
Effect of Advanced Glycation End-Products and Excessive Calorie Intake on Diet-Induced Chronic Low-Grade Inflammation Biomarkers in Murine Models

 , ,
, ,  , and
, and
Abstract
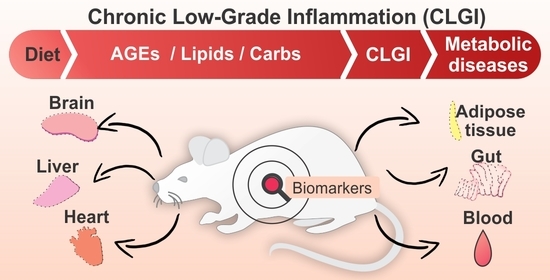
:
1. Introduction
2. Inflammation and Chronic Low-Grade Inflammation (CLGI)
3. CLGI Biomarkers
3.1. Human Studies
3.2. Murine Model Studies
4. High-AGE Diets and CLGI Initiation in Murine Models
5. Other Diet-Induced CLGI in Murine Models
5.1. Obesogenic Diet-Induced CLGI
5.2. Diet-Induced Gut Microbiota Remodeling and CLGI: A Mechanism of Metabolic Endotoxemia
6. Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Minihane, A.M.; Vinoy, S.; Russell, W.R.; Baka, A.; Roche, H.M.; Tuohy, K.M.; Teeling, J.L.; Blaak, E.E.; Fenech, M.; Vauzour, D.; et al. Low-Grade inflammation, diet composition and health: Current research evidence and its translation. Br. J. Nutr. 2015, 114, 999–1012. [Google Scholar] [CrossRef] [Green Version]
- Jandeleit-Dahm, K.; Cooper, M.E. The role of AGEs in cardiovascular disease. Curr. Pharm. Des. 2008, 14, 979–986. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, É. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef] [PubMed]
- Dalby, M.J.; Aviello, G.; Ross, A.W.; Walker, A.W.; Barrett, P.; Morgan, P.J. Diet Induced obesity is independent of metabolic endotoxemia and TLR4 Signalling, but markedly increases hypothalamic expression of the acute phase protein, SerpinA3N. Sci. Rep. 2018, 8, 15648. [Google Scholar] [CrossRef] [PubMed]
- Lainez, N.M.; Jonak, C.R.; Nair, M.G.; Ethell, I.M.; Wilson, E.H.; Carson, M.J.; Coss, D. Diet-Induced obesity elicits macrophage infiltration and reduction in spine density in the hypothalami of male but not female Mice. Front. Immunol. 2018, 9, 1992. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Delzenne, N.M. The role of the gut microbiota in energy metabolism and metabolic disease. Curr. Pharm. Des. 2009, 15, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Calder, P.C.; Ahluwalia, N.; Albers, R.; Bosco, N.; Bourdet-Sicard, R.; Haller, D.; Holgate, S.T.; Jönsson, L.S.; Latulippe, M.E.; Marcos, A.; et al. A consideration of biomarkers to be used for evaluation of inflammation in human nutritional studies. Br. J. Nutr. 2013, 109, S1–S34. [Google Scholar] [CrossRef] [Green Version]
- Calder, P.C.; Ahluwalia, N.; Brouns, F.; Buetler, T.; Clement, K.; Cunningham, K.; Esposito, K.; Jönsson, L.S.; Kolb, H.; Lansink, M.; et al. Dietary factors and low-grade inflammation in relation to overweight and obesity. Br. J. Nutr. 2011, 106 (Suppl. S3), S5–S78. [Google Scholar] [CrossRef]
- Calder, P.C.; Bosco, N.; Bourdet-Sicard, R.; Capuron, L.; Delzenne, N.; Doré, J.; Franceschi, C.; Lehtinen, M.J.; Recker, T.; Salvioli, S.; et al. Health Relevance of the modification of low grade inflammation in ageing (Inflammageing) and the role of nutrition. Ageing Res. Rev. 2017, 40, 95–119. [Google Scholar] [CrossRef]
- Yu, H.-P.; Chaudry, I.H.; Choudhry, M.A.; Hsing, C.-H.; Liu, F.-C.; Xia, Z. Inflammatory response to traumatic injury: Clinical and animal researches in inflammation. Mediat. Inflamm. 2015, 2015, e729637. [Google Scholar] [CrossRef]
- Ward, P.A. Acute and Chronic Inflammation. In Fundamentals of Inflammation; Serhan, C.N., Gilroy, D.W., Ward, P.A., Eds.; Cambridge University Press: Cambridge, UK, 2010; pp. 1–16. [Google Scholar] [CrossRef]
- Lawrence, T.; Gilroy, D.W. Chronic Inflammation: A Failure of Resolution? Int. J. Exp. Pathol. 2007, 88, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.S.; Alvarez-Leite, J.I. Low-Grade inflammation, obesity, and diabetes. Curr. Obes. Rep. 2014, 3, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Pietrzyk, L.; Torres, A.; Maciejewski, R.; Torres, K. Obesity and obese-related chronic low-grade inflammation in promotion of colorectal cancer development. Asian Pac. J. Cancer Prev. 2015, 16, 4161–4168. [Google Scholar] [CrossRef] [Green Version]
- Schett, G.; Neurath, M.F. Resolution of chronic inflammatory disease: Universal and tissue-specific concepts. Nat. Commun. 2018, 9, 3261. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Stevens, J.R. Neuropathology of schizophrenia. Arch. Gen. Psychiatry 1982, 39, 1131–1139. [Google Scholar] [CrossRef]
- Galea, P.; D’mato, B.; Goel, K.M. Ocular complications in juvenile chronic arthritis (JCA). Scott. Med. J. 1985, 30, 164–167. [Google Scholar] [CrossRef]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci 2013, 9, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef] [Green Version]
- Vohl, M.-C.; Sladek, R.; Robitaille, J.; Gurd, S.; Marceau, P.; Richard, D.; Hudson, T.J.; Tchernof, A. A survey of genes differentially expressed in subcutaneous and visceral adipose tissue in men. Obes. Res. 2004, 12, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Models Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Kim, H.J.; Lee, Y.-S.; Kang, G.M.; Lim, H.S.; Lee, S.; Song, D.K.; Kwon, O.; Hwang, I.; Son, M.; et al. Hypothalamic macrophage inducible nitric oxide synthase mediates obesity-associated hypothalamic inflammation. Cell Rep. 2018, 25, 934–946.e5. [Google Scholar] [CrossRef] [Green Version]
- Thaler, J.P.; Yi, C.-X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bektas, A.; Schurman, S.H.; Sen, R.; Ferrucci, L. Human T cell immunosenescence and inflammation in aging. J. Leukoc. Biol. 2017, 102, 977–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir, S.; Fukushima, Y.; Sakamoto, K.; Sato, K.; Fujita, H.; Inoue, J.; Uede, T.; Hamazaki, Y.; Hattori, M.; Minato, N. A CD153+CD4+ T Follicular cell population with cell-senescence features plays a crucial role in lupus pathogenesis via osteopontin production. J. Immunol. 2015, 194, 5725–5735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, Y.; Minato, N.; Hattori, M. The impact of senescence-associated T cells on immunosenescence and age-related Disorders. Inflamm. Regen. 2018, 38, 24. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of high-fat-diet on gut microbiota: A driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Wolters, M.; Ahrens, J.; Romaní-Pérez, M.; Watkins, C.; Sanz, Y.; Benítez-Páez, A.; Stanton, C.; Günther, K. Dietary fat, the gut microbiota, and metabolic health—A systematic review conducted within the MyNewGut project. Clin. Nutr. 2019, 38, 2504–2520. [Google Scholar] [CrossRef] [Green Version]
- Margioris, A.N.; Dermitzaki, E.; Venihaki, M.; Tsatsanis, C. 4—Chronic low-grade inflammation. In Diet, Immunity and Inflammation; Calder, P.C., Yaqoob, P., Eds.; Woodhead Publishing Series in Food Science, Technology and Nutrition; Woodhead Publishing: Sawston, UK, 2013; pp. 105–120. [Google Scholar] [CrossRef]
- Giuliani, A.; Prattichizzo, F.; Micolucci, L.; Ceriello, A.; Procopio, A.D.; Rippo, M.R. Mitochondrial (dys) function in inflammaging: Do MitomiRs influence the energetic, oxidative, and inflammatory status of senescent cells? Mediat. Inflamm. 2017, 2017, 2309034. [Google Scholar] [CrossRef]
- Chassaing, B.; Gewirtz, A.T. Gut Microbiota, low-grade inflammation, and metabolic syndrome. Toxicol. Pathol. 2014, 42, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, A.; Danesi, F.; Dardevet, D.; Dupont, D.; Fernandez, A.S.; Gille, D.; Nunes Dos Santos, C.; Pinto, P.; Re, R.; Rémond, D.; et al. Dairy products and inflammation: A review of the clinical evidence. Crit. Rev. Food Sci. Nutr. 2017, 57, 2497–2525. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. Clinical application of c-reactive protein for cardiovascular disease detection and prevention. Circulation 2003, 107, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Erbaş, O.; Akseki, H.S.; Aktuğ, H.; Taşkıran, D. Low-Grade chronic inflammation induces behavioral stereotypy in rats. Metab. Brain Dis. 2015, 30, 739–746. [Google Scholar] [CrossRef]
- van der Heijden, R.A.; Sheedfar, F.; Morrison, M.C.; Hommelberg, P.P.; Kor, D.; Kloosterhuis, N.J.; Gruben, N.; Youssef, S.A.; de Bruin, A.; Hofker, M.H.; et al. High-Fat diet induced obesity primes inflammation in adipose tissue prior to liver in C57BL/6j mice. Aging 2015, 7, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Power Guerra, N.; Müller, L.; Pilz, K.; Glatzel, A.; Jenderny, D.; Janowitz, D.; Vollmar, B.; Kuhla, A. Dietary-induced low-grade inflammation in the liver. Biomedicines 2020, 8, 587. [Google Scholar] [CrossRef]
- Mastrocola, R.; Collotta, D.; Gaudioso, G.; Le Berre, M.; Cento, A.S.; Ferreira Alves, G.; Chiazza, F.; Verta, R.; Bertocchi, I.; Manig, F.; et al. Effects of exogenous dietary advanced glycation end products on the cross-talk mechanisms linking microbiota to metabolic inflammation. Nutrients 2020, 12, 2497. [Google Scholar] [CrossRef]
- Matafome, P.; Santos-Silva, D.; Crisóstomo, J.; Rodrigues, T.; Rodrigues, L.; Sena, C.M.; Pereira, P.; Seiça, R. Methylglyoxal causes structural and functional alterations in adipose tissue independently of obesity. Arch. Physiol. Biochem. 2012, 118, 58–68. [Google Scholar] [CrossRef]
- Benoit, B.; Plaisancié, P.; Awada, M.; Géloën, A.; Estienne, M.; Capel, F.; Malpuech-Brugère, C.; Debard, C.; Pesenti, S.; Morio, B.; et al. High-Fat diet action on adiposity, inflammation, and insulin sensitivity depends on the control low-fat diet. Nutr. Res. 2013, 33, 952–960. [Google Scholar] [CrossRef]
- Guo, W.A.; Davidson, B.A.; Ottosen, J.; Ohtake, P.J.; Raghavendran, K.; Mullan, B.A.; Dayton, M.T.; Knight, P.R. Effect of high advanced glycation end product diet on pulmonary inflammatory response and pulmonary function following gastric aspiration. Shock 2012, 38, 677–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teissier, T.; Quersin, V.; Gnemmi, V.; Daroux, M.; Howsam, M.; Delguste, F.; Lemoine, C.; Fradin, C.; Schmidt, A.-M.; Cauffiez, C.; et al. Knockout of receptor for advanced glycation end-products attenuates age-related renal lesions. Aging Cell 2019, 18, e12850. [Google Scholar] [CrossRef]
- Leung, C.; Herath, C.B.; Jia, Z.; Goodwin, M.; Mak, K.Y.; Watt, M.J.; Forbes, J.M.; Angus, P.W. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J. Hepatol. 2014, 60, 832–838. [Google Scholar] [CrossRef]
- Tikellis, C.; Thomas, M.C.; Harcourt, B.E.; Coughlan, M.T.; Pete, J.; Bialkowski, K.; Tan, A.; Bierhaus, A.; Cooper, M.E.; Forbes, J.M. Cardiac inflammation associated with a western diet is mediated via activation of RAGE by AGEs. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E323–E330. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-A.; Gu, W.; Lee, I.-A.; Joh, E.-H.; Kim, D.-H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- Tikellis, C.; Pickering, R.J.; Tsorotes, D.; Huet, O.; Cooper, M.E.; Jandeleit-Dahm, K.; Thomas, M.C. Dicarbonyl stress in the absence of hyperglycemia increases endothelial inflammation and atherogenesis similar to that observed in diabetes. Diabetes 2014, 63, 3915–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowndhar Rajan, B.; Manivasagam, S.; Dhanusu, S.; Chandrasekar, N.; Krishna, K.; Kalaiarasu, L.P.; Babu, A.A.; Vellaichamy, E. Diet with high content of advanced glycation end products induces systemic inflammation and weight gain in experimental mice: Protective role of curcumin and gallic acid. Food Chem. Toxicol. 2018, 114, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Grossin, N.; Auger, F.; Niquet-Leridon, C.; Durieux, N.; Montaigne, D.; Schmidt, A.M.; Susen, S.; Jacolot, P.; Beuscart, J.-B.; Tessier, F.J.; et al. Dietary CML-Enriched protein induces functional arterial aging in a RAGE-dependent manner in mice. Mol. Nutr. Food Res. 2015, 59, 927–938. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Wallenstein, S.; Striker, G.E.; Vlassara, H. Reduced oxidant stress and extended lifespan in mice exposed to a low glycotoxin diet. Am. J. Pathol. 2007, 170, 1893–1902. [Google Scholar] [CrossRef] [Green Version]
- Chatzigeorgiou, A.; Kandaraki, E.; Piperi, C.; Livadas, S.; Papavassiliou, A.G.; Koutsilieris, M.; Papalois, A.; Diamanti-Kandarakis, E. Dietary glycotoxins affect scavenger receptor expression and the hormonal profile of female rats. J. Endocrinol. 2013, 218, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Diao, N.; Yuan, R.; Chen, K.; Geng, S.; Li, M.; Li, L. Subclinical-Dose endotoxin sustains low-grade inflammation and exacerbates steatohepatitis in high-fat diet-fed mice. J. Immunol. 2016, 196, 2300–2308. [Google Scholar] [CrossRef] [Green Version]
- Shangari, N.; Depeint, F.; Furrer, R.; Bruce, W.R.; Popovic, M.; Zheng, F.; O’Brien, P.J. A thermolyzed diet increases oxidative stress, plasma α-aldehydes and colonic inflammation in the rat. Chem. Biol. Interact. 2007, 169, 100–109. [Google Scholar] [CrossRef]
- Feng, J.X.; Hou, F.F.; Liang, M.; Wang, G.B.; Zhang, X.; Li, H.Y.; Xie, D.; Tian, J.W.; Liu, Z.Q. Restricted intake of dietary advanced glycation end products retards renal progression in the remnant kidney model. Kidney Int. 2007, 71, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, K.; Merhi, Z.; Jindal, S.; Goldsammler, M.; Charron, M.J.; Buyuk, E. Dietary advanced glycation end products (AGEs) could alter ovarian function in mice. Mol. Cell. Endocrinol. 2020, 510, 110826. [Google Scholar] [CrossRef]
- Chassaing, B.; Srinivasan, G.; Delgado, M.A.; Young, A.N.; Gewirtz, A.T.; Vijay-Kumar, M. Fecal Lipocalin 2, a Sensitive and Broadly Dynamic Non-Invasive Biomarker for Intestinal Inflammation. PLoS ONE 2012, 7, e44328. [Google Scholar] [CrossRef] [Green Version]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Bergé, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C.; et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Rorato, R.; Borges, B.; de Carvalho Borges, B.; Uchoa, E.T.; Antunes-Rodrigues, J.; Elias, C.F.; Elias, L.L.K. LPS-Induced low-grade inflammation increases hypothalamic JNK expression and causes central insulin resistance irrespective of body weight changes. Int. J. Mol. Sci 2017, 18, 1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Song, S.; Wang, Y.; Huang, C.; Zhang, F.; Liu, J.; Hong, J.-S. Low-Grade inflammation aggravates rotenone neurotoxicity and disrupts circadian clock gene expression in rats. Neurotox. Res. 2019, 35, 421–431. [Google Scholar] [CrossRef]
- Zhu, Y.; Lan, F.; Wei, J.; Chong, B.; Chen, P.; Huynh, L.; Wong, N.; Liu, Y. Influence of dietary advanced glycation end products on wound healing in nondiabetic mice. J. Food Sci. 2011, 76, T5–T10. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-F.; Chiu, S.-Y.; Huang, H.-W.; Cheng, B.-H.; Pan, H.-M.; Huang, W.-L.; Chang, H.-H.; Liao, C.-C.; Jiang, S.-T.; Su, Y.-C. A reporter mouse for non-invasive detection of toll-like receptor ligands induced acute phase responses. Sci. Rep. 2019, 9, 19065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M. Glycation of proteins. In Glycoscience: Biology and Medicine; Taniguchi, N., Endo, T., Hart, G.W., Seeberger, P.H., Wong, C.-H., Eds.; Springer: Tokyo, Japan, 2015; pp. 1339–1345. [Google Scholar] [CrossRef]
- Rahbar, S. The Discovery of Glycated Hemoglobin: A Major Event in the Study of Nonenzymatic Chemistry in Biological Systems. Ann. N. Y. Acad. Sci. 2005, 1043, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Brás, I.C.; König, A.; Outeiro, T.F. Glycation in Huntington’s Disease: A possible modifier and target for intervention. J. Huntingt. Dis. 2019, 8, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. the role of advanced glycation end products in aging and metabolic diseases: Bridging association and causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Park, S.; Lakatta, E.G. RAGE signaling in inflammation and arterial aging. Front. Biosci 2009, 14, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.-S.; Park, S.; Kim, J. The role of glycation in the pathogenesis of aging and its prevention through herbal products and physical exercise. J. Exerc. Nutr. Biochem. 2017, 21, 55–61. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Cai, W.; Sandu, O.; Peppa, M.; Goldberg, T.; Vlassara, H. Diet-Derived advanced glycation end products are major contributors to the body’s age pool and induce inflammation in healthy subjects. Ann. N. Y. Acad. Sci. 2005, 1043, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Bekedam, E.K.; Schols, H.A.; van Boekel, M.A.J.S.; Smit, G. High Molecular Weight Melanoidins from Coffee Brew. J. Agric. Food Chem. 2006, 54, 7658–7666. [Google Scholar] [CrossRef]
- Hellwig, M.; Henle, T. Baking, ageing, diabetes: A short history of the Maillard Reaction. Angew. Chem. Int. Ed. 2014, 53, 10316–10329. [Google Scholar] [CrossRef]
- Nguyen, H.T.; van der Fels-Klerx, H.J.; van Boekel, M.A.J.S. Nϵ-(Carboxymethyl)Lysine: A review on analytical methods, formation, and occurrence in processed food, and health impact. Food Rev. Int. 2014, 30, 36–52. [Google Scholar] [CrossRef]
- Tessier, F.J.; Birlouez-Aragon, I. Health effects of dietary Maillard Reaction products: The results of ICARE and other studies. Amino Acids 2012, 42, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, K.C.W.; Linkens, A.M.A.; Wetzels, S.M.W.; Wouters, K.; Vanmierlo, T.; van de Waarenburg, M.P.H.; Scheijen, J.L.J.M.; de Vos, W.M.; Belzer, C.; Schalkwijk, C.G. Dietary advanced glycation endproducts (AGEs) increase their concentration in plasma and tissues, result in inflammation and modulate gut microbial composition in mice; evidence for reversibility. Food Res. Int. 2021, 147, 110547. [Google Scholar] [CrossRef] [PubMed]
- Snelson, M.; Tan, S.M.; Clarke, R.E.; de Pasquale, C.; Thallas-Bonke, V.; Nguyen, T.-V.; Penfold, S.A.; Harcourt, B.E.; Sourris, K.C.; Lindblom, R.S.; et al. Processed foods drive intestinal barrier permeability and microvascular diseases. Sci. Adv. 2021, 7, eabe4841. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Yuan, X.; Zhao, J.; Zhang, Y.; Hu, J.; Wang, J.; Li, J. Dietary Advanced Glycation End Products Modify Gut Microbial Composition and Partially Increase Colon Permeability in Rats. Mol. Nutr. Food Res. 2017, 61, 1700118. [Google Scholar] [CrossRef] [PubMed]
- Seiquer, I.; Rubio, L.A.; Peinado, M.J.; Delgado-Andrade, C.; Navarro, M.P. Maillard Reaction products modulate gut microbiota composition in adolescents. Mol. Nutr. Food Res. 2014, 58, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Andrade, C.; Pastoriza de la Cueva, S.; Peinado, M.J.; Rufián-Henares, J.Á.; Navarro, M.P.; Rubio, L.A. Modifications in bacterial groups and short chain fatty acid production in the gut of healthy adult rats after long-term consumption of dietary Maillard Reaction products. Food Res. Int. 2017, 100, 134–142. [Google Scholar] [CrossRef]
- Patel, R.; Baker, S.S.; Liu, W.; Desai, S.; Alkhouri, R.; Kozielski, R.; Mastrandrea, L.; Sarfraz, A.; Cai, W.; Vlassara, H.; et al. Effect of dietary advanced glycation end products on mouse liver. PLoS ONE 2012, 7, e35143. [Google Scholar] [CrossRef]
- Harcourt, B.E.; Sourris, K.C.; Coughlan, M.T.; Walker, K.Z.; Dougherty, S.L.; Andrikopoulos, S.; Morley, A.L.; Thallas-Bonke, V.; Chand, V.; Penfold, S.A.; et al. Targeted reduction of advanced glycation improves renal function in obesity. Kidney Int. 2011, 80, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.-Y.; Choudhury, R.P.; Cai, W.; Lu, M.; Fallon, J.T.; Fisher, E.A.; Vlassara, H. Dietary glycotoxins promote diabetic atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 2003, 168, 213–220. [Google Scholar] [CrossRef]
- Peppa, M.; Brem, H.; Ehrlich, P.; Zhang, J.-G.; Cai, W.; Li, Z.; Croitoru, A.; Thung, S.; Vlassara, H. Adverse effects of dietary glycotoxins on wound healing in genetically diabetic mice. Diabetes 2003, 52, 2805–2813. [Google Scholar] [CrossRef] [Green Version]
- Alexiou, P.; Chatzopoulou, M.; Pegklidou, K.; Demopoulos, V.J. RAGE: A multi-ligand receptor unveiling novel insights in health and disease. Curr. Med. Chem. 2010, 17, 2232–2252. [Google Scholar] [CrossRef]
- Boulanger, E.; Grossin, N.; Wautier, M.-P.; Taamma, R.; Wautier, J.-L. Mesothelial RAGE Activation by AGEs enhances VEGF release and potentiates capillary tube formation. Kidney Int. 2007, 71, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toprak, C.; Yigitaslan, S. Alagebrium and complications of diabetes mellitus. Eurasian J. Med. 2019, 51, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Goldsammler, M.; Merhi, Z.; Thornton, K.; Charron, M.J.; Buyuk, E. Ovarian rage expression changes with follicular development and superovulation. Fertil. Steril. 2018, 110, e122. [Google Scholar] [CrossRef]
- Tessier, F.J.; Niquet-Léridon, C.; Jacolot, P.; Jouquand, C.; Genin, M.; Schmidt, A.-M.; Grossin, N.; Boulanger, E. Quantitative assessment of organ distribution of dietary protein-bound 13C-labeled Nɛ-carboxymethyllysine after a chronic oral exposure in mice. Mol. Nutr. Food Res. 2016, 60, 2446–2456. [Google Scholar] [CrossRef]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “RAGE” in lung disease: The receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef]
- Sanders, K.A.; Delker, D.A.; Huecksteadt, T.; Beck, E.; Wuren, T.; Chen, Y.; Zhang, Y.; Hazel, M.W.; Hoidal, J.R. RAGE is a critical mediator of pulmonary oxidative stress, alveolar macrophage activation and emphysema in response to cigarette smoke. Sci. Rep. 2019, 9, 231. [Google Scholar] [CrossRef]
- Van Putte, L.; De Schrijver, S.; Moortgat, P. The effects of advanced glycation end products (AGEs) on dermal wound healing and scar formation: A systematic review. Scars Burns Heal. 2016, 2. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Nam, J.-H. Insight into the Relationship between obesity-induced low-level chronic inflammation and COVID-19 Infection. Int. J. Obes. 2020, 44, 1541–1542. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, A.M.; Macedo-de la Concha, L.E.; Pantoja-Meléndez, C.A. Low-Grade inflammation and its relation to obesity and chronic degenerative diseases. Revista Médica Hospital General México 2017, 80, 101–105. [Google Scholar] [CrossRef]
- Wang, J.; Chen, W.-D.; Wang, Y.-D. The relationship between gut microbiota and inflammatory diseases: The role of macrophages. Front. Microbiol. 2020, 11, 1065. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, W.; Song, G.; Pang, S.; Peng, Z.; Li, Y.; Wang, P. High-Fructose diet increases inflammatory cytokines and alters gut microbiota composition in rats. Mediat. Inflamm. 2020, 2020, e6672636. [Google Scholar] [CrossRef] [PubMed]
- Powell-Wiley, T.M.; Paul, P.; Lora, L.E.; Després, J.-P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Prashanthan, S.; et al. Obesity and cardiovascular disease: A scientific statement from the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, A.; Hernandez, M.A.; Moore, K.J.; Schmidt, A.M.; Fisher, E.A. Leukocyte heterogeneity in adipose tissue, including in obesity. Circ. Res. 2020, 126, 1590–1612. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Feldstein, A.E. Adipocyte cell death, fatty liver disease and associated metabolic disorders. Dig. Dis. 2014, 32, 579–585. [Google Scholar] [CrossRef] [Green Version]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Öhman, M.K.; Wright, A.P.; Wickenheiser, K.J.; Luo, W.; Russo, H.M.; Eitzman, D.T. Mcp-1 Deficiency protects against visceral fat-induced atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, V.; Ferrante, A.W., Jr. Obesity, inflammation, and macrophages. In Nestlé Nutrition Institute Workshop Series: Pediatric Program; Kalhan, S.C., Prentice, A.M., Yajnik, C.S., Eds.; Karger: Basel, Switzerland, 2009; Volume 63, pp. 151–162. [Google Scholar] [CrossRef] [Green Version]
- Ahima, R.S.; Antwi, D.A. Brain regulation of appetite and satiety. Endocrinol. Metab. Clin. N. Am. 2008, 37, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [Green Version]
- Arroyo-Espliguero, R.; Avanzas, P.; Jeffery, S.; Kaski, J.C. CD14 and toll-like receptor 4: A link between infection and acute coronary events? Heart 2004, 90, 983–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takiishi, T.; Fenero, C.I.M.; Câmara, N.O.S. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers 2017, 5, e1373208. [Google Scholar] [CrossRef]
- Putignani, L.; Del Chierico, F.; Petrucca, A.; Vernocchi, P.; Dallapiccola, B. The human gut microbiota: A dynamic interplay with the host from birth to senescence settled during childhood. Pediatr. Res. 2014, 76, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, V.K.; Yong, V.C.; Chong, P.P.; Amin Nordin, S.; Basir, R.; Abdullah, M. Mycobiome in the Gut: A multiperspective review. Mediat. Inflamm. 2020, 2020, e9560684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimarăes, V.; Sokol, H.; Doré, J.; Corthier, G.; Furet, J.-P. The firmicutes/bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 2015, 74, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.L.A.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Models Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. mBio 2014, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet–induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.P.; Halaka, D.A.; Hayouka, Z.; Tirosh, O. High-Fat diet induced alteration of mice microbiota and the functional ability to utilize fructooligosaccharide for ethanol production. Front. Cell. Infect. Microbiol. 2020, 10, 376. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.P.B.; Texeira, T.F.S.; Ferreira, A.B.; Peluzio, M.d.C.G.; Alfenas, R.d.C.G. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br. J. Nutr. 2012, 108, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, J.; Guo, X.; Huang, X.; Huang, Q. RAGE Plays a Role in LPS-Induced NF-ΚB activation and endothelial hyperpermeability. Sensors 2017, 17, 722. [Google Scholar] [CrossRef] [PubMed]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The firmicutes/bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Chen, K.; Yuan, R.; Peng, L.; Maitra, U.; Diao, N.; Chen, C.; Zhang, Y.; Hu, Y.; Qi, C.-F.; et al. The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat. Commun. 2016, 7, 13436. [Google Scholar] [CrossRef]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferriéres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.J.; Magness, S.; Jobin, C.; Lund, P.K. High-Fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef] [Green Version]
- Chakraborti, C.K. New-Found link between microbiota and obesity. World J. Gastrointest. Pathophysiol. 2015, 6, 110–119. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Northrop, W.; Melton, P.E.; Ellison, G.C.; Newsholme, P.; Mamotte, C.D.S. Butyrate generated by gut microbiota and its therapeutic role in metabolic syndrome. Pharmacol. Res. 2020, 160, 105174. [Google Scholar] [CrossRef]
- Daniel, H.; Gholami, A.M.; Berry, D.; Desmarchelier, C.; Hahne, H.; Loh, G.; Mondot, S.; Lepage, P.; Rothballer, M.; Walker, A.; et al. High-Fat Diet Alters Gut Microbiota Physiology in Mice. Multidiscip. J. Microb. Ecol. 2014, 8, 295–308. [Google Scholar] [CrossRef]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative effects of a high-fat diet on intestinal permeability: A review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Al-Sadi, R.; Guo, S.; Dokladny, K.; Smith, M.A.; Ye, D.; Kaza, A.; Watterson, D.M.; Ma, T.Y. Mechanism of interleukin-1β induced-increase in mouse intestinal permeability in vivo. J. Interferon Cytokine Res. 2012, 32, 474–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Li, H.; Qu, X.; Huang, L.; Kong, C.; Qin, H.; Sun, Z.; Yan, X. High fat diet exacerbates intestinal barrier dysfunction and changes gut microbiota in intestinal-specific ACF7 knockout mice. Biomed. Pharmacother. 2019, 110, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.B.; Canuto, L.P.; Collares-Buzato, C.B. Intestinal luminal content from high-fat-fed prediabetic mice changes epithelial barrier function in vitro. Life Sci. 2019, 216, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Willenberg, I.; Rund, K.; Rong, S.; Shushakova, N.; Gueler, F.; Schebb, N.H. Characterization of changes in plasma and tissue oxylipin levels in LPS and CLP induced murine sepsis. Inflamm. Res. 2016, 65, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Maitra, U.; Deng, H.; Glaros, T.; Baker, B.; Capelluto, D.G.S.; Li, Z.; Li, L. Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J. Immunol. 2012, 189, 1014–1023. [Google Scholar] [CrossRef] [Green Version]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2016, 6, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Adolph, T.E.; Gerner, R.R.; Wieser, V.; Tilg, H. Lipocalin-2: A master mediator of intestinal and metabolic inflammation. Trends Endocrinol. Metab. 2017, 28, 388–397. [Google Scholar] [CrossRef]
- del Castillo, M.D.; Iriondo-DeHond, A.; Iriondo-DeHond, M.; Gonzalez, I.; Medrano, A.; Filip, R.; Uribarri, J. Healthy eating recommendations: Good for reducing dietary contribution to the body’s advanced glycation/lipoxidation end products pool? Nutr. Res. Rev. 2021, 34, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Joung, H.; Chu, J.; Kim, B.-K.; Choi, I.-S.; Kim, W.; Park, T.-S. Probiotics ameliorate chronic low-grade inflammation and fat accumulation with gut microbiota composition change in diet-induced obese mice models. Appl. Microbiol. Biotechnol. 2021, 105, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.A.; Lira, F.S.; Rosa Neto, J.C.; Pimentel, G.D.; Souza, G.I.H.; da Silva, C.M.G.; de Souza, C.T.; Ribeiro, E.B.; Sawaya, A.C.H.F.; Oller do Nascimento, C.M.; et al. Green tea extract supplementation induces the lipolytic pathway, attenuates obesity, and reduces low-grade inflammation in mice fed a high-fat diet. Mediat. Inflamm. 2013, 2013, 635470. [Google Scholar] [CrossRef] [PubMed]
- Heyman-Lindén, L.; Kotowska, D.; Sand, E.; Bjursell, M.; Plaza, M.; Turner, C.; Holm, C.; Fåk, F.; Berger, K. Lingonberries alter the gut microbiota and prevent low-grade inflammation in high-fat diet fed mice. Food Nutr. Res. 2016, 60, 29993. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
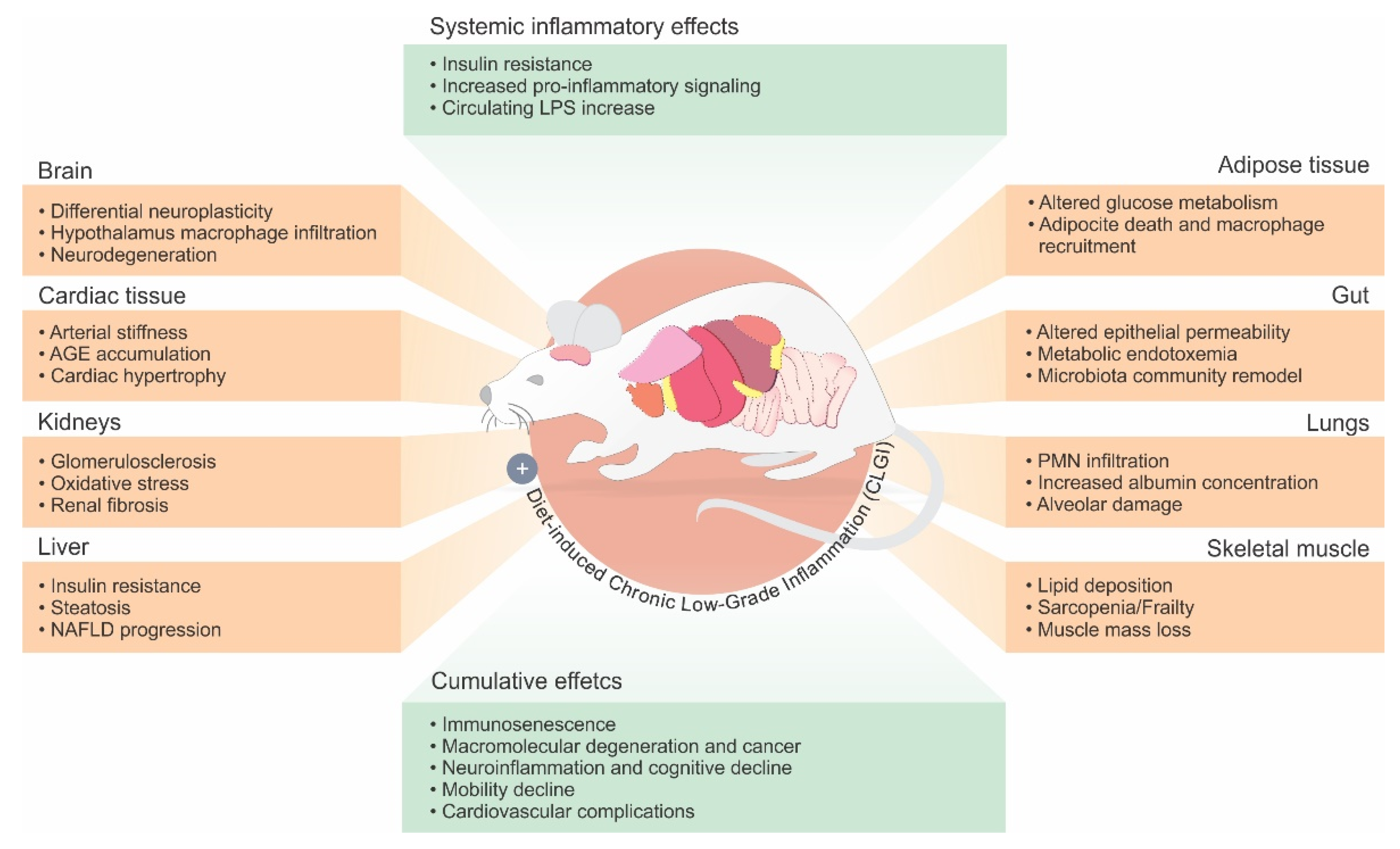{kind=link}
| Role in CLGI | Class | Biomarker | Sample | Biomarker Levels between Control and Experimental Conditions (Arrows Show Expression Tendency) | References |
|---|---|---|---|---|---|
| Anti-inflammatory | Cytokine | Interleukin 2 (IL-2) | Brain | Control: 10 pg/mg Affected: 120 pg/mg (↑) (Protein) | [36] |
| Liver | Control: 100 pg/mg Affected: 150 pg/mg (↑) (Protein) | ||||
| Interleukin 10 (IL-10) | Adipose tissue | No change (→) (mRNA) | [37] | ||
| Liver | No change (→) (mRNA) | [38] | |||
| [37] | |||||
| Plasma | Control:10 pg/mL Affected: 20 pg/mL (↑) (Protein) | [37] | |||
| Control: 15 pg/mL Affected: 9 pg/mL (↓) (Protein) | [39] | ||||
| Pro and anti-inflammatory | Adipokine | Adiponectin | Plasma | Control: 90 μg/mL Affected: 60 μg/mL (↓) (Protein) | [40] |
| Control: 30 µg/mL Affected: 45 µg/mL (↑) (Protein) | [41] | ||||
| Control: 8 ng/mL (→) (Protein) | [37] | ||||
| Cytokine | Interleukin 6 (IL-6) | Adipose Tissue | 2-fold change (↑) (mRNA) | [41] | |
| BAL | No change (→) (Protein) | [42] | |||
| Kidney | 15-fold change (↑) (mRNA) | [43] | |||
| Liver | No change (→) (mRNA) | [38] | |||
| 8-fold change (↑) (mRNA) | [44] | ||||
| Myocardium | Control: 21 ng/µg Affected: 28 ng/µg (↑) (Protein) | [45] | |||
| 18-fold change (↑) (mRNA) | |||||
| Plasma | 3-fold change (↑) (mRNA) | [46] | |||
| Control: 4 pg/mL Affected: 2 pg/mL (↓) (Protein) | [39] | ||||
| No change (→) (mRNA) | [39] | ||||
| Pro-inflammatory | Adhesion molecule | Intercellular adhesion molecule 1 (ICAM-1) | Aorta | 1.4-fold change (↑) (mRNA) | [47] |
| Myocardium | 5.6-fold (↑) (mRNA) | [45] | |||
| Plasma | 3.5-fold change (↑) (Protein) | [48] | |||
| Vascular cell adhesion molecule 1 (VCAM-1) | Aorta | 2.5-fold change (↑) (Protein) | [49] | ||
| 1.4-fold change (↑) (mRNA) | |||||
| 2-fold change (↑) (mRNA) | [47] | ||||
| Kidney | 4-fold change (↑) (mRNA) | [43] | |||
| Cell receptor | Receptor for advanced glycation end-products (RAGE) | Aorta | 2-fold change (↑) (Protein) | [49] | |
| No change (→) (mRNA) | |||||
| Kidney | 3-fold change (↑) (mRNA) | [50] | |||
| Liver | 3-fold change (↑) (mRNA) | [50] | |||
| 1.5-fold change (↑) (mRNA) | [44] | ||||
| PBMC | 2-fold change (↓) (Protein) | [51] | |||
| Spleen | 3-fold change (↑) (mRNA) | [50] | |||
| Chemokine | Keratinocyte chemoattractant (KC ou CXCL1) | BAL | Control: 10 pg/mL Affected: 600 pg/mL (↑) (Protein) | [42] | |
| Macrophage Inflammatory Protein 2 (MIP-2) | BAL | No change (→) (Protein) | [42] | ||
| Liver | Control: 20 pg/mL Affected: 180 pg/mL (↑) (Protein) | [52] | |||
| Plasma | Control: 900 pg/mL Affected: 1500 pg/mL (↑) (Protein) | [52] | |||
| Cluster of differentiation | CD68 Clone ED1 (ED1) | Colon | 4-fold (↑) (Histochemistry) | [53] | |
| Kidney | 2.4-fold (↑) (Histochemistry) | [54] | |||
| Cluster of differentiation 11 (CD11) | Ovary | 1.2-fold change (↑) (mRNA) | [55] | ||
| Cluster of differentiation 11 c (CD11c) | Adipose Tissue | No change (→) (mRNA) | [41] | ||
| Cluster of differentiation 14 (CD14) | Plasma | Control: 400 ng/mL Affected: 800 ng/mL (↓) (Protein) | [41] | ||
| Cluster of differentiation 206 (CD206) | Ovary | 2.3-fold change (↓) (mRNA) | [55] | ||
| Cluster of differentiation 43 (CD43) | Liver | 4-fold change (↑) (mRNA) | [44] | ||
| Cluster of differentiation 68 (CD68) | Adipose Tissue | No change (→) (mRNA) | [41] | ||
| Plasma | 4-fold change (↑) (mRNA) | [46] | |||
| Cluster of differentiation 95 (CD95l or FAS Ligand) | Liver | 2.5-fold change (↑) (mRNA) | [52] | ||
| Plasma | 4-fold change (↑) (mRNA) | [46] | |||
| Complement system | Complement component 5a (c5a) | Plasma | 3.5-fold change (↑) (Protein) | [48] | |
| Cytokine | Interferon gamma (IFN-γ) | Plasma | No change (→) (Protein) | [39] | |
| Interleukin 1 alpha (IL-1α) | Plasma | 3.5-fold change (↑) (Protein) | [48] | ||
| Interleukin 1 beta (IL-1β) | Adipose tissue | No change (→) (mRNA) | [37] | ||
| BAL | No change (→) (Protein) | [42] | |||
| Colon | 2-fold change (↑) (mRNA) | [56] | |||
| Liver | No change (→) (mRNA) | [38] | |||
| 2-fold change (↑) (mRNA) | [37] | ||||
| Plasma | Control: 1 pg/mL Affected: 5 pg/mL (↑) (Protein) | [39] | |||
| 2-fold change (↑) (mRNA) | [46] | ||||
| Interleukin 16 (IL-16) | Plasma | 3.5-fold change (↑) (Protein) | [48] | ||
| No change (→) (Protein) | [37] | ||||
| Interleukin 17 (IL-17) | Plasma | Control: 11 pg/mL Affected: 18 pg/mL (↑) (Protein) | [39] | ||
| Tumor necrosis factor alpha (TNF-α) | Adipose tissue | 3-fold change (↑) (mRNA) | [37] | ||
| BAL | No change (→) (Protein) | [42] | |||
| Brain | Control: 0.2 ng/mg Affected: 0.7 ng/mg (↑) (Protein) | [36] | |||
| Kidney | 10-fold change (↑) (mRNA) | [43] | |||
| Liver | 3-fold change (↑) (mRNA) | [38] | |||
| No change (→) (mRNA) | [37] | ||||
| Control: 0.7 ng/mg Affected: 1 ng/mg (↑) (Protein) | [36] | ||||
| Myocardium | 2-fold change (↑) (mRNA) | [45] | |||
| Plasma | Control: 4 pg/mL Affected: 10 pg/mL (↑) (Protein) | [39] | |||
| No change (→) (Protein) | [57] | ||||
| Tumor necrosis factor soluble receptor II (TNF-sRII) | BAL | Control: 150 pg/ml Affected: 849.4 pg/ml (↑) (Protein) | [42] | ||
| Histologic hallmark (Dyong adipocytes and macrophages) | Crown-like structures (CLS) | Adipose tissue | Control: 0.4 CLS/mm2 Affected: 1.8 CLS/mm2 (↑) (Histochemistry) | [37] | |
| Inflammatory cell chemoattractant | Monocyte chemoattractant protein-1 (MCP-1) | Adipose tissue | 2-fold change (↑) (mRNA) | [41] | |
| 4-fold change (↑) (mRNA) | [37] | ||||
| Aorta | 1.8-fold change (↑) (mRNA) | [47] | |||
| BAL | Control: 10 ng/mL Affected: 90 pg/mL (↑) (Protein) | [42] | |||
| Liver | 2-fold change (↑) (mRNA) | [44] | |||
| No change (→) (mRNA) | [37] | ||||
| Myocardium | 3-fold change (↑) (mRNA) | [45] | |||
| Plasma | 4-fold change (↑) (Protein) | [48] | |||
| Intracellular cell-signal | Factor nuclear kappa B (NF-κβ) | Brain | Control: 15 pg/mL Affected: 50 pg/mL (↑) (Protein) | [36] | |
| Liver | Control: 10 pg/mL (→) (Protein) | [36] | |||
| IκB kinase (pIKK) | Peritone | 4-fold change (↑) (mRNA) | [46] | ||
| Mechanistic target of rapamycin (mTOR) | Liver | Control: 3 ng/mg Affected: 6 ng/mg (↑) (Protein) | [52] | ||
| Protein kinase B (AKT) | Adipose tissue | 1.5-fold (↓) (mRNA) | [41] | ||
| No change (→) (Protein) | [58] | ||||
| LPS presenting protein | Lipopolysaccharide binding protein (LBP) | Plasma | Control: 13 ng/mL Affected: 15 ng/mL (↑) (Protein) | [4] | |
| Macrophage biomarker | Chloroacetate esterase (CAE) | Liver | 6-fold change (↑) (Histochemistry) | [38] | |
| Macrophage glycoprotein (MOMA-2) | Liver | Control: 57 Cells/Area Affected: 70 Cells/Area (↑) (Histochemistry) | [52] | ||
| Macrophage receptor | EGF-like module-containing mucin-like hormone receptor-like 1 (EMR1 or F4/80) | Adipose tissue | 4-fold change (↑) (mRNA) | [37] | |
| Liver | 2-fold (↑) (Histochemistry) | [38] | |||
| 2-fold change (↑) (mRNA) | [37] | ||||
| Ovary | 2.3-fold change (↑) (mRNA) | [55] | |||
| Plasma | 2.5-fold (↑) mRNA | [46] | |||
| Macrophage scavenger receptor | Scavenger Receptor A (SR-a) | PBMC | 2.5-fold change (↓) (Protein) | [51] | |
| Microglia marker | Ionized Calcium-Binding Adaptor Molecule 1 (IBA1) | Brain | 1.25-fold change (↑) (Histochemistry) | [59] | |
| Mitogen-activated protein kinase | p-p38MAPK | Liver | 2.67-fold change (↑) (Protein) | [52] | |
| c-Jun N-terminal kinase (pJNK) | Brain | 2-fold change (↑) (mRNA) | [58] | ||
| Liver | 1.84-fold change (↑) (mRNA) | [52] | |||
| Neutrophil gelatinase-associated | Lipocalin-2 (LCN-2) | Colon | 95-fold change (↑) (mRNA) | [56] | |
| Feces | Control: 5 ng/mL Affected: 80 ng/mL (↑) (Protein) | [56] | |||
| Plasma | Control: 100 ng/mL Affected: 300 ng/mL (↑) (Protein) | [56] | |||
| NF-κβ p65 subunit | Transcription factor p65 (p65) | Myocardio | 1.5-fold change (↑) (mRNA) | [45] | |
| Peritone | 2-fold change (↑) (mRNA) | [42] | |||
| Oxidative stress biomarker | Myeloperoxidase (MPO) | Colon | No change (→) (Protein) | [56] | |
| Plasminogen regulation | Plasminogen activator inhibitor-1 (PAI-1) | Plasma | Control: 15 pg/mL Affected: 2 pg/mL (↓) (Protein) | [39] | |
| Control: 2900 pg/mL Affected: 3100 pg/mL (↑) (Protein) | [57] | ||||
| RAGE ligand | High–mobility group box 1 (HMGB1) | Plasma | 4 ng/mL (↑) (Protein) | [60] | |
| S100 A8/A10 | Myocardio | Control: 0.3 ng/mg Affected: 0.7 ng/mg (↑) (Protein) | [45] | ||
| Secretory serine protease | Serine protease inhibitor A3N (Serpina3n) | Brain | Control: 180 IOD Affected: 220 IOD (↑) (In situ hybridization) | [4] | |
| Signaling adapter | Insulin receptor substrate 1 (IRS-1) | Brain | No change (→) (Protein) | [58] | |
| Transport protein | Fatty-acid-binding Proteins (FABP) | Plasma | 2.5-fold change (↑) (mRNA) | [46] |
| Diet | AGE Levels in the Diets (Technique) | Time of Exposure (Weeks) | CLGI Biomarkers | Target Organ/Animal Model/Sex | Reference |
|---|---|---|---|---|---|
| High-AGE diet | Control: CML: Free: 3.0 μg/g; protein-bound: 10.0 μg/g Carboxyethyllysine (CEL): Free: 0.4 μg/g; protein-bound: 2.1 μg/g MG-H1: Free: 0.4 μg/g; protein-bound: 89.0 μg/g Baked chow diet: CML: Free: 1.0 μg/g; protein-bound: 38.0 μg/g CEL: Free: 0.5 μg/g; protein-bound: 30.5 μg/g MG-H1: Free: 1.6 μg/g; protein-bound: 137 μg/g (UPLC-MS/MS) | 10 | CRP, TNF-α, IFN-δ, IL-6, IL-10 | Plasma, fecal microbiota/C57BL/6/Females | [74] |
| High-AGE diet | Control: CML: 2.58 μg/g CEL: 0.89 μg/g MG-H1: 34.51 μg/g Baked chow diet: CML: 4.87 μg/g CEL: 1.38 μg/g MG-H1: 43.49 μg/g (QTRAP LC-MS/MS) | 24 | MCP1, LPS, C3a, C5a, occludin | Plasma and gut/Sprague-Dawley and C57BL/6/Males | [75] |
| MG-H1-enriched diet | 3420 μg/g (HPLC-MS/MS) | 22 | IL-1β, IL-17, IFN-γ, TNF-α, PAI-1 | Plasma, fecal microbiota/C57BL/6/Males | [39] |
| CML-enriched diet | Control: 61.9 μg/g CML diet: 605 μg/g (ELISA) | 13 | F4/80, CD11c, CD206 | Ovary/C57BL/6/Females | [55] |
| CML-enriched diet | Commercial CML 0.1% w/w | 24 | C5a, ICAM, IFN-δ, IL-1α, IL-1β, IL-1ra, IL-6, IL-10, IL-12, IL-13, IL-16, IL-17, IL23, TNF-α | Plasma/Swiss/Males | [48] |
| High-AGE diet | Control: CML: 2.79 μg/g Baked chow diet: CML: 14.45 μg/g (HPLC) | 6, 12, 18 | Microbiota | Gut/Sprague-Dawley/Males | [76] |
| CML-enriched diet | Control: 17.5 µg/g CML diet: 200 μg/g (HPLC-LTQ) | 36 | VCAM-1, RAGE | Aorta/RAGE KO/Males | [49] |
| High-AGE diet | Control: Furosine: 28.80 μg/g Hydroxymethylfurfural (HMF): 0.44 μg/g CML: 2.20 μg/g Baked chow diet: Furosine: 1787.08 μg/g HMF: 5.15 μg/g CML: 12.46 μg/g (ND) | 22 | Microbiota | Gut/Wistar/Males | [77] |
| High-AGE diet | Control: Furosine: 28.8 μg/g HMF: 0.44 μg/g Bread crust diet: Furosine: 49.5 μg/g HMF: 4.26 μg/g (HPLC) | 22 | Microbiota | Gut/Wistar/Males | [78] |
| High-AGE methionine choline-deficient diet | Control: 31 nmol/glysine CML diet: 137 nmol/glysine (ELISA) | 12 | Il-6, MCP-1, RAGE, CD43 | Liver/Sprague–Dawley/Males | [44] |
| High-AGE diet | Control: CML diet: 13 μg/g Fructoselysine: 104 μg/g Furosine: 268 μg/g H-AGE: CML diet: 760 μg/g Fructoselysine: 205 μg/g Furosine: 526 μg/g (ND) | 12 | RAGE, SR-A | PBMC/Wistar/Females | [51] |
| High-AGE diet | Control: 23 μg/g AGE diet: 110 μg/g (ELISA) | 4 | TNFα, TNF sRII, IL-1β, IL-6, IL-10, CXC, KC, MIP-2, CINC-1, MCP-1 | Bronchoalveolar lavage/CD-1/Mixed | [42] |
| High-AGE diet | Control: CML: 60649 U/g H-AGE CML: 197305 U/g (ELISA) | 39 | Neutrophil infiltration | Liver/C57BL/6NHsd/Males | [79] |
| HFD-High-AGE diet | Control: 20.90 nmol CML/mol lysine/g AGE diet: 101.90 nmol CML/mol lysine/g (ND) | 16 | MCP-1, MIF (macrophage migration inhibitory factor), RAGE | Kidney/C57BL/6 (RAGEKO)/Males | [80] |
| Market bought High-AGE diet | 53–1473 AU/g | 1 | HMGB1 | Wound healing/Kunming mice/Males | [60] |
| High-AGE diet | Control: 1 µmol CML/lysine/day AGE diet: 4 µmol CML/lysine/day (ELISA) | 16 | IL-6, TNFα, ICAM-1, MCP-1, p65, RAGE, S1—A8/A9 | Myocardio/RAGE KO/Males | [45] |
| High-AGE diet | Control: 112 µg/g CML diet: 785 µg/g (ELISA) | 5, 9, and 13 | Macrophage infiltration (ED1-positive), MCP-1 | Kidney/Sprague-Dawley/Males | [54] |
| High-AGE diet | Control: 119,000 µg/g CML diet: 930,000 µg/g (ELISA) | 11 | Macrophage infiltration | Colon/Sprague-Dawley/Males | [53] |
| High-AGE diet | Control CML: 2700 U/mg CML diet: 12,500 U/mg Control MG: 0.65 U/mg MG diet: 2.5 U/mg (ELISA) | 8 | VCAM-1, RAGE, MOMA-2 | Aorta/ApoE KO/Males | [81] |
| High-AGE diet | Control CML: 107 U/mg CML diet: 535 U/mg Control MG: 3.6 U/mg MG diet: 18 U/mg (ELISA) | 28 | Inflammatory cell infiltration | Skin/db/db/Females | [82] |
| CLGI Trigger | Target Organ | Time of Exposure (Weeks) | CLGI Biomarkers | Animal/Sex | Reference |
|---|---|---|---|---|---|
| HFD | Liver | 24 | TNF-α, IL-1β, IL-6, IL-10, CAE+, F4/80+ | C57BL/6J/Females | [38] |
| Adipose tissue, liver | 24, 40, and 52 | TNF-α, IL-1β, MCP-1, F4/80+, crown-like structures | C57BL/6/Males | [37] | |
| Adipose tissue | 11 | CD14, AKT, CD68, C11c, MCP-1, IL-6 | C57BL/6/Males | [41] | |
| Hypothalamus | 8 | Serpina3n | C57BL/6J, TLR4 KO, CD14 KO/Males | [4] | |
| Gut microbiota | 12 | NF-kB, mTOR, AKT | C57BL/6/Males | [57] | |
| Gut microbiota | 8 | PPARγ, C/EBPa, FAZ, aFABP, CD68, F4/80, p-IKK β, p65, TNF-α, IL 1β, IL-6 | C57BL/6J, TLR4 KO C57BL/10ScNJ/Males | [46] | |
| High-calorie diet (30% fructose) | Liver, brain | 8 | TNF-α, IL-2, NF-κB, HVA | Sprague-Dawley/Males | [36] |
| Intragastric fructose injection | Serum, liver, pancreas | 20 | IL-6, TNF-α, MIP-2, IL-10 | Sprague-Dawley/Males | [96] |
| CLGI Trigger | Target Organ | Time of Exposure (Weeks) | CLGI Assessment Biomarkers | Strain/Sex | Reference |
|---|---|---|---|---|---|
| LPS | Hypothalamus | 12 | Iba1, TH | Sprague-Dawley/Males | [59] |
| Hypothalamus | 1 | IRS1, AKT, JNK | Wistar/Males | [58] | |
| Liver | 4 | p38 MAPK, MPO, TNF-α, MCP-1, IL-6 | ApoE KO, C57BL/6J/Males | [52] | |
| Plasma | 8 | TNF-α, TNF-β, MCP-1, IL-6 | ApoE KO/Male | [120] | |
| DSS | Colon, feces, plasma | 1 | Lipocalin-2 | C57BL/6/Males; IL-10 KO/Females | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogueira Silva Lima, M.T.; Howsam, M.; Anton, P.M.; Delayre-Orthez, C.; Tessier, F.J. Effect of Advanced Glycation End-Products and Excessive Calorie Intake on Diet-Induced Chronic Low-Grade Inflammation Biomarkers in Murine Models. Nutrients 2021, 13, 3091. https://doi.org/10.3390/nu13093091
Nogueira Silva Lima MT, Howsam M, Anton PM, Delayre-Orthez C, Tessier FJ. Effect of Advanced Glycation End-Products and Excessive Calorie Intake on Diet-Induced Chronic Low-Grade Inflammation Biomarkers in Murine Models. Nutrients. 2021; 13(9):3091. https://doi.org/10.3390/nu13093091
Chicago/Turabian StyleNogueira Silva Lima, Matheus Thomaz, Michael Howsam, Pauline M. Anton, Carine Delayre-Orthez, and Frédéric J. Tessier. 2021. "Effect of Advanced Glycation End-Products and Excessive Calorie Intake on Diet-Induced Chronic Low-Grade Inflammation Biomarkers in Murine Models" Nutrients 13, no. 9: 3091. https://doi.org/10.3390/nu13093091
APA StyleNogueira Silva Lima, M. T., Howsam, M., Anton, P. M., Delayre-Orthez, C., & Tessier, F. J. (2021). Effect of Advanced Glycation End-Products and Excessive Calorie Intake on Diet-Induced Chronic Low-Grade Inflammation Biomarkers in Murine Models. Nutrients, 13(9), 3091. https://doi.org/10.3390/nu13093091