Dietary Advanced Glycation Endproducts Decrease Glucocorticoid Sensitivity In Vitro

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Preparation of Dietary AGEs
2.3. Determination of Endotoxin in Dietary AGEs
2.4. Cell Culture and Exposure
2.5. Quantification of Interleukin-8 (IL8) Release by ELISA
2.6. Quantification of Glucocorticoid Receptor Protein Levels by Western Blot
2.7. Quantification of ROS Levels by DCFH-DA Assay
2.8. Statistics
3. Results
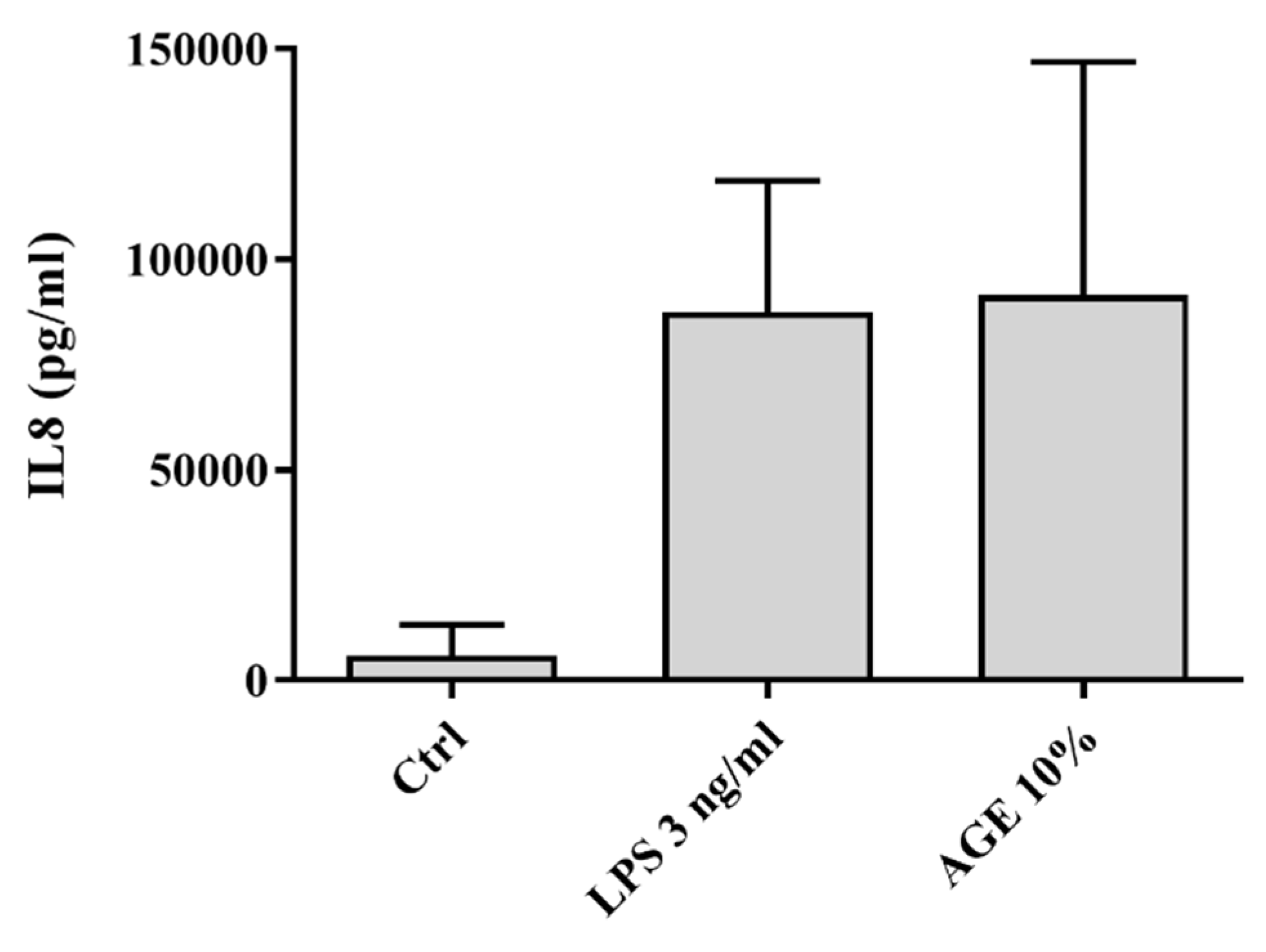
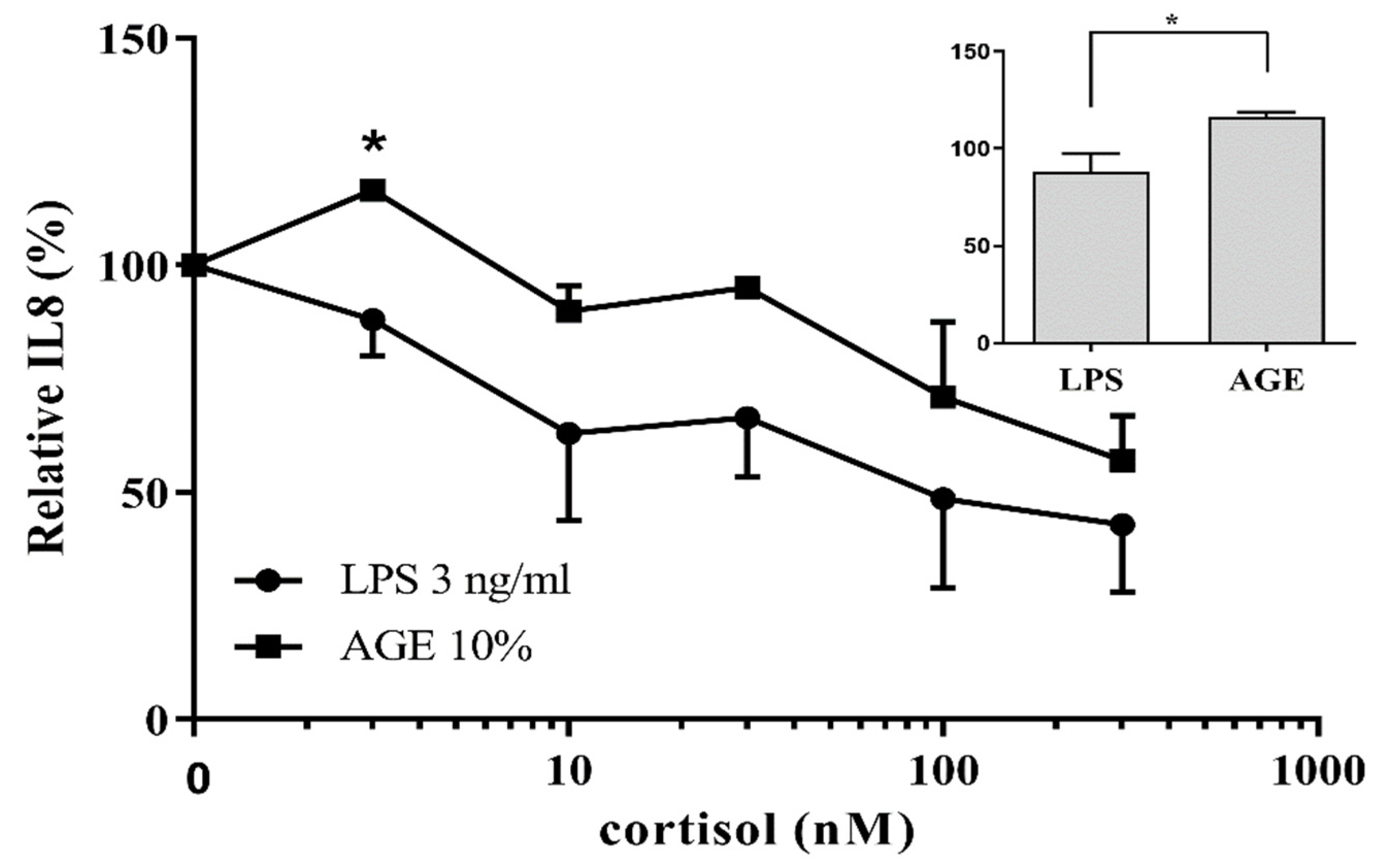
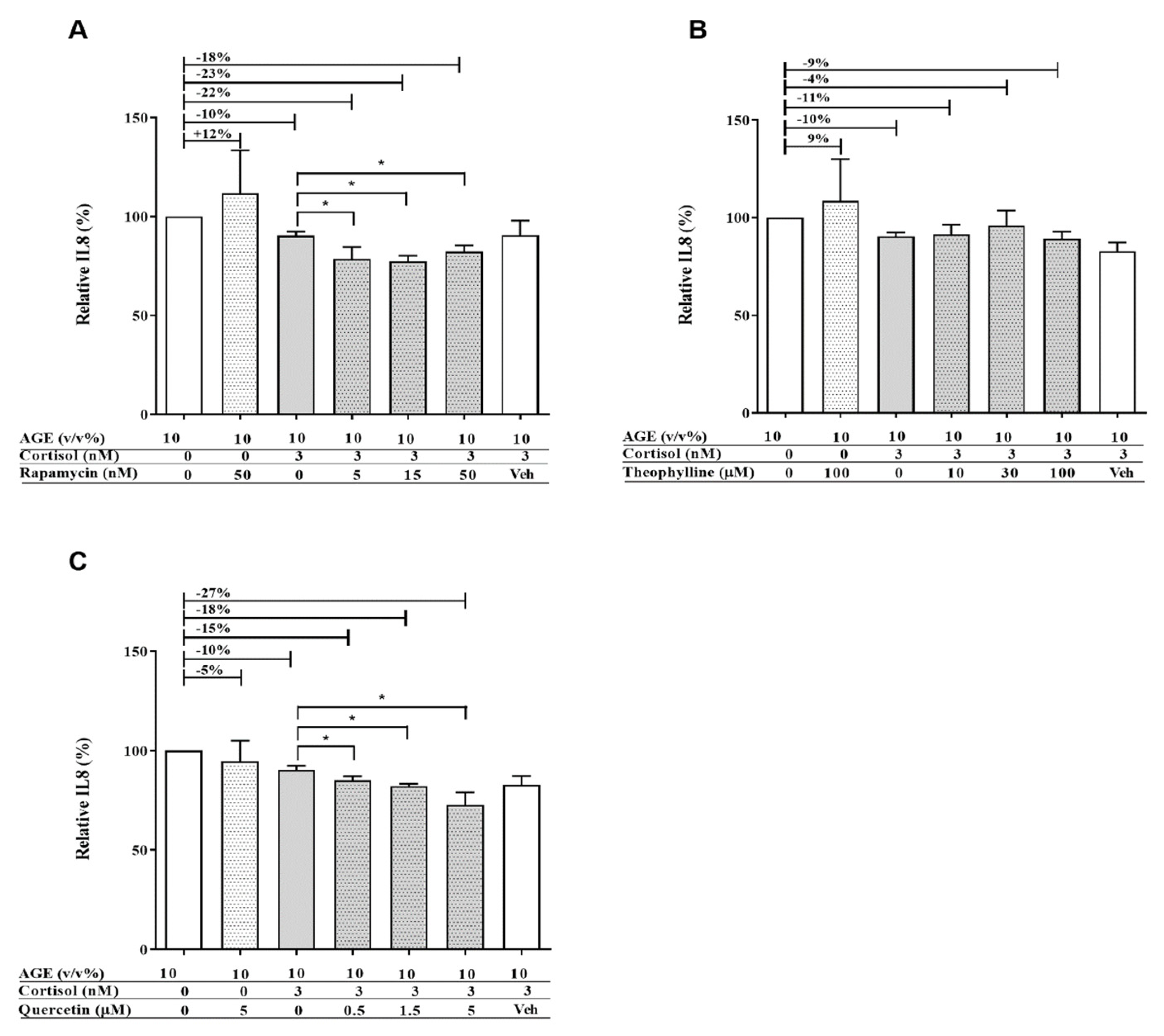
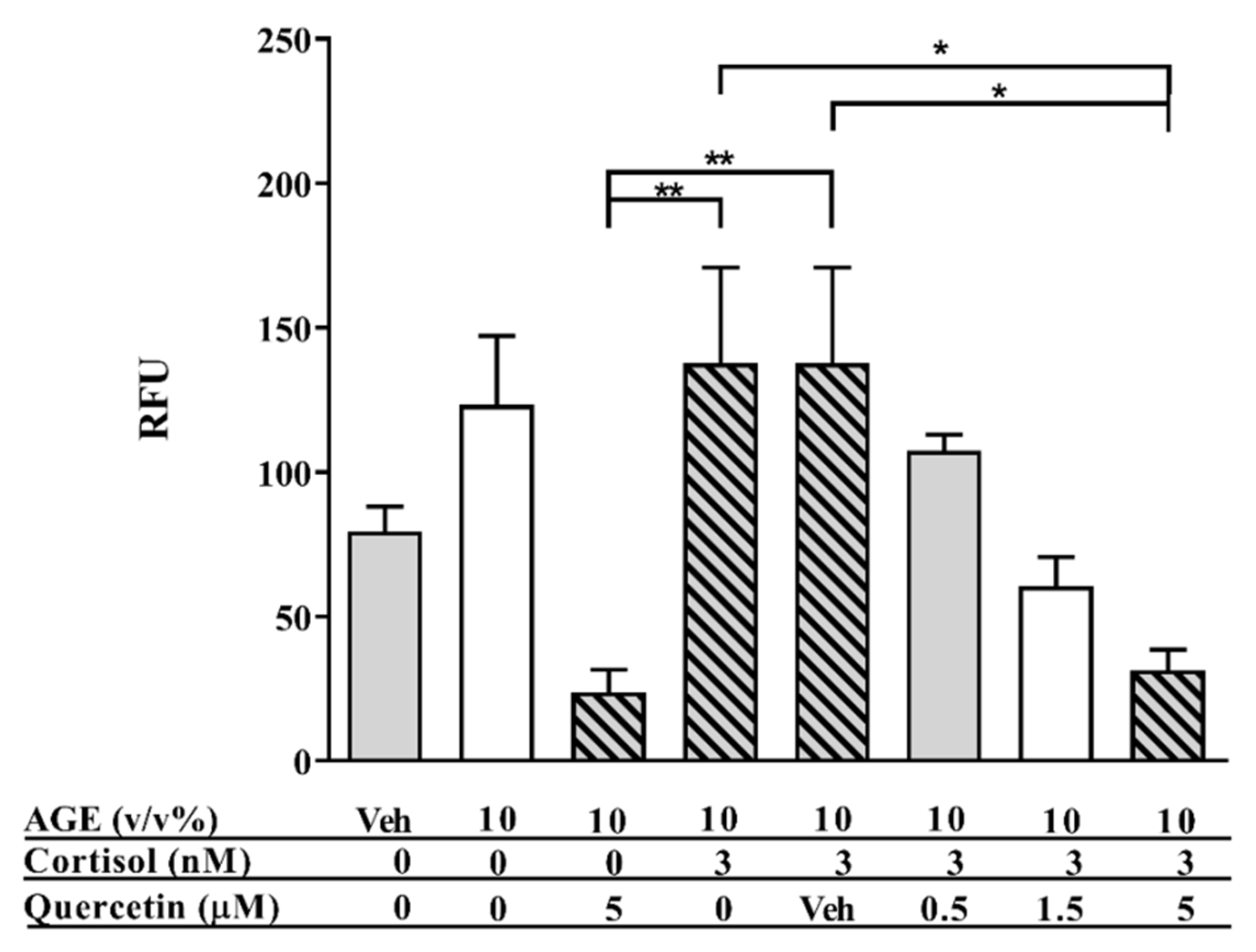
3.1. AGE-Induced Glucocorticoid Resistance
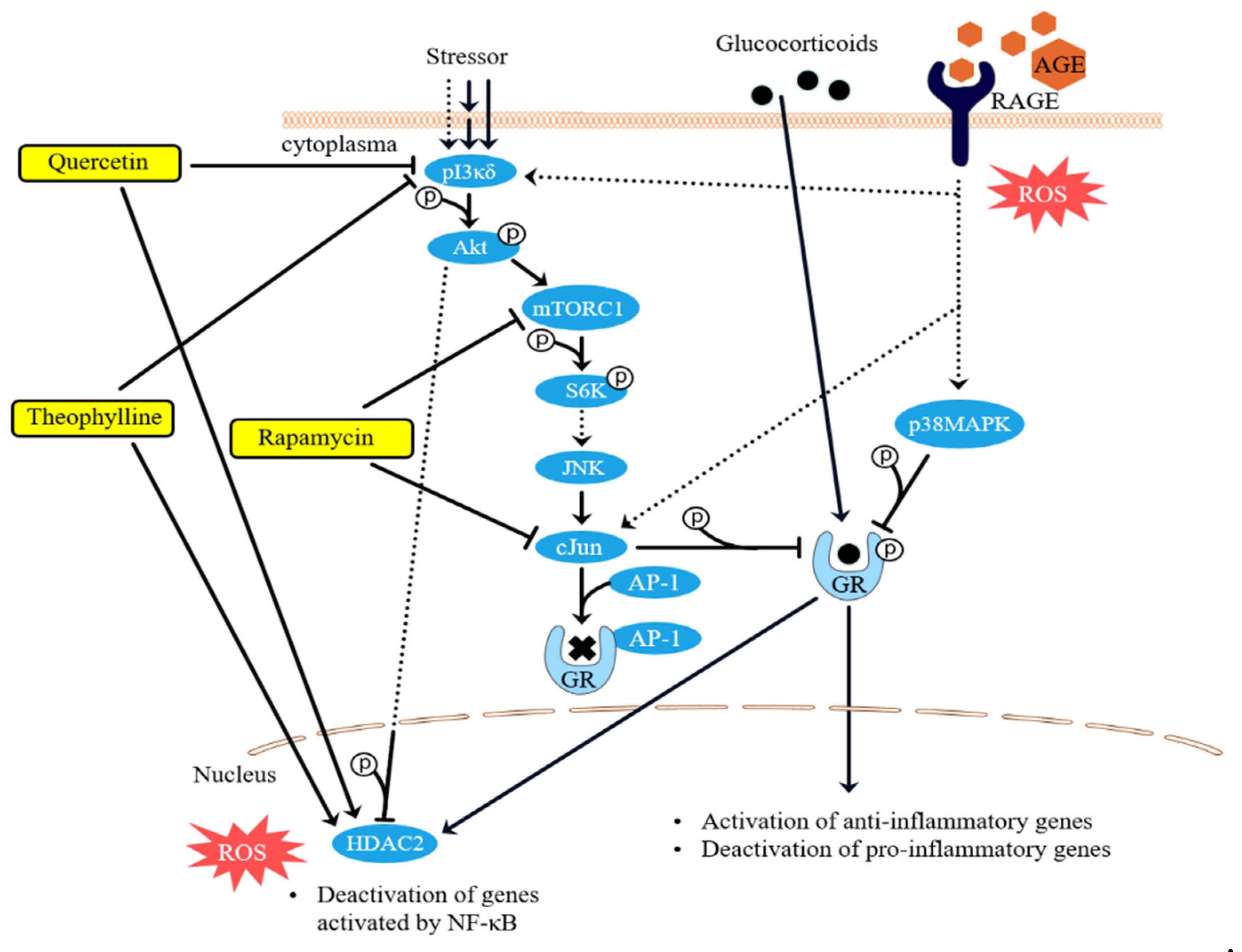
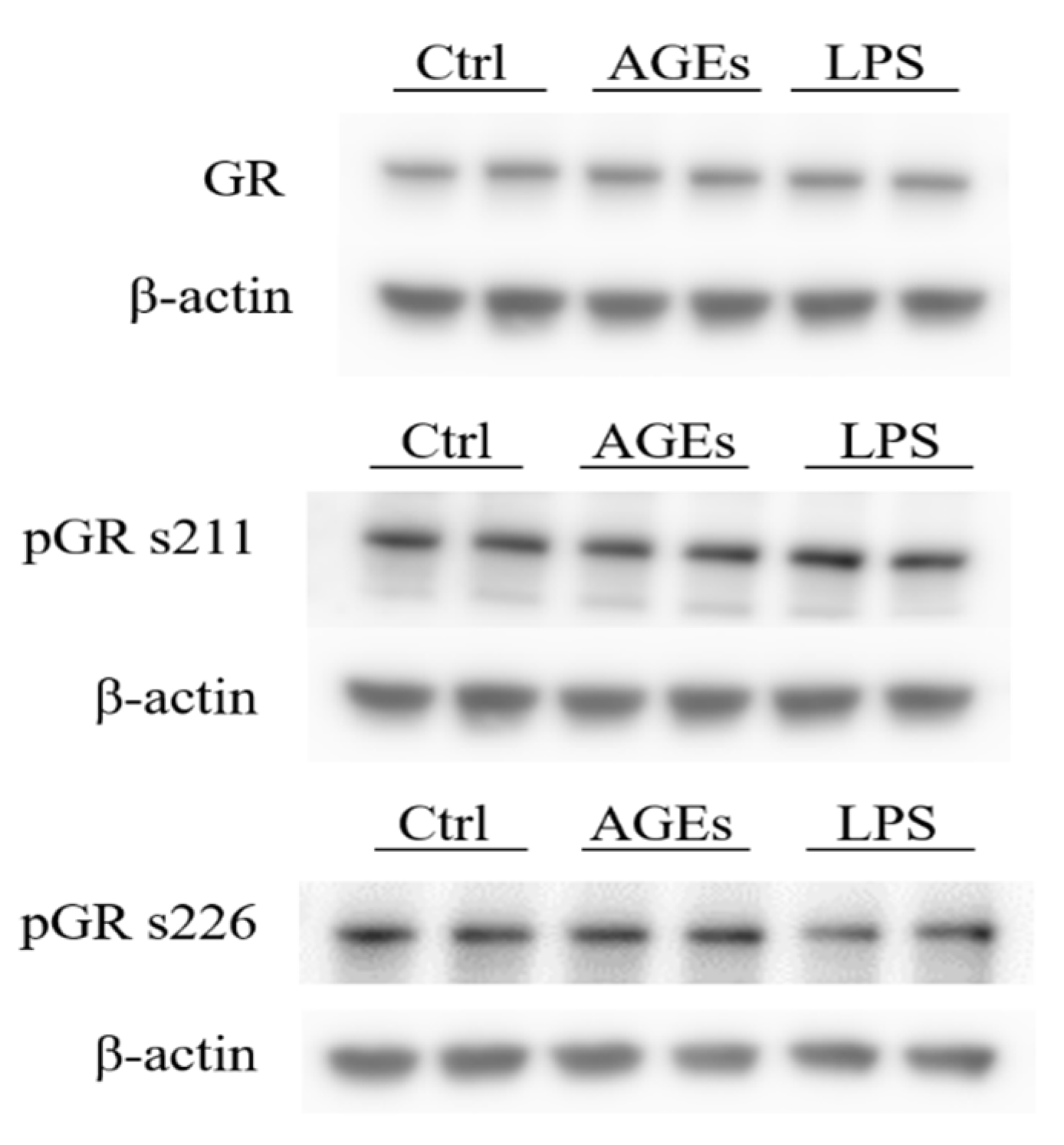
3.2. Phosphorylation of the Glucocorticoid Receptor
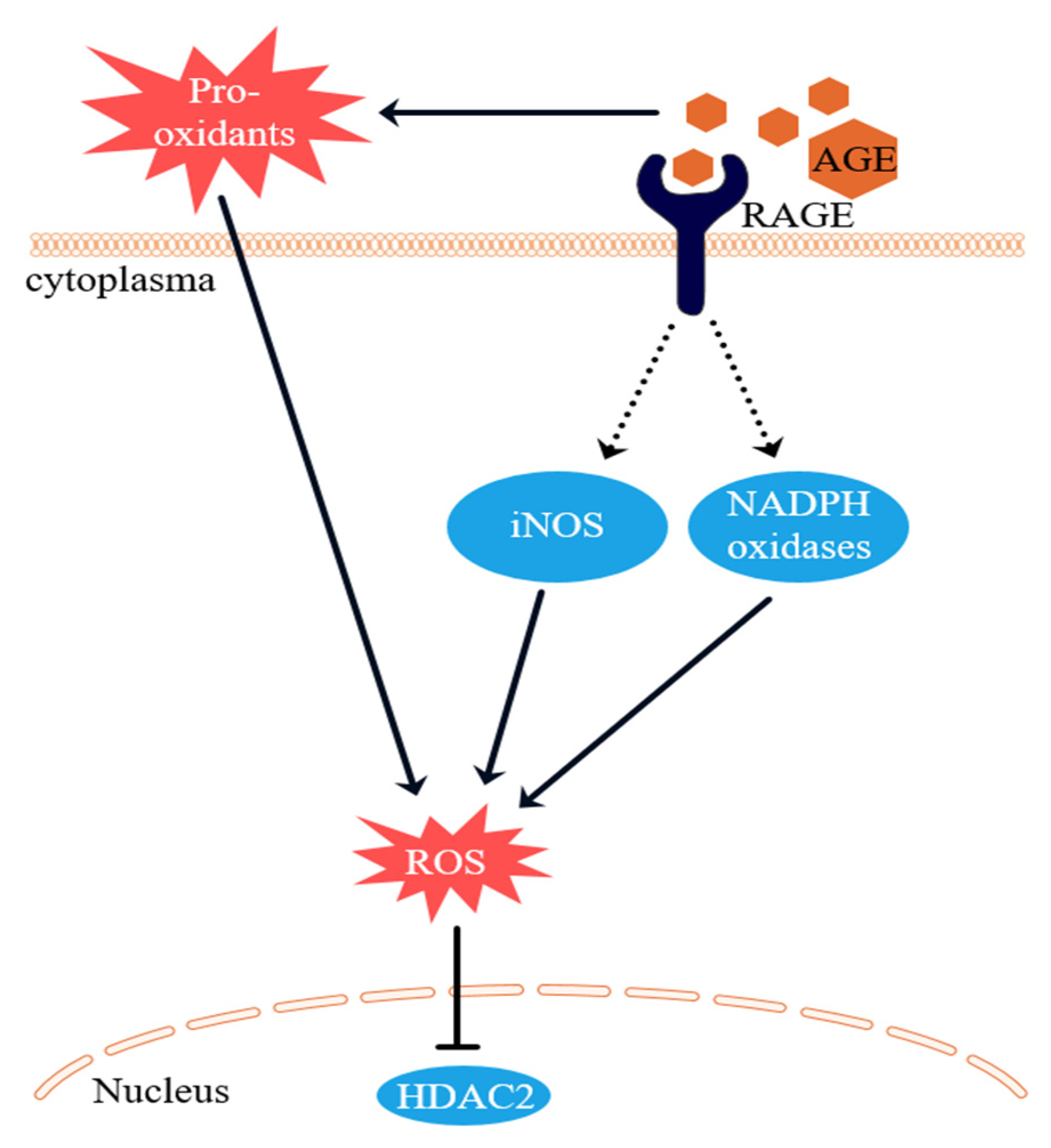
3.3. Intracellular ROS Levels
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tung, J.; Loftus, E.V., Jr.; Freese, D.K.; El-Youssef, M.; Zinsmeister, A.R.; Melton, L.J., 3rd; Harmsen, W.S.; Sandborn, W.J.; Faubion, W.A., Jr. A population-based study of the frequency of corticosteroid resistance and dependence in pediatric patients with Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2006, 12, 1093–1100. [Google Scholar] [CrossRef]
- Munkholm, P.; Langholz, E.; Davidsen, M.; Binder, V. Frequency of glucocorticoid resistance and dependency in Crohn’s disease. Gut 1994, 35, 360–362. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Mechanisms and resistance in glucocorticoid control of inflammation. J. Steroid Biochem. Mol. Biol. 2010, 120, 76–85. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Barnes, P.J.; Adcock, I.M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 2000, 20, 6891–6903. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.R. MAP kinase phosphatase 1: A novel mediator of biological effects of glucocorticoids? J. Endocrinol. 2003, 178, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Corticosteroid effects on cell signalling. Eur. Respir. J. 2006, 27, 413–426. [Google Scholar] [CrossRef]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, M.W.; Staples, K.J.; Smith, S.J.; Barnes, P.J.; Newton, R. Glucocorticoid inhibition of granulocyte macrophage-colony-stimulating factor from T cells is independent of control by nuclear factor-kappaB and conserved lymphokine element 0. Am. J. Respir. Cell Mol. Biol. 2004, 30, 555–563. [Google Scholar] [CrossRef]
- Weigel, N.L.; Moore, N.L. Steroid receptor phosphorylation: A key modulator of multiple receptor functions. Mol. Endocrinol. 2007, 21, 2311–2319. [Google Scholar] [CrossRef] [Green Version]
- Matthews, J.G.; Ito, K.; Barnes, P.J.; Adcock, I.M. Defective glucocorticoid receptor nuclear translocation and altered histone acetylation patterns in glucocorticoid-resistant patients. J. Allergy Clin. Immunol. 2004, 113, 1100–1108. [Google Scholar] [CrossRef]
- Szatmary, Z.; Garabedian, M.J.; Vilcek, J. Inhibition of glucocorticoid receptor-mediated transcriptional activation by p38 mitogen-activated protein (MAP) kinase. J. Biol. Chem. 2004, 279, 43708–43715. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.L.; Webb, M.S.; Copik, A.J.; Wang, Y.; Johnson, B.H.; Kumar, R.; Thompson, E.B. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: Correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol. Endocrinol. 2005, 19, 1569–1583. [Google Scholar] [CrossRef] [Green Version]
- Irusen, E.; Matthews, J.G.; Takahashi, A.; Barnes, P.J.; Chung, K.F.; Adcock, I.M. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: Role in steroid-insensitive asthma. J. Allergy Clin. Immunol. 2002, 109, 649–657. [Google Scholar] [CrossRef]
- Ito, K.; Hanazawa, T.; Tomita, K.; Barnes, P.J.; Adcock, I.M. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: Role of tyrosine nitration. Biochem. Biophys. Res. Commun. 2004, 315, 240–245. [Google Scholar] [CrossRef]
- Kirkham, P.; Rahman, I. Oxidative stress in asthma and COPD: Antioxidants as a therapeutic strategy. Pharmacol. Ther. 2006, 111, 476–494. [Google Scholar] [CrossRef]
- Ruijters, E.J.; Haenen, G.R.; Willemsen, M.; Weseler, A.R.; Bast, A. Food-Derived Bioactives Can Protect the Anti-Inflammatory Activity of Cortisol with Antioxidant-Dependent and -Independent Mechanisms. Int. J. Mol. Sci. 2016, 17, 239. [Google Scholar] [CrossRef] [Green Version]
- van der Lugt, T.; Weseler, A.; Gebbink, W.; Vrolijk, M.; Opperhuizen, A.; Bast, A. Dietary Advanced Glycation Endproducts Induce an Inflammatory Response in Human Macrophages In Vitro. Nutrients 2018, 10, 1868. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Mendez, J.D.; Mendez-Valenzuela, V.; Aguilar-Hernandez, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bugel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef]
- Ruiz-Leal, M.; George, S. An In Vitro procedure for evaluation of early stage oxidative stress in an established fish cell line applied to investigation of PHAH and pesticide toxicity. Mar. Environ. Res. 2004, 58, 631–635. [Google Scholar] [CrossRef]
- Bansal, S.; Siddarth, M.; Chawla, D.; Banerjee, B.D.; Madhu, S.V.; Tripathi, A.K. Advanced glycation end products enhance reactive oxygen and nitrogen species generation in neutrophils In Vitro. Mol. Cell. Biochem. 2012, 361, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917. [Google Scholar] [CrossRef]
- Ruijters, E.J.; Haenen, G.R.; Weseler, A.R.; Bast, A. The cocoa flavanol (-)-epicatechin protects the cortisol response. Pharmacol. Res. 2014, 79, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Mitani, A.; Azam, A.; Vuppusetty, C.; Ito, K.; Mercado, N.; Barnes, P.J. Quercetin restores corticosteroid sensitivity in cells from patients with chronic obstructive pulmonary disease. Exp. Lung Res. 2017, 43, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Role of HDAC2 in the pathophysiology of COPD. Annu. Rev. Physiol. 2009, 71, 451–464. [Google Scholar] [CrossRef]
- To, Y.; Ito, K.; Kizawa, Y.; Failla, M.; Ito, M.; Kusama, T.; Elliott, W.M.; Hogg, J.C.; Adcock, I.M.; Barnes, P.J. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Randall, M.J.; Haenen, G.R.; Bouwman, F.G.; van der Vliet, A.; Bast, A. The tobacco smoke component acrolein induces glucocorticoid resistant gene expression via inhibition of histone deacetylase. Toxicol. Lett. 2016, 240, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Marwick, J.A.; Wallis, G.; Meja, K.; Kuster, B.; Bouwmeester, T.; Chakravarty, P.; Fletcher, D.; Whittaker, P.A.; Barnes, P.J.; Ito, K.; et al. Oxidative stress modulates theophylline effects on steroid responsiveness. Biochem. Biophys. Res. Commun. 2008, 377, 797–802. [Google Scholar] [CrossRef]
- Ito, K.; Lim, S.; Caramori, G.; Cosio, B.; Chung, K.F.; Adcock, I.M.; Barnes, P.J. A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 8921–8926. [Google Scholar] [CrossRef] [Green Version]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Hohn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meja, K.K.; Rajendrasozhan, S.; Adenuga, D.; Biswas, S.K.; Sundar, I.K.; Spooner, G.; Marwick, J.A.; Chakravarty, P.; Fletcher, D.; Whittaker, P.; et al. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am. J. Respir. Cell Mol. Biol. 2008, 39, 312–323. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Lugt, T.; Weseler, A.R.; Vrolijk, M.F.; Opperhuizen, A.; Bast, A. Dietary Advanced Glycation Endproducts Decrease Glucocorticoid Sensitivity In Vitro. Nutrients 2020, 12, 441. https://doi.org/10.3390/nu12020441
van der Lugt T, Weseler AR, Vrolijk MF, Opperhuizen A, Bast A. Dietary Advanced Glycation Endproducts Decrease Glucocorticoid Sensitivity In Vitro. Nutrients. 2020; 12(2):441. https://doi.org/10.3390/nu12020441
Chicago/Turabian Stylevan der Lugt, Timme, Antje R. Weseler, Misha F. Vrolijk, Antoon Opperhuizen, and Aalt Bast. 2020. "Dietary Advanced Glycation Endproducts Decrease Glucocorticoid Sensitivity In Vitro" Nutrients 12, no. 2: 441. https://doi.org/10.3390/nu12020441
APA Stylevan der Lugt, T., Weseler, A. R., Vrolijk, M. F., Opperhuizen, A., & Bast, A. (2020). Dietary Advanced Glycation Endproducts Decrease Glucocorticoid Sensitivity In Vitro. Nutrients, 12(2), 441. https://doi.org/10.3390/nu12020441