
Genetic Variation in Angiotensin II Type 1 Receptor Is Linked to Lipid Levels and Hepatic Steatosis in Alcohol-Associated Liver Disease, but Not to Cirrhosis or Hepatocellular Carcinoma
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Determination of AGTR1 and PNPLA3 Genotypes
2.3. Statistical Analysis
3. Results
3.1. Study Cohorts
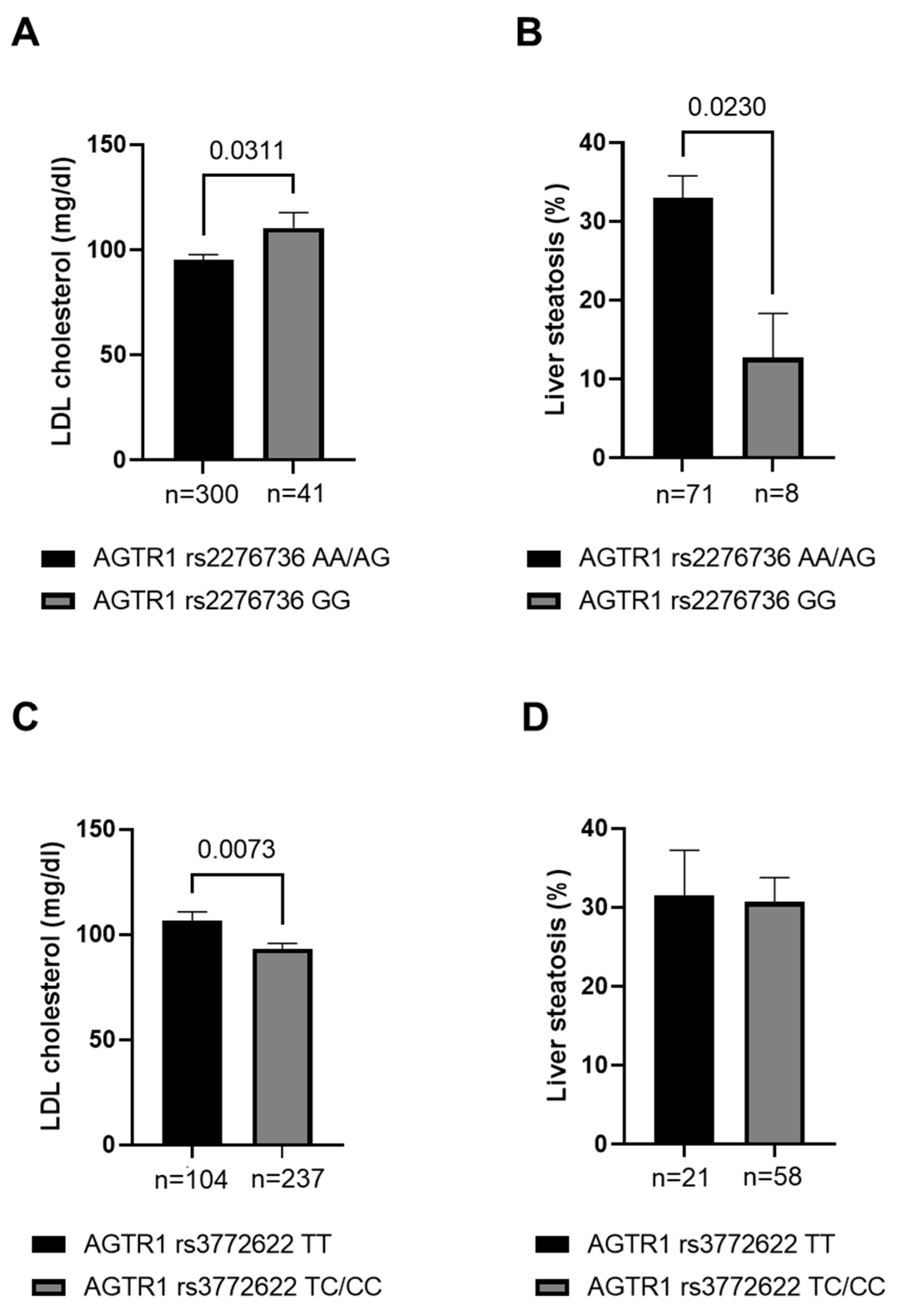
3.2. Association of Genetic Variation at AGTR1 to Lipid Parameters
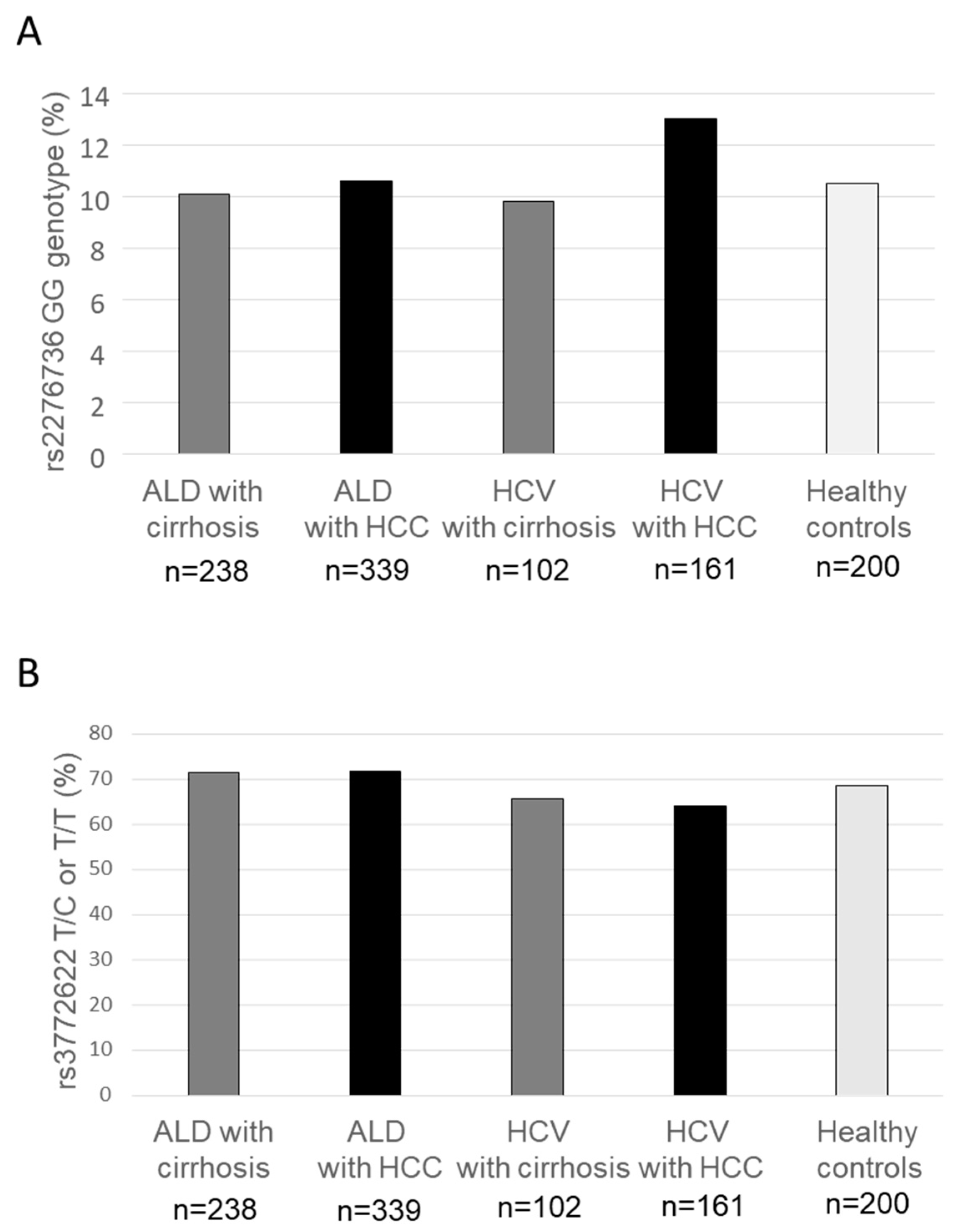
3.3. Genotype Distribution Concerning Clinical Endpoints
3.4. Gene-Gene Interaction
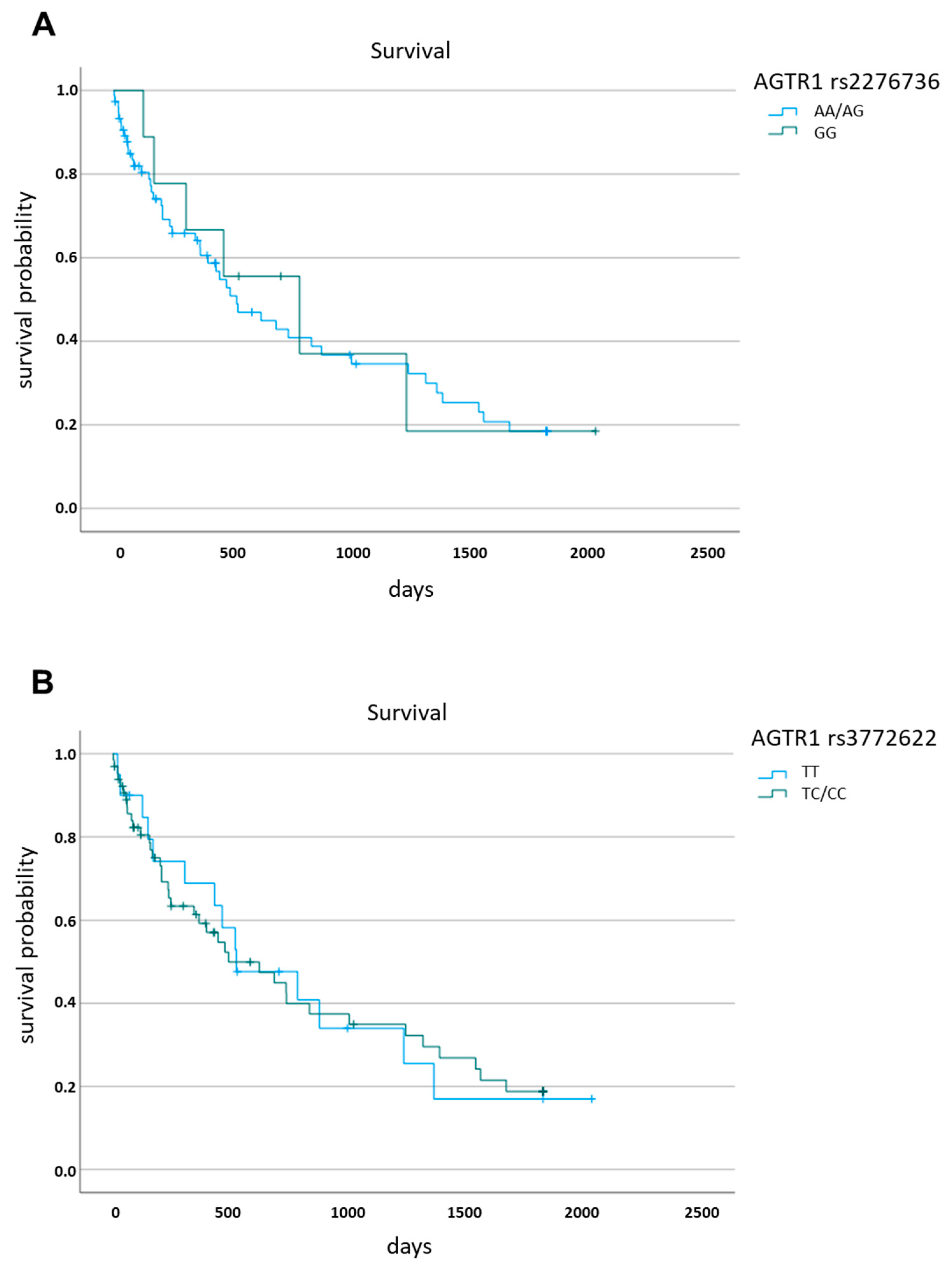
3.5. Survival
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global Burden of Liver Disease: 2023 Update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic Liver Disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Vishnubhotla, R.; Kulkarni, A.V.; Sharma, M.; Rao, P.N.; Reddy, D.N. An Update on the Genetics of Alcoholic Liver Disease. Front. Gastroenterol. 2022, 1, 1030399. [Google Scholar] [CrossRef]
- Eslam, M.; George, J. Genetic Contributions to NAFLD: Leveraging Shared Genetics to Uncover Systems Biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Hotta, K.; Nozaki, Y.; Endo, H.; Uchiyama, T.; Mawatari, H.; Iida, H.; Kato, S.; Fujita, K.; Takahashi, H.; et al. Association between Angiotensin II Type 1 Receptor Polymorphisms and the Occurrence of Nonalcoholic Fatty Liver Disease. Liver Int. 2009, 29, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Zain, S.M.; Mohamed, Z.; Mahadeva, S.; Rampal, S.; Basu, R.C.; Cheah, P.-L.; Salim, A.; Mohamed, R. Susceptibility and Gene Interaction Study of the Angiotensin II Type 1 Receptor (AGTR1) Gene Polymorphisms with Non-Alcoholic Fatty Liver Disease in a Multi-Ethnic Population. PLoS ONE 2013, 8, e58538. [Google Scholar] [CrossRef]
- Fung, M.M.; Rao, F.; Poddar, S.; Mahata, M.; Khandrika, S.; Mahata, S.K.; O’Connor, D.T. Early Inflammatory and Metabolic Changes in Association with AGTR1 Polymorphisms in Prehypertensive Subjects. Am. J. Hypertens. 2011, 24, 225–233. [Google Scholar] [CrossRef]
- Musso, G.; Saba, F.; Cassader, M.; Paschetta, E.; De Michieli, F.; Pinach, S.; Framarin, L.; Berrutti, M.; Leone, N.; Parente, R.; et al. Angiotensin II Type 1 Receptor Rs5186 Gene Variant Predicts Incident NAFLD and Associated Hypertension: Role of Dietary Fat-Induced Pro-Inflammatory Cell Activation. Am. J. Gastroenterol. 2019, 114, 607–619. [Google Scholar] [CrossRef]
- Rajapaksha, I.G.; Gunarathne, L.S.; Angus, P.W.; Herath, C.B. Update on New Aspects of the Renin-Angiotensin System in Hepatic Fibrosis and Portal Hypertension: Implications for Novel Therapeutic Options. J. Clin. Med. 2021, 10, 702. [Google Scholar] [CrossRef]
- Rinella, M.E.; Neuschwander-Tetri, B.A.; Siddiqui, M.S.; Abdelmalek, M.F.; Caldwell, S.; Barb, D.; Kleiner, D.E.; Loomba, R. AASLD Practice Guidance on the Clinical Assessment and Management of Nonalcoholic Fatty Liver Disease. Hepatology 2023, 77, 1797–1835. [Google Scholar] [CrossRef]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.-L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of Hepatocellular Carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- Nischalke, H.D.; Schmalz, F.; Buch, S.; Fischer, J.; Möller, C.; Matz-Soja, M.; Krämer, B.; Langhans, B.; Klüners, A.; Soyka, M.; et al. Genetic Variation of SAMM50 Is Not an Independent Risk Factor for Alcoholic Hepatocellular Carcinoma in Caucasian Patients. Int. J. Mol. Sci. 2022, 23, 15353. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Genetics in Non-Alcoholic Fatty Liver Disease: The Role of Risk Alleles through the Lens of Immune Response. Clin. Mol. Hepatol. 2023, 29, S184–S195. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Moreno, C.; Hampe, J.; Morgan, M.Y. The Genetics of Alcohol Dependence and Alcohol-Related Liver Disease. J. Hepatol. 2017, 66, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.S.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The Renin-Angiotensin System: Going beyond the Classical Paradigms. Am. J. Physiol.-Heart Circ. Physiol. 2019, 316, H958–H970. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1–7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [PubMed]
- Shim, K.Y.; Eom, Y.W.; Kim, M.Y.; Kang, S.H.; Baik, S.K. Role of the Renin-Angiotensin System in Hepatic Fibrosis and Portal Hypertension. Korean J. Intern. Med. 2018, 33, 453–461. [Google Scholar] [CrossRef]
- Bataller, R.; Gäbele, E.; Parsons, C.J.; Morris, T.; Yang, L.; Schoonhoven, R.; Brenner, D.A.; Rippe, R.A. Systemic Infusion of Angiotensin II Exacerbates Liver Fibrosis in Bile Duct-Ligated Rats. Hepatology 2005, 41, 1046–1055. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, L.-L.; Yuan, D.-X.; Geng, N.; Xuan, S.-Y.; Xin, Y.-N. AGTR1 Rs3772622 Gene Polymorphism Increase the Risk of Nonalcoholic Fatty Liver Disease Patients Suffer Coronary Artery Disease in Northern Chinese Han Population. Lipids Health Dis. 2016, 15, 113. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef]
- Francque, S.; Szabo, G.; Abdelmalek, M.F.; Byrne, C.D.; Cusi, K.; Dufour, J.-F.; Roden, M.; Sacks, F.; Tacke, F. Nonalcoholic Steatohepatitis: The Role of Peroxisome Proliferator-Activated Receptors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 24–39. [Google Scholar] [CrossRef]
- Park, J.G.; Mok, J.S.; Han, Y.I.; Park, T.S.; Kang, K.W.; Choi, C.S.; Park, H.D.; Park, J. Connectivity Mapping of Angiotensin-PPAR Interactions Involved in the Amelioration of Non-Alcoholic Steatohepatitis by Telmisartan. Sci. Rep. 2019, 9, 4003. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Smith, R.; Levy, P.; Strawn, W. The Hypertension-Lipid Connection: Insights into the Relation between Angiotensin II and Cholesterol in Atherogenesis. Am. J. Med. Sci. 2002, 323, 17–24. [Google Scholar] [CrossRef]
- Chen, Y.; Du, X.; Kuppa, A.; Feitosa, M.F.; Bielak, L.F.; O’Connell, J.R.; Musani, S.K.; Guo, X.; Kahali, B.; Chen, V.L.; et al. Genome-Wide Association Meta-Analysis Identifies 17 Loci Associated with Nonalcoholic Fatty Liver Disease. Nat. Genet. 2023, 55, 1640–1650. [Google Scholar] [CrossRef]
- Nischalke, H.D.; Berger, C.; Luda, C.; Berg, T.; Müller, T.; Grünhage, F.; Lammert, F.; Coenen, M.; Krämer, B.; Körner, C.; et al. The PNPLA3 Rs738409 148M/M Genotype Is a Risk Factor for Liver Cancer in Alcoholic Cirrhosis but Shows No or Weak Association in Hepatitis C Cirrhosis. PLoS ONE 2011, 6, e27087. [Google Scholar] [CrossRef]
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A Genome-Wide Association Study Confirms PNPLA3 and Identifies TM6SF2 and MBOAT7 as Risk Loci for Alcohol-Related Cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Stickel, F.; Buch, S.; Nischalke, H.D.; Weiss, K.H.; Gotthardt, D.; Fischer, J.; Rosendahl, J.; Marot, A.; Elamly, M.; Casper, M.; et al. Genetic Variants in PNPLA3 and TM6SF2 Predispose to the Development of Hepatocellular Carcinoma in Individuals with Alcohol-Related Cirrhosis. Am. J. Gastroenterol. 2018, 113, 1475–1483. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Erconi, V.; Carli, F.; Fortunato, F.; Ronchi, D.; Piciotti, R.; Sabatini, S.; Macchi, C.; et al. TM6SF2/PNPLA3/MBOAT7 Loss-of-Function Genetic Variants Impact on NAFLD Development and Progression Both in Patients and in In Vitro Models. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 759–788. [Google Scholar] [CrossRef]
- Stickel, F.; Lutz, P.; Buch, S.; Nischalke, H.D.; Silva, I.; Rausch, V.; Fischer, J.; Weiss, K.H.; Gotthardt, D.; Rosendahl, J.; et al. Genetic Variation in HSD17B13 Reduces the Risk of Developing Cirrhosis and Hepatocellular Carcinoma in Alcohol Misusers. Hepatology 2020, 72, 88–102. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant Rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Kogiso, T.; Taniai, M.; Hashimoto, E.; Tokushige, K. Differences in the Genetic Backgrounds of Patients with Alcoholic Liver Disease and Non-alcoholic Fatty Liver Disease. JGH Open 2019, 3, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Barzi, A.; Zhou, K.; Wang, S.; Dodge, J.L.; El-Khoueiry, A.; Setiawan, V.W. Etiology and Outcomes of Hepatocellular Carcinoma in an Ethnically Diverse Population: The Multiethnic Cohort. Cancers 2021, 13, 3476. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.S.; Ruhl, J.; Flynn, G.; Graubard, B.I.; McGlynn, K.A. Trends in Hepatocellular Carcinoma Stage by Racial-Ethnic Group in the United States, 1992–2019. JHEP Rep. 2023, 5, 100868. [Google Scholar] [CrossRef]
- Thylur, R.P.; Roy, S.K.; Shrivastava, A.; LaVeist, T.A.; Shankar, S.; Srivastava, R.K. Assessment of Risk Factors, and Racial and Ethnic Differences in Hepatocellular Carcinoma. JGH Open 2020, 4, 351–359. [Google Scholar] [CrossRef]
- Berberich, A.J.; Hegele, R.A. A Modern Approach to Dyslipidemia. Endocr. Rev. 2022, 43, 611–653. [Google Scholar] [CrossRef]
- Bianco, C.; Jamialahmadi, O.; Pelusi, S.; Baselli, G.; Dongiovanni, P.; Zanoni, I.; Santoro, L.; Maier, S.; Liguori, A.; Meroni, M.; et al. Non-Invasive Stratification of Hepatocellular Carcinoma Risk in Non-Alcoholic Fatty Liver Using Polygenic Risk Scores. J. Hepatol. 2021, 74, 775–782. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Alcohol-Associated Cirrhosis | HCV Infected Patients | Healthy Controls | |||
|---|---|---|---|---|---|
| without HCC | with HCC | with Cirrhosis | with HCC | ||
| Total number | 238 | 339 | 102 | 161 | 200 |
| Age, mean (range) | 58.4 (27–92) | 63.5 (36–87) | 56.6 (28–88) | 60.4 (38–82) | 62.6 (28–87) |
| Sex (% male/female) | 62.4/37.6 | 89.2/10.8 | 62.2/37.8 | 61.7/38.3 | 46.0/54.0 |
| Bilirubin [mg/dL], (Mean ± SD) | 4.18 ± 6.80 | 2.52 ± 3.93 | 2.24 ± 4.28 | 2.34 ± 2.96 | - |
| ALT [IU/L], (Mean ± SD) | 48.6 ± 114.1 | 48.9 ± 59.2 | 92.3 ± 102.7 | 102.5 ± 191.1 | - |
| AST [IU/L], (Mean ± SD) | 72.5 ± 143.4 | 84.8 ± 109.4 | 82.9 ± 82.4 | 109.1 ± 120.5 | - |
| GGT [IU/L], (Mean ± SD) | 211.9 ± 223.6 | 259.6 ± 331.2 | 122.4 ± 132.3 | 127.8 ± 116.1 | - |
| Genotype | Alcohol-Associated Cirrhosis (n = 577) | HCV Infected Patients (n = 263) | Healthy Controls (n = 200) | ||
|---|---|---|---|---|---|
| AGTR1 rs2276736 | without HCC n = 238 | with HCC n = 339 | with cirrhosis n = 102 | with HCC n = 161 | |
| AA | 99 (41.6%) | 145 (42.8%) | 48 (47.1%) | 60 (37.3%) | 91 (45.5%) |
| AG | 113 (47.5%) | 158 (46.6%) | 44 (43.1%) | 80 (49.7%) | 88 (44.0%) |
| GG MAF | 26 (10.9%) 35% | 36 (10.6%) 34% | 10 (9.8%) 31% | 21 (13.0%) 38% | 21 (10.5%) 32% |
| AGTR1 rs3772622 | |||||
| TT | 68 (28.6%) | 96 (28.3%) | 35 (34.3%) | 58 (36.0%) | 63 (31.5%) |
| TC | 123 (51.7%) | 181 (53.4%) | 47 (46.1%) | 80 (49.7%) | 100 (50.0%) |
| CC MAF | 47 (19.8%) 46% | 62 (18.3%) 45% | 20 (19.6%) 43% | 23 (14.3%) 39% | 37 (18.5%) 44% |
| A | ||||
| Parameter | p | OR | 95% CI | |
| Lower | Upper | |||
| AGTR1 AG/GG | 0.661 | 1.130 | 0.653 | 1.956 |
| PNPLA3 148 IM/MM | 0.022 | 1.633 | 1.072 | 2.488 |
| AGTR1 AG/GG by PNPLA3 IM/MM | 0.600 | 1.215 | 0.587 | 2.518 |
| Constant | 0.117 | 0.542 | ||
| B | ||||
| Parameter | p | OR | 95% CI | |
| Lower | Upper | |||
| AGTR1 AG/GG | 0.849 | 0.947 | 0.540 | 1.661 |
| PNPLA3 148 IM/MM | 0.000 | 2.663 | 1.783 | 3.977 |
| AGTR1 AG/GG by PNPLA3 IM/MM | 0.129 | 1.899 | 0.800 | 3.285 |
| Constant | 0.003 | 0.317 | ||
| A | ||||
| Parameter | p | OR | 95% CI | |
| Lower | Upper | |||
| AGTR1 TC/CC | 0.969 | 1.011 | 0.577 | 1.773 |
| PNPLA3 148 IM/MM | 0.022 | 1.633 | 1.072 | 2.488 |
| AGTR1 AG/GG by PNPLA3 IM/MM | 0.678 | 1.166 | 0.564 | 2.412 |
| Constant | 0.155 | 0.555 | ||
| B | ||||
| Parameter | p | OR | 95% CI | |
| Lower | Upper | |||
| AGTR1 TC/CC | 0.845 | 1.060 | 0.593 | 1.894 |
| PNPLA3 148 IM/MM | 0.000 | 2.663 | 1.783 | 3.977 |
| AGTR1 AG/GG by PNPLA3 IM/MM | 0.637 | 1.187 | 0.583 | 2.418 |
| Constant | 0.002 | 0.260 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nischalke, H.D.; Schmalz, F.; Fischer, J.; Möller, C.; Matz-Soja, M.; Krämer, B.; Langhans, B.; Nattermann, J.; Berg, T.; Strassburg, C.P.; et al. Genetic Variation in Angiotensin II Type 1 Receptor Is Linked to Lipid Levels and Hepatic Steatosis in Alcohol-Associated Liver Disease, but Not to Cirrhosis or Hepatocellular Carcinoma. Gastroenterol. Insights 2024, 15, 19-31. https://doi.org/10.3390/gastroent15010002
Nischalke HD, Schmalz F, Fischer J, Möller C, Matz-Soja M, Krämer B, Langhans B, Nattermann J, Berg T, Strassburg CP, et al. Genetic Variation in Angiotensin II Type 1 Receptor Is Linked to Lipid Levels and Hepatic Steatosis in Alcohol-Associated Liver Disease, but Not to Cirrhosis or Hepatocellular Carcinoma. Gastroenterology Insights. 2024; 15(1):19-31. https://doi.org/10.3390/gastroent15010002
Chicago/Turabian StyleNischalke, Hans Dieter, Franziska Schmalz, Janett Fischer, Christine Möller, Madlen Matz-Soja, Benjamin Krämer, Bettina Langhans, Jacob Nattermann, Thomas Berg, Christian P. Strassburg, and et al. 2024. "Genetic Variation in Angiotensin II Type 1 Receptor Is Linked to Lipid Levels and Hepatic Steatosis in Alcohol-Associated Liver Disease, but Not to Cirrhosis or Hepatocellular Carcinoma" Gastroenterology Insights 15, no. 1: 19-31. https://doi.org/10.3390/gastroent15010002
APA StyleNischalke, H. D., Schmalz, F., Fischer, J., Möller, C., Matz-Soja, M., Krämer, B., Langhans, B., Nattermann, J., Berg, T., Strassburg, C. P., & Lutz, P. (2024). Genetic Variation in Angiotensin II Type 1 Receptor Is Linked to Lipid Levels and Hepatic Steatosis in Alcohol-Associated Liver Disease, but Not to Cirrhosis or Hepatocellular Carcinoma. Gastroenterology Insights, 15(1), 19-31. https://doi.org/10.3390/gastroent15010002