
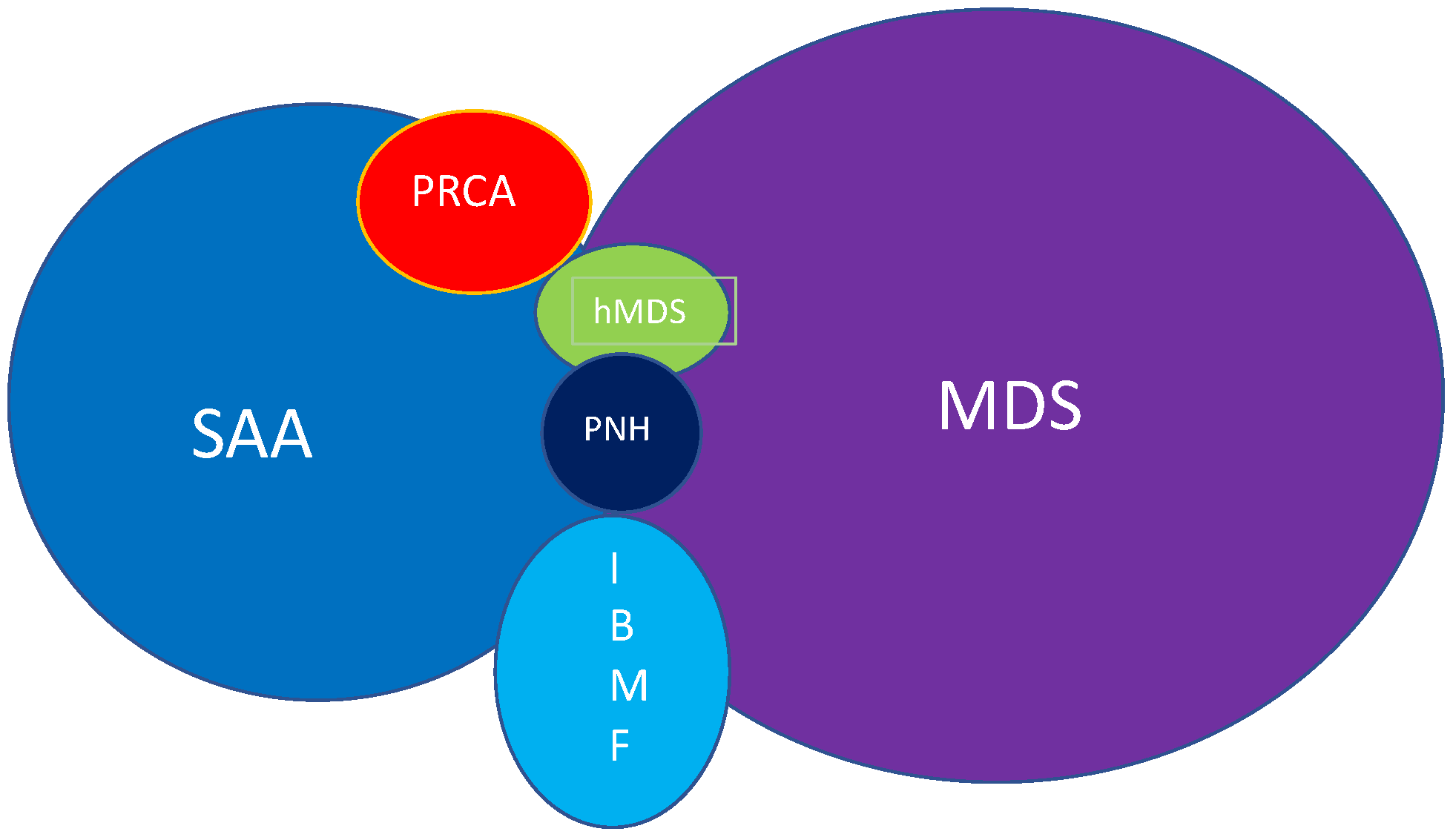
The Benign Clone Causing Aplastic Anaemia


{kind=link}
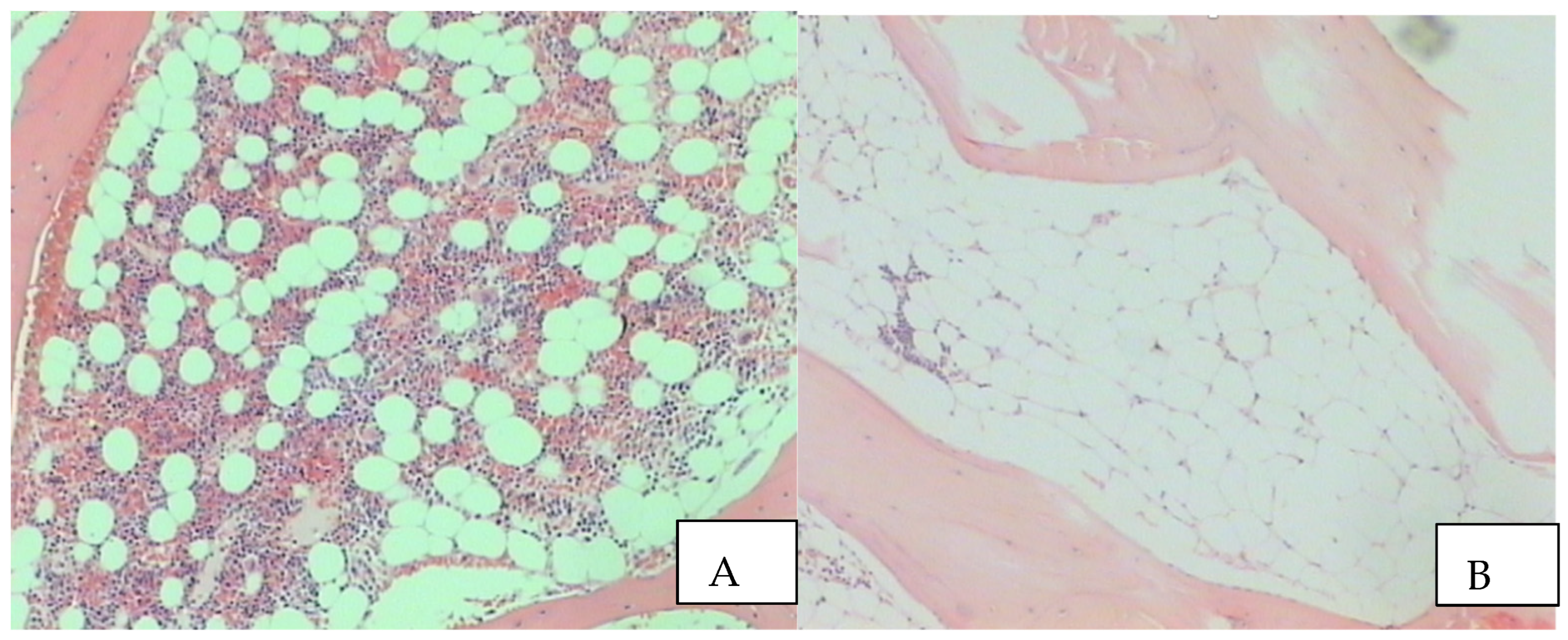{kind=link}
Abstract
:1. The Benign Clone
2. Insight in the Disease
3. Paroxysmal Nocturnal Hemoglobinuria and Aplastic Anaemia
4. Clonal Haematopoiesis and Malignancy
5. SAA Treatment
6. Chimerism Issues in SAA
7. SAA Guidelines
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brzezniakiewicz-Janus, K.; Rupa-Matysek, J.H.; Hoppe, A.; Gil, L. Impact of clonal hematopoiesis on outcomes in patients with aplastic anemia. Acta Haematol. Pol. 2021, 52, 543–551. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Baumann, I. Classification of childhood aplastic anemia and myelodysplastic syndrome. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strahm, B.; Locatelli, F.; Bader, P.; Ehlert, K.; Kremens, B.; Zintl, F.; Führer, M.; Stachel, D.; Sykora, K.W.; Sedlacek, P.; et al. Reduced intensity conditioning in unrelated donor transplantation for refractory cytopenia in childhood. Bone Marrow Transpl. 2007, 40, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socié, G.; Rosenfeld, S.; Frickhofen, N.; Gluckman, E.; Tichelli, A. Late clonal diseases of treated aplastic anemia. Seminal. Hematol. 2000, 37, 91–101. [Google Scholar] [CrossRef]
- Babushok, D.V.; Perdigones, N.; Perin, J.C.; Olson, T.S.; Ye, W.; Roth, J.J.; Lind, C.; Cattier, C.; Li, Y.; Hartung, H.; et al. Emergence of clonal hematopoiesis in the majority of patients with acquired aplastic anemia. Cancer Genet. 2015, 208, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Negoro, E.; Negata, Y.; Clemente, M.J.; Hosono, N.; Shen, W.; Nazha, S.; Yoshizato, T.; Hirsch, C.; Przychodzen, B.; Mahfouz, R.Z.; et al. Origins of myelodysplastic syndrome after aplastic anemia. Blood 2017, 130, 1953–1957. [Google Scholar] [CrossRef] [Green Version]
- Gadalla, S.M.; Wang, T.; Dagnall, C.; Spellman, S.R.; Lee, S.J.; Williams, K.M.; Wong, J.Y.; De Vivo, I.; Savage, S.A. Association between donor leukocyte telomere length and survival after unrelated allogeneic hematopoietic cell transplantation for severe aplastic anemia. JAMA 2015, 313, 594–602. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Lee, J.W.; Lee, S.E.; Cho, B.S.; Kim, M.; Eom, K.S.; Kim, Y.J.; Kim, H.J.; Lee, S.; Min, C.K.; et al. The characteristics and clinical outcome of adult patients with aplastic anemia and abnormal cytogenetics at diagnosis. Genes. Chromosomes Cancer 2010, 49, 844–850. [Google Scholar] [CrossRef]
- Norasetthada, L.; Wongkhantee, S.; Chaipokam, J.; Charoenpeasset, K.; Chuncharunee, S.; Rojnuckarin, P.; Sirijerechai, C.; Wanachiwanawin, W.; Issaragisil, S. Thai Aplastic Anemia Study Group. Adult aplastic anemia in Thailand: Incidence and treatment outcome from a prospective nationwide population-based study. Ann. Hematol. 2021, 1000, 2443–2452. [Google Scholar] [CrossRef]
- Young, N.S. Aplastic Anemia. N. Engl. J. Med. 2018, 379, 1843–1856. [Google Scholar] [CrossRef]
- Guidice, V.; Selleri, C. Aplastic anemia: Pathophysiology. Semin. Hematol. 2022, 59, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Camitta, B.M.; Rappeport, J.M.; Parkman, R.; Nathan, D.G. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood 1975, 45, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejewski, J.P.; Risitano, A.; Sloand, E.M.; Nunez, O.; Young, N.S. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood 2002, 99, 3129–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeZem, A.E.; Churpek, J.E. Approach to the diagnosis of aplastic anemia. Blood Adv. 2021, 5, 2660–2671. [Google Scholar] [CrossRef]
- Durrani, J.; Maciejewski, J.P. Idiopathic aplastic anemia vs hypocellular myelodysplastic syndrome. Hematol. Am. Soc. Hematol. Educ. Program. 2019, 2019, 97–104. [Google Scholar] [CrossRef]
- Votavova, H.; Belickova, M. Hypoplastic myelodysplastic syndrome and acquired aplastic anemia: Immune-mediated bone marrow failure syndromes. Int. J. Oncol. 2022, 60, 7. [Google Scholar] [CrossRef]
- Yoshizato, T.; Dumitriu, B.; Hosokawa, K.; Makishima, H.; Yoshida, K.; Townsley, D.; Sato-Otsubo, A.; Sato, Y.; Liu, D.; Suzuki, H.; et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N. Engl. J. Med. 2015, 373, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, T.; Normitsu, I. Relationship between Aplastic Anemia and Paroxysmal Nocturnal Hemoglobinuria. Int. J. Hematol. 2002, 75, 117–122. [Google Scholar] [CrossRef]
- Babushok, D.V. When does a PNH clone have clinical significance? Hematol. Am. Soc. Educ. Program. 2021, 2021, 143–152. [Google Scholar] [CrossRef]
- McCann, S.R. Blood Matters; Interview with Lucio Luzzatto: Stockholm, Sweden, 2013; ISBN 978-90-823759-1-6. [Google Scholar]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef]
- Afable, M.G.; Tiu, R.V.; Maciejewski, J.P. Clonal evolution in aplastic anemia. Am. Soc. Hematol. Educ. Program. 2011, 128, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marnell, C.S.; Bick, A.; Nataragjan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cell. Cardiol. 2021, 161, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Locasciulli, A.; Oneto, R.; Bacigalupo, A.; Socié, G.; Korthof, E.; Bekassy, A.; Schrezenmeier, H.; Passweg, J.; Fuhrer, M. Severe Aplastic Anemia Working Party of the European Blood and Marrow Transplant Group. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: A report from the European Group for Blood and Marrow Transplantation (EBMT). Haematologica 2007, 92, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Gale, R.P.; Eapen, M.; Logan, B.; Zhang, M.; Lazarus, H.M. Are there roles for observational database studies and structured quantification of expert opinion to answer therapy controversies in Transplants? Bone Marrow Transplant. 2009, 43, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Tsugi, N.; Hosokawa, K.; Urushihara, R.; Tanabe, M.; Katagiri, T.; Ozawa, T.; Takamatsu, H.; Ishiyama, K.; Yamazaki, H.; Kishi, H.; et al. Frequent HLA-DR loss on hemopoietic stem progenitor cells in patients with cyclosporine-dependent aplastic anemia carrying HLA-DR15. Leukemia 2022, 36, 1666–1675. [Google Scholar] [CrossRef]
- Zaimoku Patel, B.A.; Adams, S.; Shaloub, R.; Groarke, E.; Lee, A.I.C.; Kajigaya, S.; Feng, X.; Rios, O.J.; Eager, H.; Alemu, L.; et al. HLA associations, somatic loss of HLA expression and clinical outcomes in immune aplastic anemia. Blood 2021, 138, 2781–2898. [Google Scholar] [CrossRef]
- Young, N.L. Current concepts in the pathophysiology and treatment of aplastic anemia. Am. Soc. Hematol. Educ. Program. 2013, 76, 76–78. [Google Scholar] [CrossRef] [Green Version]
- Peffault de Latour, P.; Kulasekarara, A.; Iacobelli, S.; Trewel, S.R.; Cook, R.; Griffin, M.; Halkes, C.J.M.; Recher, C.; Barraco, F.; Forcade, E.; et al. for the Severe Aplastic Anemia Working Party of the European Society for Blood and marrow Transplantation. Eltrombopag Added to Immunosuppression in Severe Aplastic Anemia. N. Engl. J. Med. 2022, 286, 11–23. [Google Scholar] [CrossRef]
- Bacigalupo, A. How I treat acquired aplastic anemia. Blood 2017, 129, 1428–1436. [Google Scholar] [CrossRef]
- Cesaro, S.; Peffault de Latour, R.; Tridello, G.; Pillon, M.; Carlson, K.; Fagioli, F.; Jouet, J.-P.; Koh, M.B.C.; Panizzolo, I.R.; Kyrcz-Krzemien, S.; et al. Second allogeneic stem cell transplant for aplastic anaemia; a retrospective study by the severe aplastic anaemia Working Party of the EBMT. Br. J. Haematol. 2015, 171, 606–614. [Google Scholar] [CrossRef] [PubMed]
- McCann, S.R.; Bacigalupo, A.; Gluckman, E.; Hinterberger, W.; Hows, J.; Ljungman, P.; Marin, P.; Nissen, C.; van’t Veer Kerthof, E.; Raghavachar, A.; et al. Graft rejection and second bone marrow transplantation for acquired aplastic anaemia: A report from the aplastic anaemia Working Party of the EBMT. Bone Marrow Transplant. 1994, 13, 233–237. [Google Scholar]
- Dufour, C.; Veys, P.; Carraro, E.; Bhatnagar, N.; Pillon, M.; Wynn, R.; Gibson, B.; Vora, A.J.; Steward, C.G.; Ewins, A.M.; et al. Similar outcomes of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on behalf of the UK Paediatric BMT Working Party, Paediatric Diseases and Severe Aplastic Working Party of the EBMT. Br. J. Haematol. 2015, 171, 585–594. [Google Scholar] [CrossRef]
- Young, M.E.; Potter, V.; Kulasekararaj, A.G.; Mufti, G.J.; Marsh, J.C. Haematopoietic stem cell transplantation for acquired aplastic anaemia. Curr. Opin. Hematol. 2013, 20, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Sociè, G.; Henry-Amar, M.; Cossett, J.M.; Devergie, A.; Grinsky, T.; Gluckman, E. Increased Incidence of Solid Malignant tumors after Bone Marrow Transplantation for Severe Aplastic Anemia. Blood 1991, 78, 277–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawler, M.; McCann, S.R.; March, J.C.; Ljungman, P.; Hows, J.; Vandenberghe, E.; O’Riordan, J.; Locasciulli, A.; Socié, G.; Kelly, A.; et al. Severe Aplastic Anaemia Working Party of the European Blood and Marrow Transplant Group. Serial chimerism analyses indicate that mixed hematopoietic chimerism influences the probability of graft rejection and disease recurrence following allogeneic stem cell transplantation (SCT) for severe aplastic anaemia (SAA): Indication for routine assessment of chimerism post SCT for SAA. Br. J. Haematol. 2009, 144, 933–934. [Google Scholar] [PubMed]
- Piccin, A.; McCann, S.; Socié, G.; Oneto, R.; Bacigalupo, A.; Locasciulli, A.; Marsh, J.; Schrezenmeier, H.; Tichelli, A.; Hand, E.; et al. Survival of patients with documented autologous recovery after SCT for severe aplastic anaemia: A study by the WPSAA of the EBMT. Bone Marrow Transpl. 2010, 45, 1008–1013. [Google Scholar] [CrossRef] [Green Version]
- Marsh, J.C.; Ball, S.E.; Cavenagh, J.; Darbyshire, P.; Dokal, I.; Gordon-Smith, E.C.; Keidan, J.; Laurie, A.; Martin, A.; Merciera, J.; et al. Guidelines for the diagnosis and management of aplastic anaemia. Br. J. Haematol. 2009, 147, 43–70. [Google Scholar] [CrossRef] [PubMed]
- Killick, S.B.; Brown, N.; Cavenagh, J.; Dokal, I.; Foukaneli, T.; Hill, A.; Hillmen, P.; Ireland, R.; Kulasekaraj, A.; Mufti, G.; et al. Wood Guidelines for the diagnosis and management of adult aplastic anaemia. British Society for Standards in Haematology. Br. J. Haematol. 2016, 172, 187–207. [Google Scholar] [CrossRef]
- McCann, S.R. Can guidelines inhibit innovation and critical thinking? Bone Marrow Transplant. 2020, 55, 1217–1219. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCann, S.R.; Piccin, A. The Benign Clone Causing Aplastic Anaemia. Thalass. Rep. 2023, 13, 157-164. https://doi.org/10.3390/thalassrep13020015
McCann SR, Piccin A. The Benign Clone Causing Aplastic Anaemia. Thalassemia Reports. 2023; 13(2):157-164. https://doi.org/10.3390/thalassrep13020015
Chicago/Turabian StyleMcCann, Shaun R., and Andrea Piccin. 2023. "The Benign Clone Causing Aplastic Anaemia" Thalassemia Reports 13, no. 2: 157-164. https://doi.org/10.3390/thalassrep13020015
APA StyleMcCann, S. R., & Piccin, A. (2023). The Benign Clone Causing Aplastic Anaemia. Thalassemia Reports, 13(2), 157-164. https://doi.org/10.3390/thalassrep13020015