
JAK2 Mutations Are Rare and Diverse in Myelodysplastic Syndromes: Case Series and Review of the Literature


Abstract
:1. Introduction
- JAK2 V617F mutation is rare in myelodysplastic syndromes and in its presence a myeloproliferative disease needs to be excluded.
- JAK2 mutations are diverse and JAK2 variant mutations may lead to a myelodysplastic syndrome phenotype.
- JAK2 R564L and JAK2 I670V are reported as JAK2 mutation variants in association with a myelodysplastic phenotype.
- Further studies are recommended to investigate the relationship between and significance of JAK2 mutations variants and their clinico-morphologic phenotypes.
2. Materials and Methods
3. Results
3.1. Cases Presentation
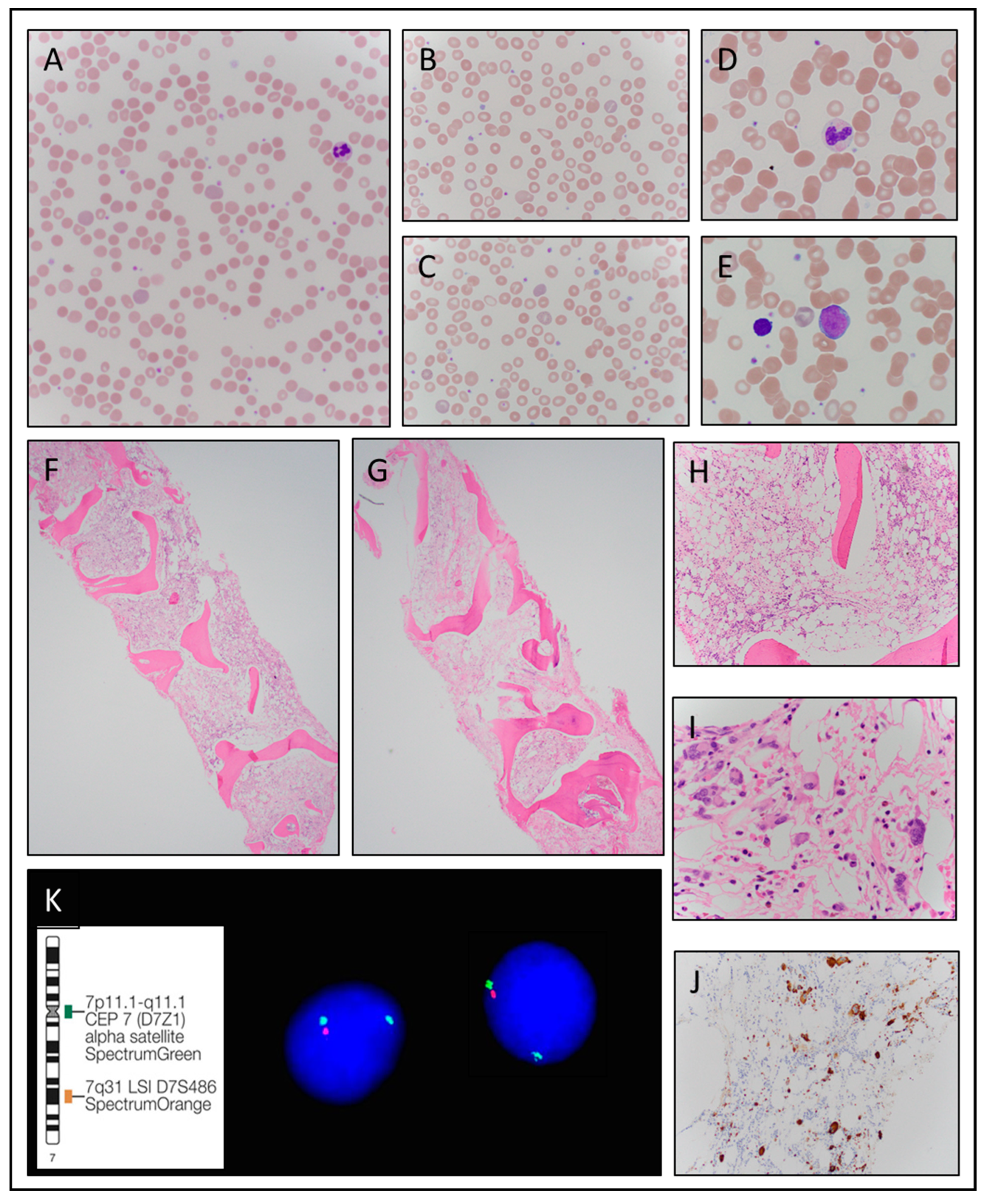
3.1.1. Case 1
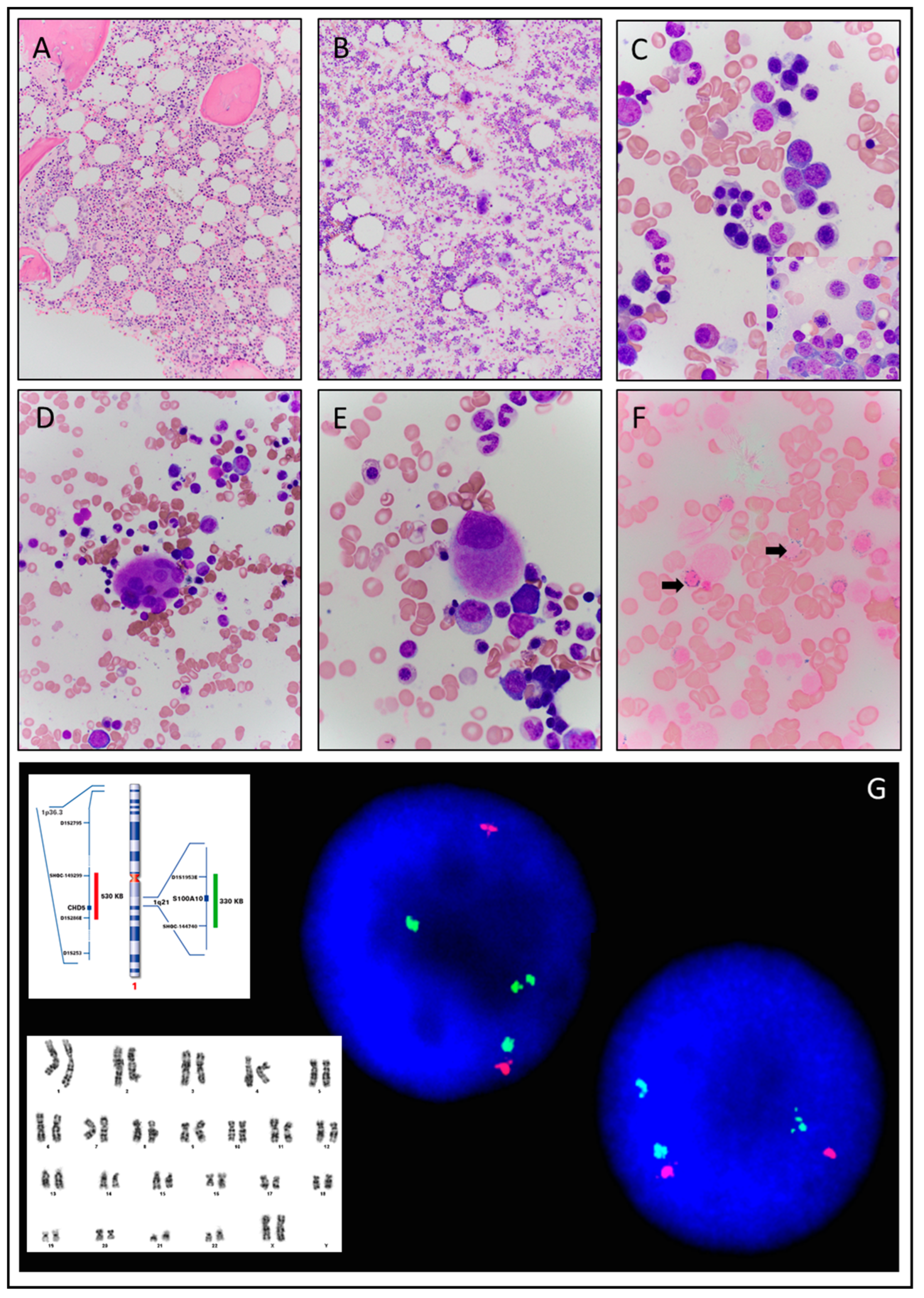
3.1.2. Case 2
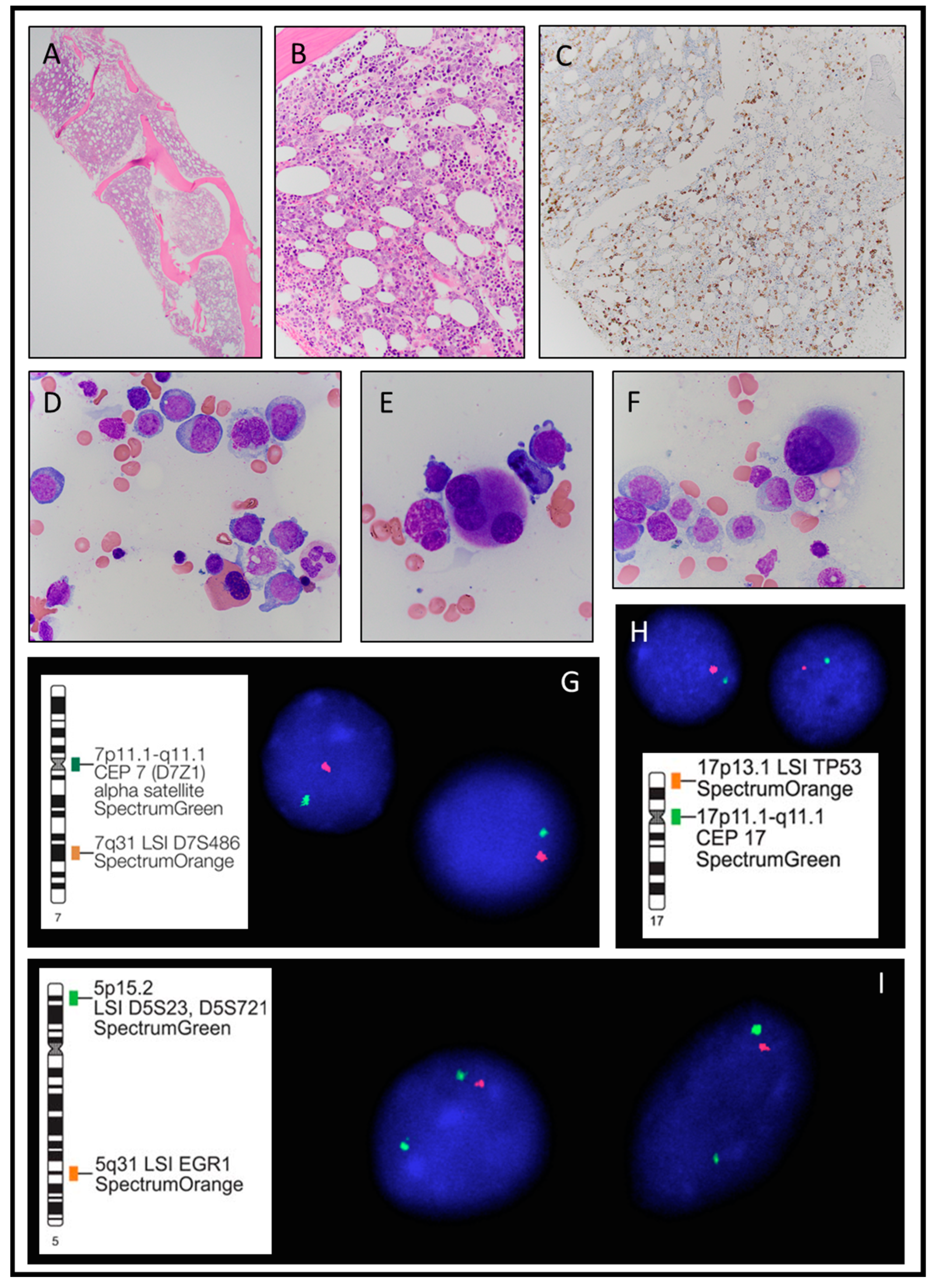
3.1.3. Case 3
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brooks, J.A.; Putoczki, T. JAK-STAT Signalling Pathway in Cancer. Cancers 2020, 12, 1971. [Google Scholar] [CrossRef]
- Nielsen, C.; Birgens, H.S.; Nordestgaard, B.G.; Kjær, L.; Bojesen, S.E. The JAK2 V617F somatic mutation, mortality and cancer risk in the general population. Haematologica 2011, 96, 450–453. [Google Scholar] [CrossRef]
- WHO. Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC Press: Lyon, France, 2017; pp. 115–116. [Google Scholar]
- Nielsen, C.; Birgens, H.S.; Nordestgaard, B.G.; Bojesen, S.E. Diagnostic value of JAK2 V617F somatic mutation for myeloproliferative cancer in 49 488 individuals from the general population. Br. J. Haematol. 2012, 160, 70–79. [Google Scholar] [CrossRef]
- Trelinski, J.; Chojnowski, K.; Cebula-Obrsut, B.; Smolewski, P. Impaired apoptosis of megakaryocytes and bone marrow mononuclear cells in essential thrombocythemia: Correlation with JAK2V617F mutational status and cytoreductive therapy. Med. Oncol. 2012, 29, 2388–2395. [Google Scholar] [CrossRef] [Green Version]
- Raivola, J.; Haikarainen, T.; Abraham, B.G.; Silvennoinen, O. Janus Kinases in Leukemia. Cancers 2021, 13, 800. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Benton, B.C.; Boddu, P.C.; DiNardo, C.D.; Bose, P.; Wang, F.; Assi, R.; Pemmaraju, N.; Devendra, K.C.; Pierce, S.; Patel, K.; et al. Janus Kinase 2 Variants Associated with the Transformation of Myeloproliferative Neoplasms into Acute Myeloid Leukemia. Cancer 2019, 125, 1855–1866. [Google Scholar] [CrossRef]
- Schulze, S.; Stengel, R.; Jaekel, N.; Wang, S.-Y.; Franke, G.-N.; Roskos, M.; Schneider, M.; Niederwieser, D.; Al-Ali, H.K. Concomitant and noncanonical JAK2 and MPL mutations in JAK2V617F- and MPLW515 L-positive myelofibrosisGenes Chromosomes. Cancer 2019, 58, 747–755. [Google Scholar]
- Fermo, E.; Zaninoni, A.; Imperiali, F.G.; Bianchi, P.; Colombi, M.; Barcellini, W.; Zanella, A. Analysis of JAK2 V167F Mutation in Myelodysplastic Syndromes. Blood 2007, 110, 4591. [Google Scholar] [CrossRef]
- Zhang, L.; Padron, E.; Lancet, J. The molecular basis and clinical significance of genetic mutations identified in myelodysplastic syndromes. Leuk. Res. 2015, 39, 6–17. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Sangiorgio, V.F.; Geyer, J.T.; Margolskee, E.; Kawaaz, M.A.; Mathew, S.; Tam, W.; Orazi, A. Myeloid neoplasms with isolated del(5q) and JAK2 V617F mutation: A “grey zone” combination of myelodysplastic and myeloproliferative features? Haematologica 2019, 105, e276–e279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, W.; Lea, N.C.; Cervera, J.; Germing, U.; Fenaux, P.; Cassinat, B.; Kiladjian, J.-J.; Varkonyi, J.; Antunovic, P.; Westwood, N.B.; et al. The JAK2 V617F mutation identifies a subgroup of MDS patients with isolated deletion 5q and a proliferative bone marrow. Leukemia 2006, 20, 1319–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellstro<monospace>̈</monospace>m-Lindberg, E.; Cazzola, M. The Role of JAK2 Mutations in RARS and Other MDS. Hematology 2008, 2008, 52–59. [Google Scholar]
- Patnaik, M.M.; Lasho, T.L.; Finke, C.M.; Gangat, N.; Caramazza, D.; Holtan, S.G.; Pardanani, A.; Knudson, R.A.; Ketterling, R.P.; Chen, D.; et al. WHO-defined ‘myelodysplastic syndrome with isolated del(5q)’ in 88 consecutive patients: Survival data, leukemic transformation rates and prevalence of JAK2, MPL and IDH mutations. Leukemia 2010, 24, 1283–1289. [Google Scholar] [CrossRef] [Green Version]
- Ohyashiki, K. The JAK2 V617F tyrosine kinase mutation in myelodysplastic syndromes (MDS) developing myelofibrosis indicates the myeloproliferative nature in a subset of MDS patients. Leukemia 2005, 19, 2359–2360. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Han, B. Comparison and Implications of Mutational Profiles of Myelodysplastic Syndromes, Myeloproliferative Neoplasms, and Myelodysplastic/Myeloproliferative Neoplasms: A Meta-Analysis. Front. Oncol. 2020, 10, 579221. [Google Scholar] [CrossRef]
- Steensma, D.P.; Dewald, G.W.; Lasho, T.L.; Powell, H.L.; McClure, R.F.; Levine, R.L.; Gilliland, D.G.; Tefferi, A. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event inboth “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood 2005, 106, 4. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.J.; Dunphy, C.H.; O’Malley, D.P.; Rice, L.; Ewton, A.A.; Chang, C.C. The implication of identifying JAK2V617F in myeloproliferative neoplasms and myelodysplastic syndromes with bone marrow fibrosis. J. Hematopathol. 2008, 1, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Yip, S.-F.; So, C.-C.; Chan, A.-Y.; Liu, H.-Y.; Wan, T.-K.; Chan, L.-C. The lack of association between JAK2 V617F mutation and myelodysplastic syndrome with or without myelofibrosis. Leukemia 2006, 20, 1165. [Google Scholar] [CrossRef] [Green Version]
- de Renzis, B.; Mansat-De Mas, V.; Wattel, E.; Beyne-Rauzy, O.; Knoops, L.; Cabrespine, A.; Azgui, Z.; Ades, L.; Kiladjian, J.J.; Fenaux, P.; et al. Prognostic impact of JAK2V617F mutation in myelodysplatic syndromes: A matched case control study. Leuk. Res. Rep. 2013, 2, 64–66. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluteau, O.; Sebert, M.; Leblanc, T.; Peffault de Latour, R.; Quentin, S.; Lainey, E.; Hernandez, L.; Dalle, J.H.; Sicre de Fontbrune, F.; Lengline, E.; et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood J. Am. Soc. Hematol. 2018, 131, 717–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, Y.; Narumi, S.; Guan, Y.; Przychodzen, B.P.; Hirsch, C.M.; Makishima, H.; Shima, H.; Aly, M.; Pastor, V.; Kuzmanovic, T.; et al. Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood 2018, 132, 2309–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidsson, J.; Puschmann, A.; Tedgård, U.; Bryder, D.; Nilsson, L.; Cammenga, J. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 2018, 32, 1106–1115. [Google Scholar] [CrossRef]
- Weinberg, I.; Borohovitz, A.; Krichevsky, S.; Perlman, R.; Ben-Yehuda, A.; Ben-Yehuda, D. Janus Kinase V617F mutation in cigarette smokers. Am. J. Hematol. 2011, 87, 5–8. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Literature (Reference Number) | Population of Concern to Our Study | Number of Participants in Study | JAK2 Mutation/Other Studies | Frequency (Number of Cases) with JAK2 V617F Mutation/Variants | Conclusions |
|---|---|---|---|---|---|
| Nielson C. et al. [1] | General healthy | 10,507 | JAK2 V617F | 0.2% (18) | JAK2 V617F is very rare in the general healthy population and is associated with increased morbidity and mortality. |
| Nielson C. et al. [4] | General healthy | 49,488 | JAK2 V617F | 0.1% (68) | JAK2 V617F somatic mutation in general healthy participants has a high diagnostic value for myeloproliferative neoplasms when combined with hematological indices. |
| Trelinski J. et al. [5] | E.T. | 43 | JAK2 V617F | Impaired apoptosis of megakaryocytes and bone marrow mononuclear cells. | |
| Schulze et al. [9] | MF (PMF, post-ET-MF, and post-PV-MF) | 128 | JAK2 V617FMPL | 14.6% (82) JAK2R1063H (6) JAK2R893T(1) JAK2T525A(1) MPLY591D(3) MPLW515 L(2) MPLE335K(1) | Recurrent concomitant classical and/or noncanonical JAK2- and MPL-mutations detected in 15.7% of JAK2V617F- and MPLW515-positive MF patients and appear to express genotype—phenotype associations. |
| Fermo et al. [10] | MDS | 53 | JAK2 V617F | 5% (3) | In MDS, JAK2 V617F has a low prevalence and identifies a subset with proliferative characteristics. |
| Bejar et al. [12] | MDS | 439 | JAK2 | 3% (13) | JAK2 mutations are rare in MDS. |
| Haferlach et al. [13] | MDS | 944 | JAK2 | 4–5% | RARS-T usually associated with JAK2 and SF3B1 co-mutations with lower JAK2 mutation burden suggesting they evolved from RARS or RCMD-RS. |
| Sangiorgio et al. [14] | MDS with del(5q) | 47 | JAK2 V617F | 12.7% (6) | JAK2-mutated myeloid neoplasms with isolated del(5q) show overlap MPN/MDS features |
| Ingram et al. [15] | MDS with del(5q) | 97 | JAK2 V617F | 6.2% (6) | JAK2-mutated cases with deletion 5q are usually hypercellular. It is unclear whether the JAK2 mutation is an early or late event. |
| Patnaik et al. [17] | MDS with del(5q) | 88 | JAK2 V617F | 6.4% (5) | No significant difference in blood counts or clinical outcome between patients with and without JAK2 V617F. |
| Ohyashiki et al. [18]. | MDS with and without fibrosis | 38 (MDS without fibrosis) +6 (MDS with fibrosis) | JAK2 V617F | (2) | MDS with fibrosis may sometimes be associated with JAK2 V617F. |
| Wan Z. et al. [19] | MDS | 3100 | JAK2 | 2.88% | Meta-analysis with extensive literature review. JAK2 mutations are rare in MDS. |
| Steensma et al. [20] | MDS | 101 | JAK2 V617F | 5% (5) | JAK2 V617F mutation is infrequent in MDS. |
| Olsen R et al. [21] | MPN/MDS and MDS with fibrosis | 45 | JAK2 V617F | 0% in non-MPN cases | JAK2 V617F is useful in discriminating MDS with fibrosis from MPN cases. |
| S.F Yip et al. [22] | MDS with and without fibrosis | 186 include 39 assessed for JAK2 | JAK2 V617F | 0% | JAK2 V617F is unlikely to play a role in MDS with or without fibrosis biology and MPNs need to be strictly excluded. |
| Benoit de Renzis et al. [23]. | MDS | 132 | JAK2 V617F | (37). | JAK2 V617F is associated with a lower incidence of progression to AML and better overall survival. |
| Lindsley R.C. et al. [24]. | MDS before and after stem cell transplant | 1514 | JAK2 V617F | 2% (28) | JAK2 V617F mutation was associated with shorter survival and higher rate of death without relapse after transplant. High-intensity conditioning regimens may not benefit patients with JAK2 mutations. May benefit from JAK2 inhibitors. |
| C | G | Clinical Presentation | Hb g/dL | MCV fl | WBC K/µL | ANC K/µL | Plt K/µL | LDH IU/L | Pathologic Diagnosis at Presentation | Karyotype FISH | JAK2 Mutation Variant(VAF) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | Progressive anemia | 9.5 | 88 | 5.0 | 3.04 | 293 | 164 | Hypoplastic MDS with fibrosis | 46,XY,+1,der(1;7)(q10;p10)[10]/46,XY[10] FISH: Deletion 7q31 | JAK2 V617F (8%) |
| 2 | F | Progressive macrocytic anemia | 8.4 | 111 | 5.7 | 2.90 | 162 | 178 | MDS-RS-MLD | 46,XX,dup(1)(q21q32)[11]/46,XX[9] FISH: Gain of the long arm of chromosome 1 | JAK2 R564L (46%) |
| 3 | M | Pancytopenia | 7.5 | 103 | 1.0 | 0.14 | 14 | 207 | MDS-EB2 | Complex karyotype with four clones 45,XY,der(4)t(4;17)(q21;q11.2),add(5)(q11.2),-7,-17,+r[1]/43,XY,der(4)t(4;17)(q21;q11.2),add(5)(q11.2),-7,dic(7;12)(q32;q15),-17[8]/43,XY,der(4)t(4;17)(q21;q11.2),add(5)(q11.2),-7,dic(7;12)(q32;q15),-17, add(21)(p11.2)[6]/ 42,XY,der(4)t(4;17)(q21;q11.2),add(5)(q11.2),-7,dic(7;12)(q32;q15),-17,-21, add(22)(q11.2)[4]/46,XY[1] FISH: Deletion 5q31, monosomy 7, monosomy 17 with deletion of TP53. | JAK2 I670V (44%) |
| C: Case number, G: Gender, M: Male, F: Female, Hb: Hemoglobin, MCV: Mean Corpuscular Volume, WBC: White Blood Cell, ANC: Absolute Neutrophil Count, Plt: Platelet, LDH: lactate dehydrogenase | |||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delio, M.; Bryke, C.; Mendez, L.; Joseph, L.; Jassim, S. JAK2 Mutations Are Rare and Diverse in Myelodysplastic Syndromes: Case Series and Review of the Literature. Hematol. Rep. 2023, 15, 73-87. https://doi.org/10.3390/hematolrep15010008
Delio M, Bryke C, Mendez L, Joseph L, Jassim S. JAK2 Mutations Are Rare and Diverse in Myelodysplastic Syndromes: Case Series and Review of the Literature. Hematology Reports. 2023; 15(1):73-87. https://doi.org/10.3390/hematolrep15010008
Chicago/Turabian StyleDelio, Melissa, Christine Bryke, Lourdes Mendez, Loren Joseph, and Sarmad Jassim. 2023. "JAK2 Mutations Are Rare and Diverse in Myelodysplastic Syndromes: Case Series and Review of the Literature" Hematology Reports 15, no. 1: 73-87. https://doi.org/10.3390/hematolrep15010008
APA StyleDelio, M., Bryke, C., Mendez, L., Joseph, L., & Jassim, S. (2023). JAK2 Mutations Are Rare and Diverse in Myelodysplastic Syndromes: Case Series and Review of the Literature. Hematology Reports, 15(1), 73-87. https://doi.org/10.3390/hematolrep15010008