Pharm. Biopharm. 2008, 69, 364–371. [Google Scholar] [CrossRef]
- Moore, M.D.; Cogdill, R.P.; Wildfong, P.L.D. Evaluation of chemometric algorithms in quantitative X-ray powder diffraction (XRPD) of intact multi-component consolidated samples. J. Pharm. Biomed. Anal. 2009, 49, 619–626. [Google Scholar]
- Artursson, T.; Hagman, A.; Bjork, S.; Trygg, J.; Wold, S.; Jacobsson, S.P. Study of preprocessing methods for the determination of crystalline phases in binary mixtures of drug substances by X-ray powder diffraction and multivariate calibration. Appl. Spectrosc. 2000, 54, 1222–1230. [Google Scholar] [CrossRef]
- Wold, S.; Antti, H.; Lindgren, F.; Ohman, J. Orthogonal signal correction of near-infrared spectra. Chemom. Intell. Lab. Syst. 1998, 44, 175–185. [Google Scholar] [CrossRef]
- McGoverin, C.M.; Rades, T.; Gordon, K.C. Recent Pharmaceutical Applications of Raman and Terahertz Spectroscopies. J. Pharm. Sci. 2008, 97, 4598–4621. [Google Scholar] [CrossRef]
- Strachan, C.J.; Rades, T.; Gordon, K.C. A theoretical and spectroscopic study of gamma-crystalline and amorphous indometacin. J. Pharm. Pharmacol. 2007, 59, 261–269. [Google Scholar] [CrossRef]
- Cejka, J.; Kratochvil, B.; Cisarova, I.; Jegorov, A. Simvastatin. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2003, 59, 428–430. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zimper, U.; Aaltonen, J.; McGoverin, C.M.; Gordon, K.C.; Krauel-Goellner, K.; Rades, T. Quantification of Process Induced Disorder in Milled Samples Using Different Analytical Techniques. Pharmaceutics 2010, 2, 30-49. https://doi.org/10.3390/pharmaceutics2010030
Zimper U, Aaltonen J, McGoverin CM, Gordon KC, Krauel-Goellner K, Rades T. Quantification of Process Induced Disorder in Milled Samples Using Different Analytical Techniques. Pharmaceutics. 2010; 2(1):30-49. https://doi.org/10.3390/pharmaceutics2010030
Chicago/Turabian StyleZimper, Ulrike, Jaakko Aaltonen, Cushla M. McGoverin, Keith C. Gordon, Karen Krauel-Goellner, and Thomas Rades. 2010. "Quantification of Process Induced Disorder in Milled Samples Using Different Analytical Techniques" Pharmaceutics 2, no. 1: 30-49. https://doi.org/10.3390/pharmaceutics2010030
APA StyleZimper, U., Aaltonen, J., McGoverin, C. M., Gordon, K. C., Krauel-Goellner, K., & Rades, T. (2010). Quantification of Process Induced Disorder in Milled Samples Using Different Analytical Techniques. Pharmaceutics, 2(1), 30-49. https://doi.org/10.3390/pharmaceutics2010030