

Posttraumatic and Idiopathic Spike–Wave Discharges in Rats: Discrimination by Morphology and Thalamus Involvement

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
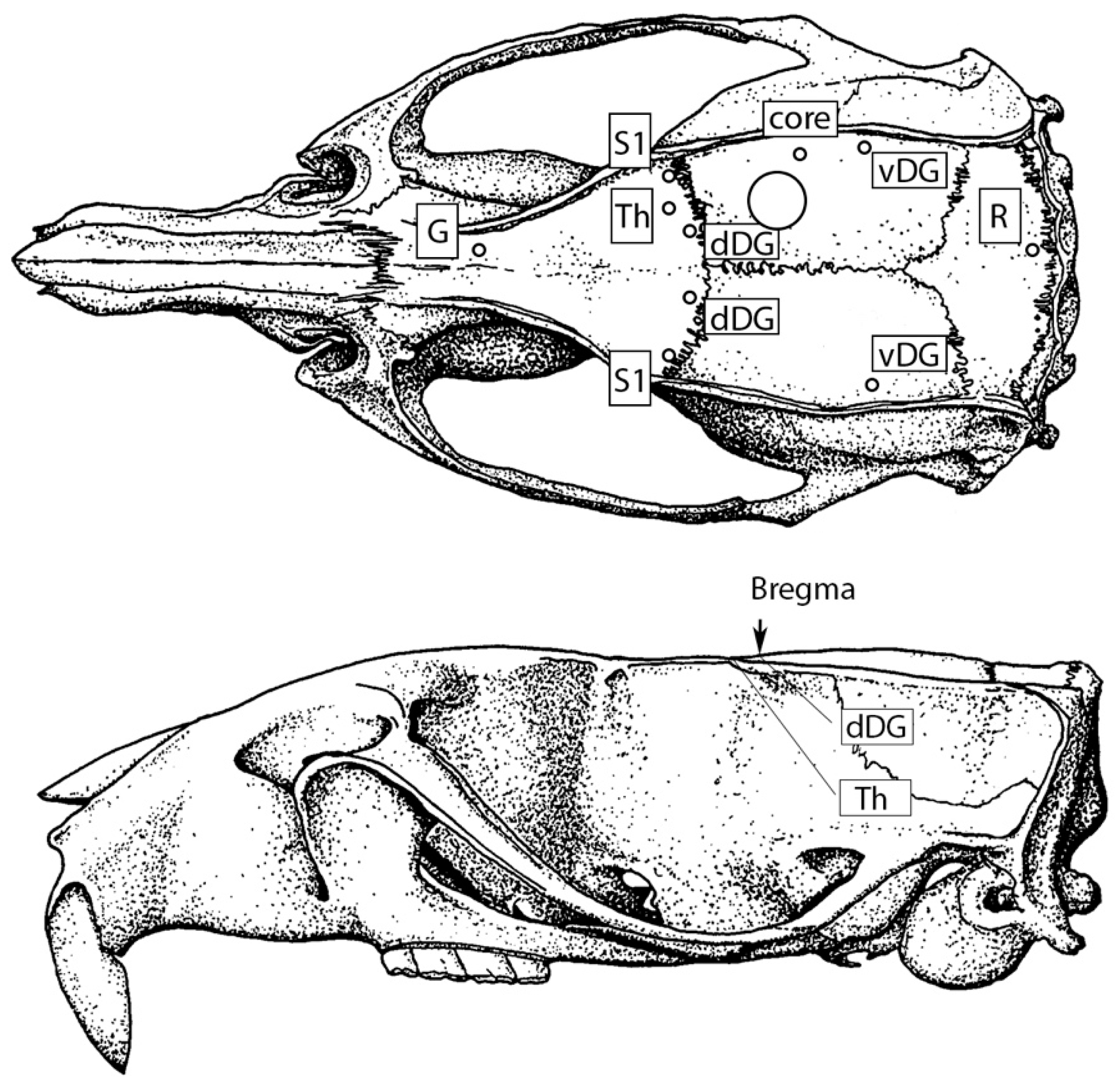
2.2. Implantation of Electrodes
2.3. TBI Modeling
2.4. In Vivo Electrophysiology
2.5. Detection of Epileptiform Activity
2.6. Statistical Methods
3. Results
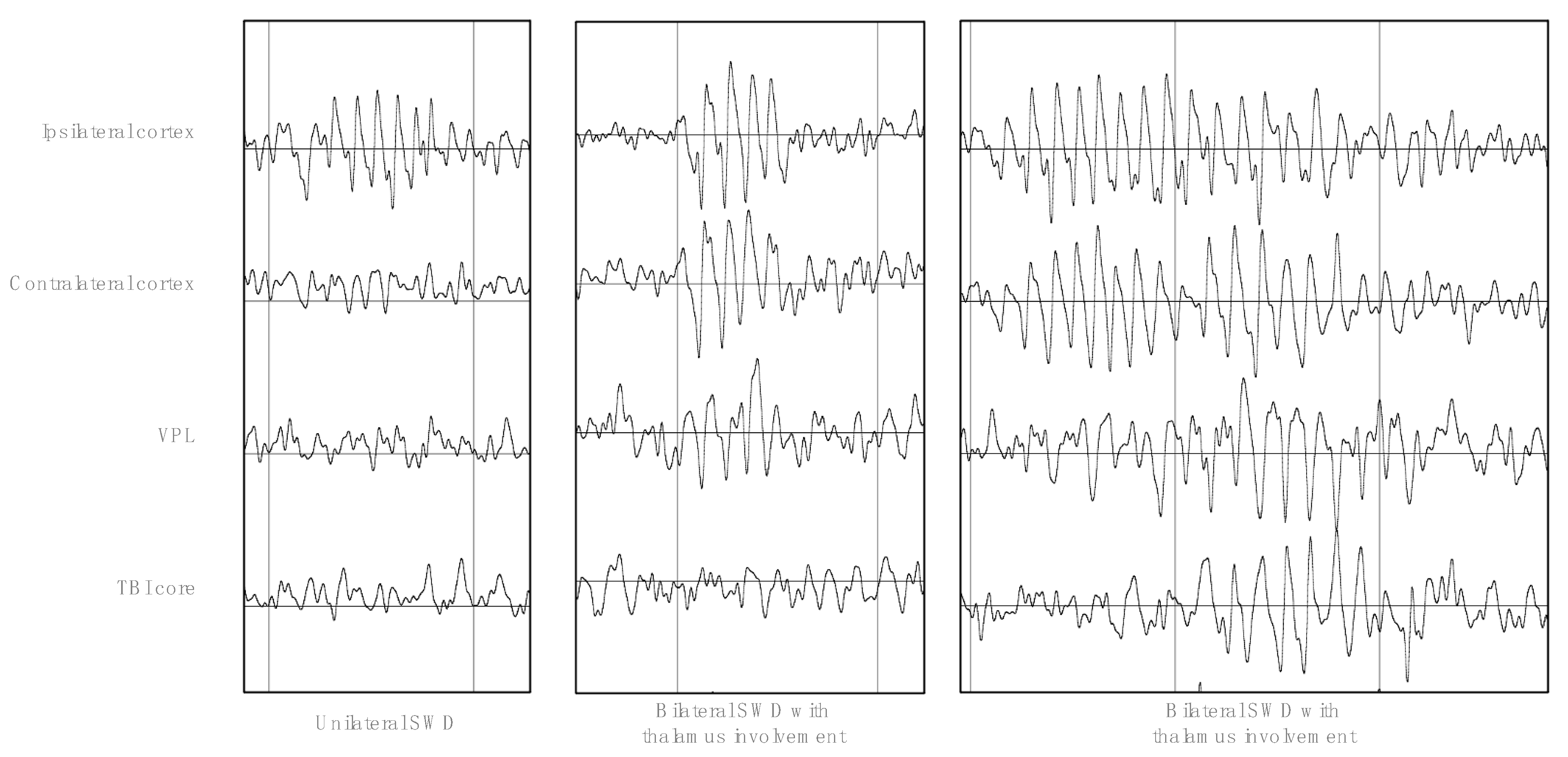
3.1. SWD Characteristics and Time Course during the Experiment
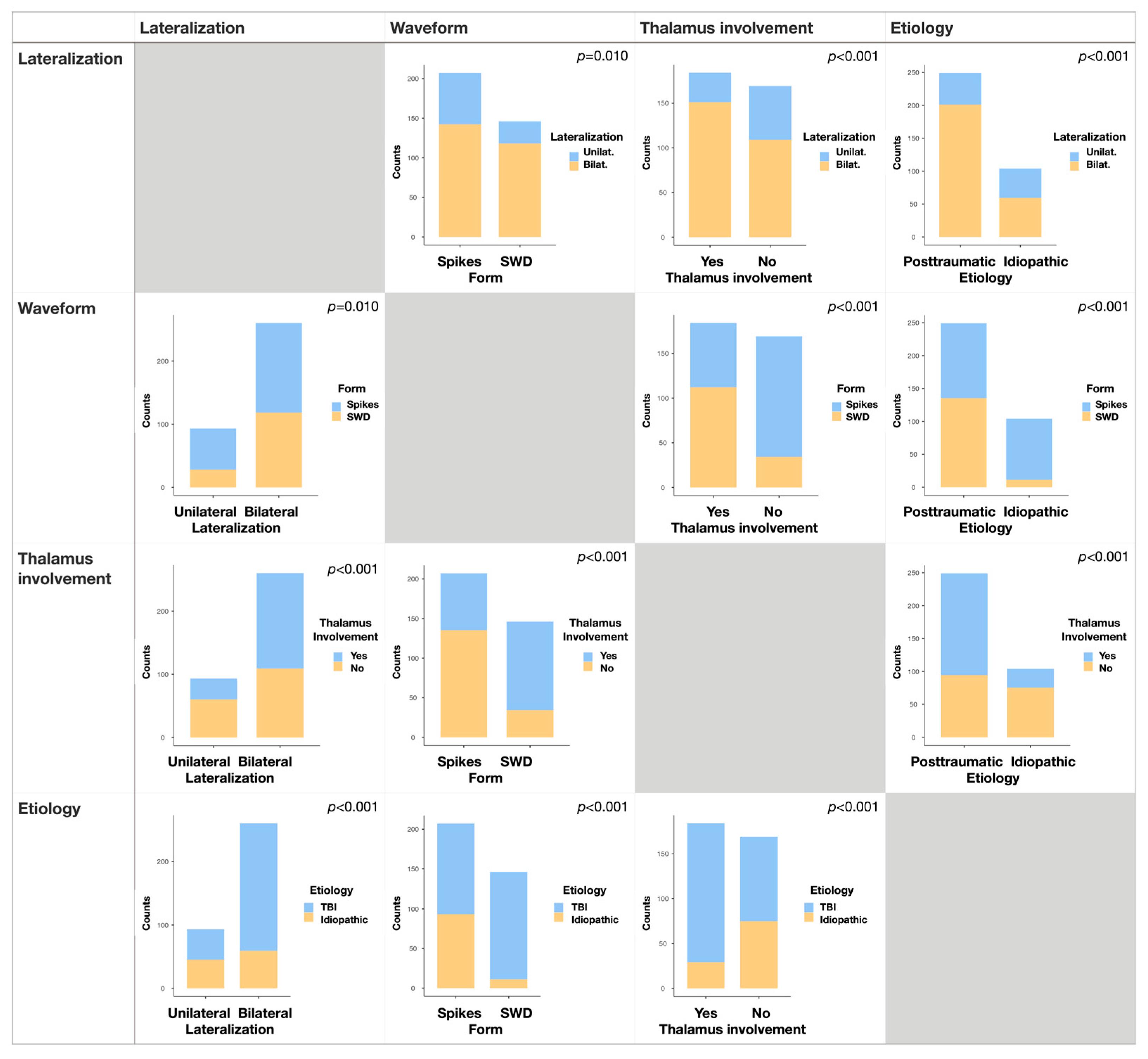
3.2. Waveform, Lateralization, and Thalamus Involvement
3.3. Posttraumatic vs. Idiopathic Etiology
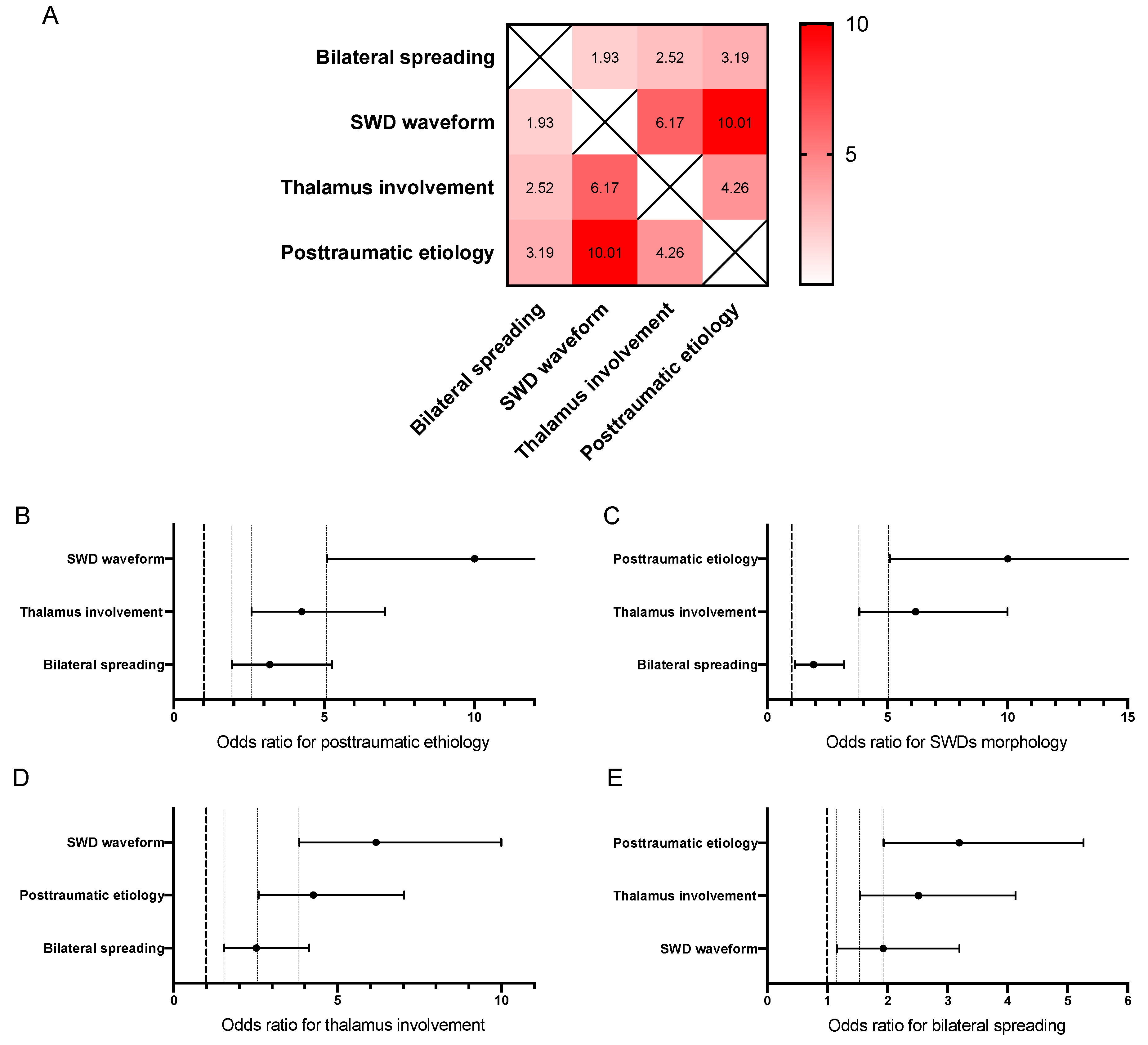
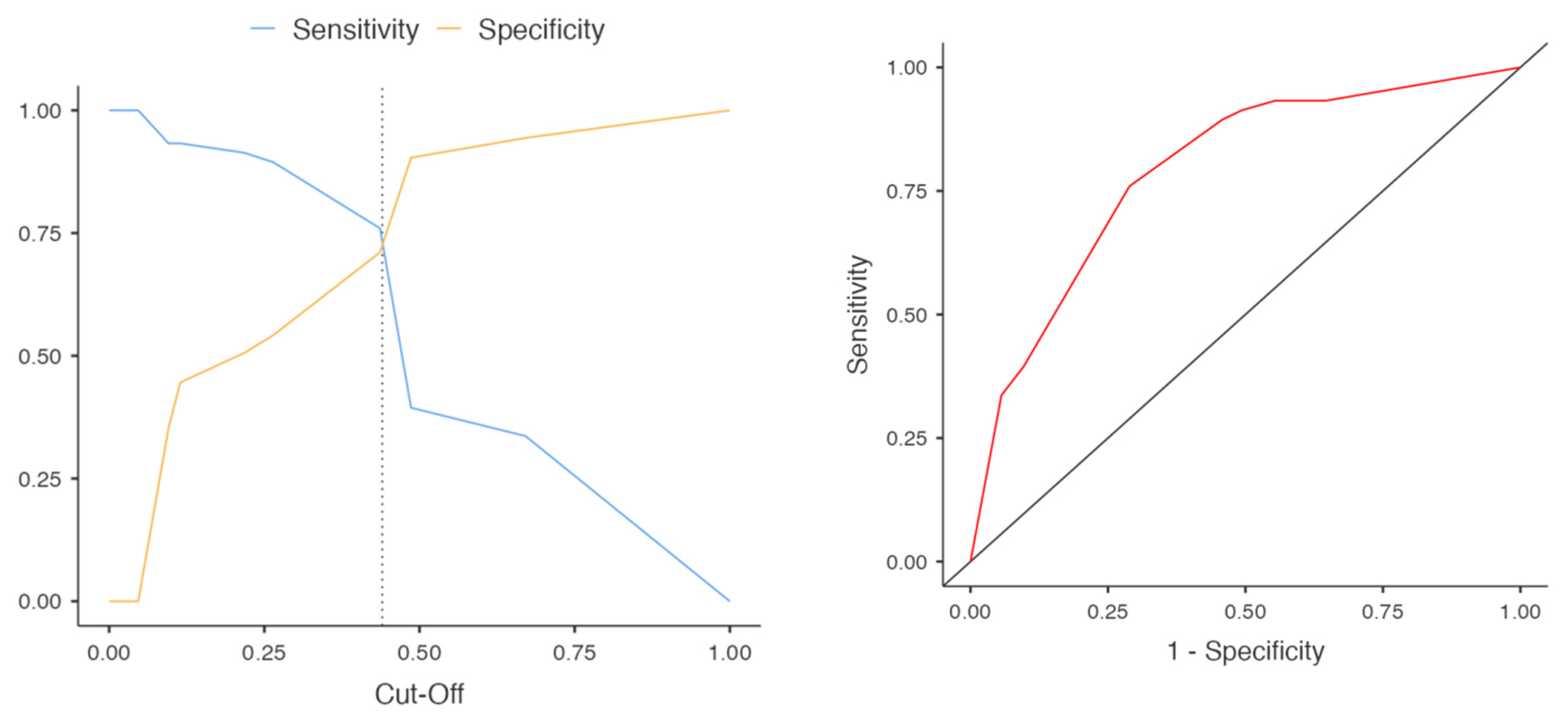
3.4. Segregation of SWDs by Etiology
4. Discussion
5. Conclusions
6. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AUC | Area under the ROC curve |
| LFPI | Lateral fluid percussion injury |
| MRI | Magnetic resonance imaging |
| OR | Odds ratio |
| PTE | Posttraumatic epilepsy |
| ROC | Receiver operating characteristic |
| RTn | Reticular thalamic nucleus |
| S1 | Somatosensory cortex |
| SWD | Spike–wave discharge |
| TBI | Traumatic brain injury |
| TC | Thalamocortical |
References
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma 2015, 32, 1834–1848. [Google Scholar] [CrossRef]
- Reilly, P. The impact of neurotrauma on society: An international perspective. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2007; Volume 161, pp. 3–9. ISBN 9780444530172. [Google Scholar]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2019, 130, 1–18. [Google Scholar] [CrossRef]
- Beghi, E.; Giussani, G.; Nichols, E.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Abraha, H.N.; Adib, M.G.; Agrawal, S.; Alahdab, F.; et al. Global, regional, and national burden of epilepsy, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 357–375. [Google Scholar] [CrossRef]
- Annegers, J.F.; Hauser, W.A.; Coan, S.P.; Rocca, W.A. A Population-Based Study of Seizures after Traumatic Brain Injuries. N. Engl. J. Med. 1998, 338, 20–24. [Google Scholar] [CrossRef]
- Englander, J.; Bushnik, T.; Duong, T.T.; Cifu, D.X.; Zafonte, R.; Wright, J.; Hughes, R.; Bergman, W. Analyzing risk factors for late posttraumatic seizures: A prospective, multicenter investigation. Arch. Phys. Med. Rehabil. 2003, 84, 365–373. [Google Scholar] [CrossRef]
- Gupta, P.K.; Sayed, N.; Ding, K.; Agostini, M.A.; Van Ness, P.C.; Yablon, S.; Madden, C.; Mickey, B.; D’Ambrosio, R.; Diaz-Arrastia, R. Subtypes of Post-Traumatic Epilepsy: Clinical, Electrophysiological, and Imaging Features. J. Neurotrauma 2014, 31, 1439–1443. [Google Scholar] [CrossRef]
- Christensen, J. The Epidemiology of Posttraumatic Epilepsy. Semin. Neurol. 2015, 35, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Temkin, N.R. Risk Factors for Posttraumatic Seizures in Adults. Epilepsia 2003, 44, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Boyle, E.J.; Wu, A.C.; Cole, A.J.; Staley, K.J.; Zafar, S.; Cash, S.S.; Westover, M.B. Epileptiform activity in traumatic brain injury predicts post-traumatic epilepsy. Ann. Neurol. 2018, 83, 858–862. [Google Scholar] [CrossRef]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE Official Report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann-Eden, B.; Bruckmeir, J. Predictors and dynamics of posttraumatic epilepsy. Acta Neurol. Scand. 1997, 95, 257–262. [Google Scholar] [CrossRef]
- Pitkänen, A.; Ekolle Ndode-Ekane, X.; Lapinlampi, N.; Puhakka, N. Epilepsy biomarkers—Toward etiology and pathology specificity. Neurobiol. Dis. 2018, 123, 42–58. [Google Scholar] [CrossRef]
- Dudek, F.E.; Bertram, E.H. Counterpoint to “what is an epileptic seizure?” by D’Ambrosio and Miller. Epilepsy Curr. 2010, 10, 91–94. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, T.K.; Noble, L.; Andrews, B.; Faden, A.I. Traumatic brain injury in the rat: Characterization of a midline fluid-percussion model. Cent. Nerv. Syst. Trauma 1987, 4, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Lifshitz, J.; Marklund, N.; Grady, M.S.; Graham, D.I.; Hovda, D.A.; McIntosh, T.K. Lateral Fluid Percussion Brain Injury: A 15-Year Review and Evaluation. J. Neurotrauma 2005, 22, 42–75. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.; Fujioka, W.; Lifshitz, J.; Crockett, D.P.; Thakker-Varia, S. Lateral Fluid Percussion: Model of Traumatic Brain Injury in Mice. J. Vis. Exp. 2011, 3063. [Google Scholar] [CrossRef]
- Marklund, N. Injury Models of the Central Nervous System. In Methods in Molecular Biology; Kobeissy, F.H., Dixon, C.E., Hayes, R.L., Mondello, S., Eds.; Springer: New York, NY, USA, 2016; Volume 1462, ISBN 978-1-4939-3814-8. [Google Scholar]
- Pitkänen, A.; Mcintosh, T.K. Animal Models of Post-Traumatic Epilepsy. J. Neurotrauma 2006, 23, 241–261. [Google Scholar] [CrossRef]
- Hicks, R.; Soares, H.; Smith, D.; McIntosh, T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996, 91, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, W.S.; Maris, D.O.; McCall, T.; Grady, M.S. Adaptation of the Fluid Percussion Injury Model to the Mouse. J. Neurotrauma 1998, 15, 217–229. [Google Scholar] [CrossRef]
- Ekolle Ndode-Ekane, X.; Kharatishvili, I.; Pitkänen, A. Unfolded Maps for Quantitative Analysis of Cortical Lesion Location and Extent after Traumatic Brain Injury. J. Neurotrauma 2016, 34, 459–474. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Onufriev, M.V.; Moiseeva, J.V.; Novikova, M.R.; Gulyaeva, N.V. Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats. Int. J. Mol. Sci. 2021, 22, 5883. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Tret’yakova, L.V.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Butuzov, A.V.; Novikova, M.R.; Kvichansky, A.A.; Moiseeva, Y.V.; Onufriev, M.V.; et al. Neuroinflammatory Cytokine Response, Neuronal Death, and Microglial Proliferation in the Hippocampus of Rats During the Early Period After Lateral Fluid Percussion-Induced Traumatic Injury of the Neocortex. Mol. Neurobiol. 2021, 59, 1151–1167. [Google Scholar] [CrossRef]
- Komoltsev, I.; Shalneva, D.; Kostyunina, O.; Volkova, A.; Frankevich, S.; Shirobokova, N.; Belikova, A.; Balan, S.; Chizhova, O.; Salyp, O.; et al. Delayed TBI-Induced Neuronal Death in the Ipsilateral Hippocampus and Behavioral Deficits in Rats: Influence of Corticosterone-Dependent Survivorship Bias? Int. J. Mol. Sci. 2023, 24, 4542. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Gulyaeva, N.V. Brain Trauma, Glucocorticoids and Neuroinflammation: Dangerous Liaisons for the Hippocampus. Biomedicines 2022, 10, 1139. [Google Scholar] [CrossRef]
- Kandratavicius, L.; Balista, P.; Lopes-Aguiar, C.; Ruggiero, R.; Umeoka, E.; Garcia-Cairasco, N.; Bueno-Junior, L.; Leite, J. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705. [Google Scholar] [CrossRef]
- Klein, P.; Dingledine, R.; Aronica, E.; Bernard, C.; Blümcke, I.; Boison, D.; Brodie, M.J.; Brooks-Kayal, A.R.; Engel, J.; Forcelli, P.A.; et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia 2018, 59, 37–66. [Google Scholar] [CrossRef]
- Becker, A.J. Review: Animal models of acquired epilepsy: Insights into mechanisms of human epileptogenesis. Neuropathol. Appl. Neurobiol. 2018, 44, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Meeren, H.K.M.; Pijn, J.P.M.; van Luijtelaar, E.L.J.M.; Coenen, A.M.L.; Lopes da Silva, F.H. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J. Neurosci. 2002, 22, 1480–1495. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D. Cellular interactions in the rat somatosensory thalamocortical system during normal epileptic 5–9 Hz oscillations. J. Physiol. 2003, 552, 881–905. [Google Scholar] [CrossRef] [PubMed]
- Sitnikova, E. Thalamo-cortical mechanisms of sleep spindles and spike-wave discharges in rat model of absence epilepsy (a review). Epilepsy Res. 2010, 89, 17–26. [Google Scholar] [CrossRef]
- Rodgers, K.M.; Dudek, F.E.; Barth, D.S. Progressive, Seizure-Like, Spike-Wave Discharges Are Common in Both Injured and Uninjured Sprague-Dawley Rats: Implications for the Fluid Percussion Injury Model of Post-Traumatic Epilepsy. J. Neurosci. 2015, 35, 9194–9204. [Google Scholar] [CrossRef]
- Pearce, P.S.; Friedman, D.; LaFrancois, J.J.; Iyengar, S.S.; Fenton, A.A.; MacLusky, N.J.; Scharfman, H.E. Spike–wave discharges in adult Sprague–Dawley rats and their implications for animal models of temporal lobe epilepsy. Epilepsy Behav. 2014, 32, 121–131. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Levshina, I.P.; Novikova, M.R.; Manolova, A.O.; Gulyaeva, N.V. Differential early effects of traumatic brain injury on spike-wave discharges in Sprague-Dawley rats. Neurosci. Res. 2021, 166, 42–54. [Google Scholar] [CrossRef]
- Kabadi, S.V.; Hilton, G.D.; Stoica, B.A.; Zapple, D.N.; Faden, A.I. Fluid-percussion-induced traumatic brain injury model in rats. Nat. Protoc. 2010, 5, 1552–1563. [Google Scholar] [CrossRef]
- McIntosh, T.K.; Vink, R.; Noble, L.; Yamakami, I.; Fernyak, S.; Soares, H.; Faden, A.L. Traumatic brain injury in the rat: Characterization of a lateral fluid-percussion model. Neuroscience 1989, 28, 233–244. [Google Scholar] [CrossRef]
- Gurkoff, G.G.; Gahan, J.D.; Ghiasvand, R.T.; Hunsaker, M.R.; Van, K.; Feng, J.-F.; Shahlaie, K.; Berman, R.F.; Lyeth, B.G.; Folkerts, M.M. Evaluation of Metric, Topological, and Temporal Ordering Memory Tasks after Lateral Fluid Percussion Injury. J. Neurotrauma 2013, 30, 292–300. [Google Scholar] [CrossRef]
- Loane, D.J.; Byrnes, K.R. Role of Microglia in Neurotrauma. Neurotherapeutics 2010, 7, 366–377. [Google Scholar] [CrossRef]
- Dudek, F.E.; Staley, K.J. The Time Course and Circuit Mechanisms of Acquired Epileptogenesis. In Jasper’s Basic Mechanisms of the Epilepsies; National Center for Biotechnology Information: Bethesda, MD, USA, 2013. [Google Scholar]
- Tauck, D.L.; Nadler, J.V. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J. Neurosci. 1985, 5, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Dudek, F.E.; Sutula, T.P. Epileptogenesis in the dentate gyrus: A critical perspective. Prog. Brain Res. 2007, 163, 755–773. [Google Scholar] [PubMed]
- Buckmaster, P.S. Mossy fiber sprouting in the dentate gyrus. Epilepsia 2010, 51, 39. [Google Scholar] [CrossRef]
- Gorter, J.A.; Van Vliet, E.A.; Aronica, E.; Lopes Da Silva, F.H. Progression of spontaneous seizures after status epilepticus is associated with mossy fibre sprouting and extensive bilateral loss of hilar parvalbumin and somatostatin-immunoreactive neurons. Eur. J. Neurosci. 2001, 13, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Lukasiuk, K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011, 10, 173–186. [Google Scholar] [CrossRef]
- Pitkänen, A.; Lukasiuk, K. Molecular biomarkers of epileptogenesis. Biomark. Med. 2011, 5, 629–633. [Google Scholar] [CrossRef]
- Pitkänen, A.; Immonen, R. Epilepsy Related to Traumatic Brain Injury. Neurotherapeutics 2014, 11, 286–296. [Google Scholar] [CrossRef]
- Kharatishvili, I.; Nissinen, J.P.; McIntosh, T.K.; Pitkänen, A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience 2006, 140, 685–697. [Google Scholar] [CrossRef]
- D’Ambrosio, R.; Perucca, E. Epilepsy after head injury. Curr. Opin. Neurol. 2004, 17, 731–735. [Google Scholar] [CrossRef]
- D’Ambrosio, R.; Fender, J.S.; Fairbanks, J.P.; Simon, E.A.; Born, D.E.; Doyle, D.L.; Miller, J.W. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain 2005, 128, 174–188. [Google Scholar] [CrossRef]
- Buzsáki, G.; Laszlovszky, I.; Lajtha, A.; Vadász, C. Spike-and-wave neocortical patterns in rats: Genetic and aminergic control. Neuroscience 1990, 38, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Lüttjohann, A.; van Luijtelaar, G. The role of thalamic nuclei in genetic generalized epilepsies. Epilepsy Res. 2022, 182, 106918. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.J. Reconsidering Network Mechanisms in Absence Seizures: Unhitching the Wave Cart from the Spike Horse. Epilepsy Curr. 2021, 21, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Terlau, J.; Yang, J.; Khastkhodaei, Z.; Seidenbecher, T.; Luhmann, H.J.; Pape, H.; Lüttjohann, A. Spike-wave discharges in absence epilepsy: Segregation of electrographic components reveals distinct pathways of seizure activity. J. Physiol. 2020, 598, 2397–2414. [Google Scholar] [CrossRef] [PubMed]
- Wiest, M.C.; Nicolelis, M.A.L. Behavioral detection of tactile stimuli during 7–12 Hz cortical oscillations in awake rats. Nat. Neurosci. 2003, 6, 913–914. [Google Scholar] [CrossRef] [PubMed]
- Fanselow, E.E.; Sameshima, K.; Baccala, L.A.; Nicolelis, M.A.L. Thalamic bursting in rats during different awake behavioral states. Proc. Natl. Acad. Sci. USA 2001, 98, 15330–15335. [Google Scholar] [CrossRef]
- McCafferty, C.; Gruenbaum, B.F.; Tung, R.; Li, J.-J.; Zheng, X.; Salvino, P.; Vincent, P.; Kratochvil, Z.; Ryu, J.H.; Khalaf, A.; et al. Decreased but diverse activity of cortical and thalamic neurons in consciousness-impairing rodent absence seizures. Nat. Commun. 2023, 14, 117. [Google Scholar] [CrossRef]
- Huusko, N.; Pitk??nen, A. Parvalbumin immunoreactivity and expression of GABAA receptor subunits in the thalamus after experimental TBI. Neuroscience 2014, 267, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Manninen, E.; Chary, K.; Lapinlampi, N.; Andrade, P.; Paananen, T.; Sierra, A.; Tohka, J.; Gröhn, O.; Pitkänen, A. Acute thalamic damage as a prognostic biomarker for post-traumatic epileptogenesis. Epilepsia 2021, 62, 1852–1864. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.A.; Ndode-Ekane, X.E.; Lehto, L.J.; Gorter, J.A.; Andrade, P.; Aronica, E.; Gröhn, O.; Pitkänen, A. Long-lasting blood-brain barrier dysfunction and neuroinflammation after traumatic brain injury. Neurobiol. Dis. 2020, 145, 105080. [Google Scholar] [CrossRef]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R.A.; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komoltsev, I.; Salyp, O.; Volkova, A.; Bashkatova, D.; Shirobokova, N.; Frankevich, S.; Shalneva, D.; Kostyunina, O.; Chizhova, O.; Kostrukov, P.; et al. Posttraumatic and Idiopathic Spike–Wave Discharges in Rats: Discrimination by Morphology and Thalamus Involvement. Neurol. Int. 2023, 15, 609-621. https://doi.org/10.3390/neurolint15020038
Komoltsev I, Salyp O, Volkova A, Bashkatova D, Shirobokova N, Frankevich S, Shalneva D, Kostyunina O, Chizhova O, Kostrukov P, et al. Posttraumatic and Idiopathic Spike–Wave Discharges in Rats: Discrimination by Morphology and Thalamus Involvement. Neurology International. 2023; 15(2):609-621. https://doi.org/10.3390/neurolint15020038
Chicago/Turabian StyleKomoltsev, Ilia, Olga Salyp, Aleksandra Volkova, Daria Bashkatova, Natalia Shirobokova, Stepan Frankevich, Daria Shalneva, Olga Kostyunina, Olesya Chizhova, Pavel Kostrukov, and et al. 2023. "Posttraumatic and Idiopathic Spike–Wave Discharges in Rats: Discrimination by Morphology and Thalamus Involvement" Neurology International 15, no. 2: 609-621. https://doi.org/10.3390/neurolint15020038
APA StyleKomoltsev, I., Salyp, O., Volkova, A., Bashkatova, D., Shirobokova, N., Frankevich, S., Shalneva, D., Kostyunina, O., Chizhova, O., Kostrukov, P., Novikova, M., & Gulyaeva, N. (2023). Posttraumatic and Idiopathic Spike–Wave Discharges in Rats: Discrimination by Morphology and Thalamus Involvement. Neurology International, 15(2), 609-621. https://doi.org/10.3390/neurolint15020038