
Neuroprotective Effects and Therapeutic Potential of the Citrus Flavonoid Hesperetin in Neurodegenerative Diseases



Abstract
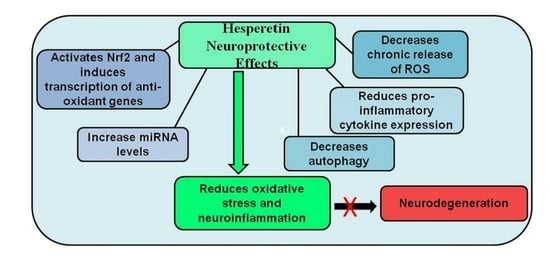
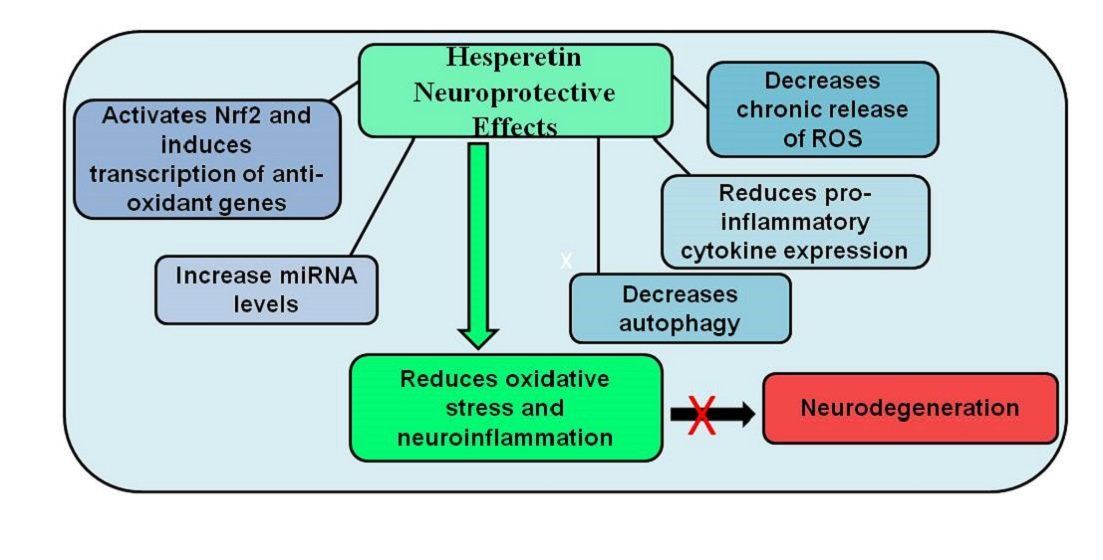
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
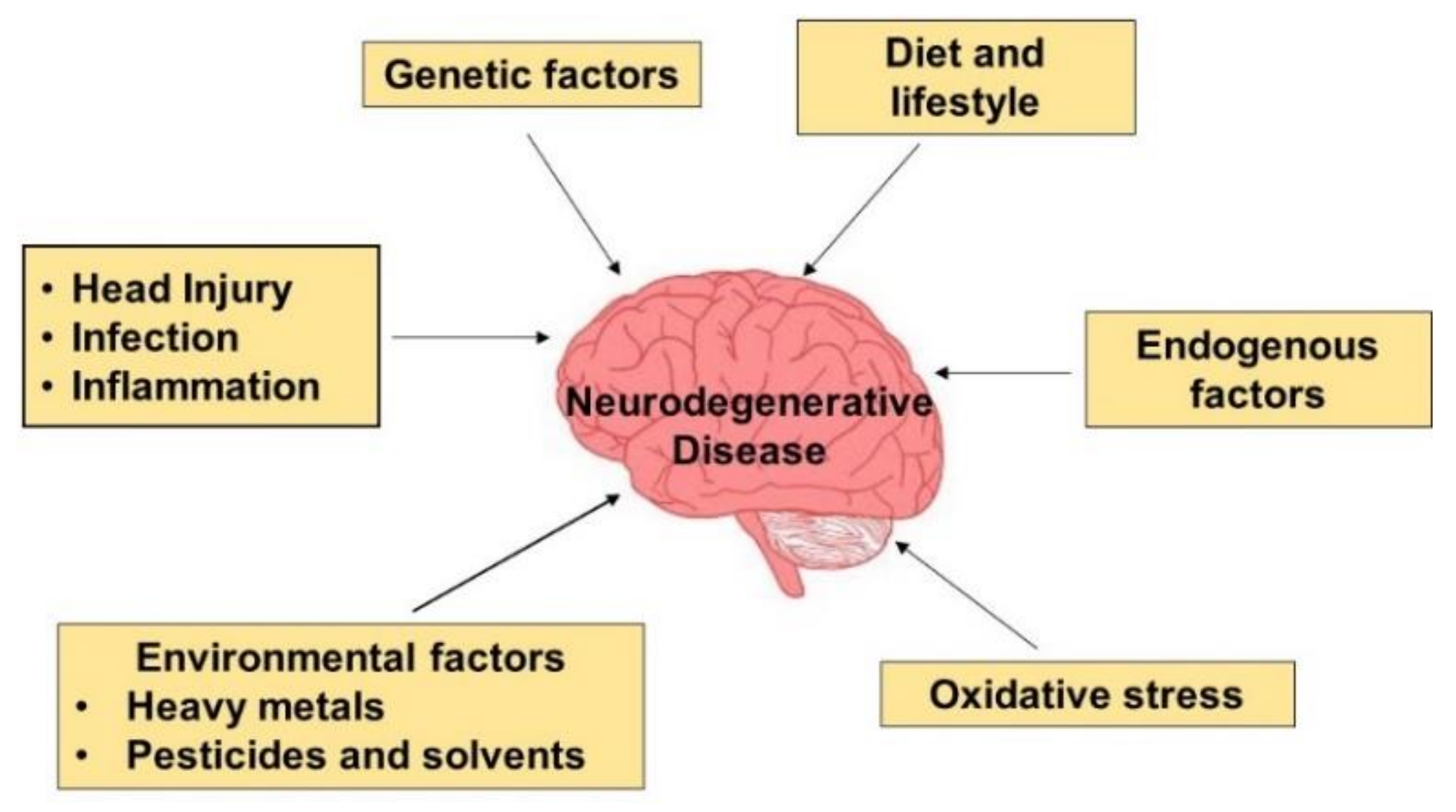
2. Neurodegenerative Etiology
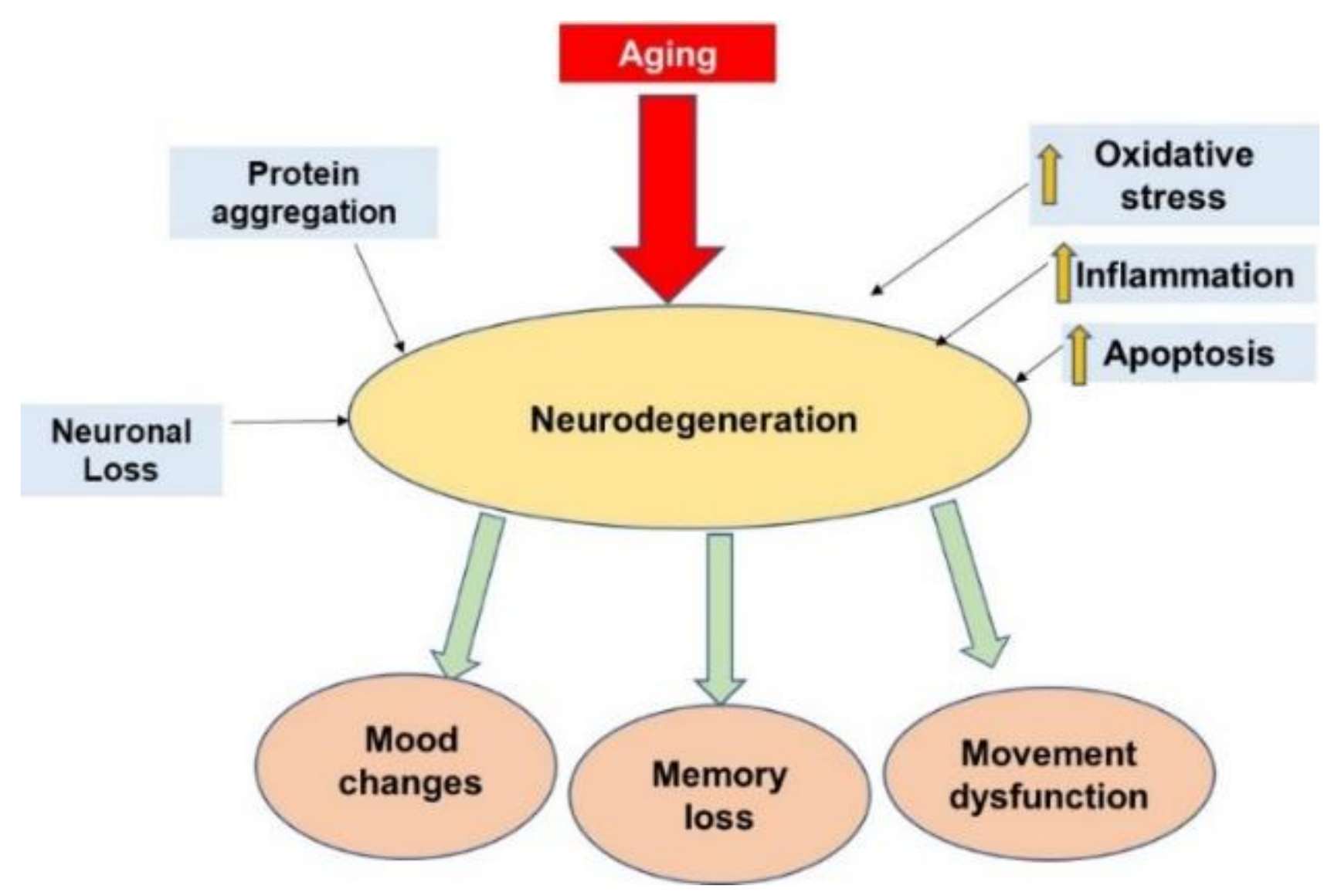
Aging and Neurodegeneration
3. The Role of Neuroinflammation in Neurodegeneration
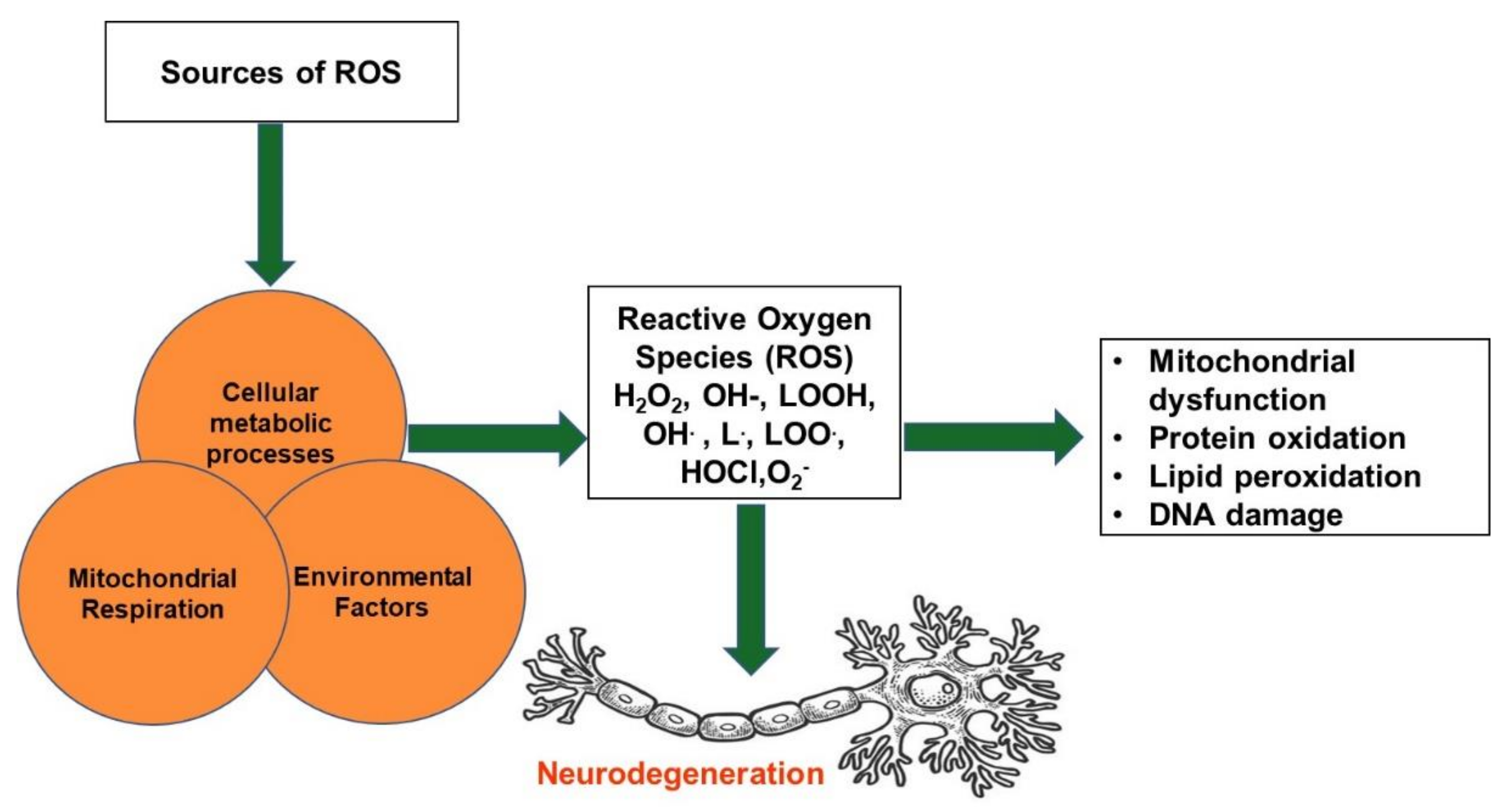
4. Oxidative Stress and Neurodegenerative Diseases
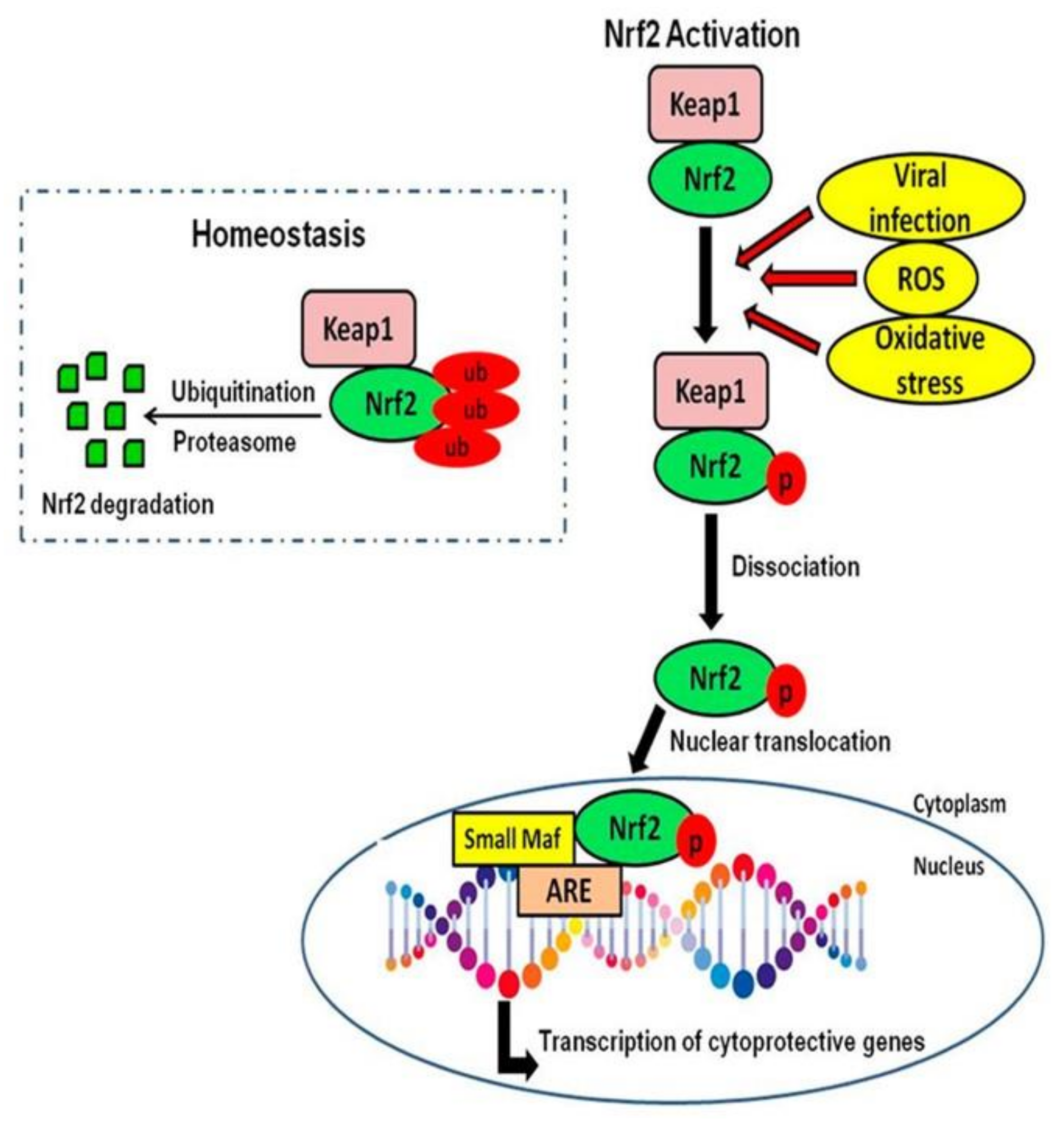
5. Nrf2 Role in Neurodegeneration and Neuroinflammation
Neuroprotective Effects of Nrf2 Activation
6. Flavonoids
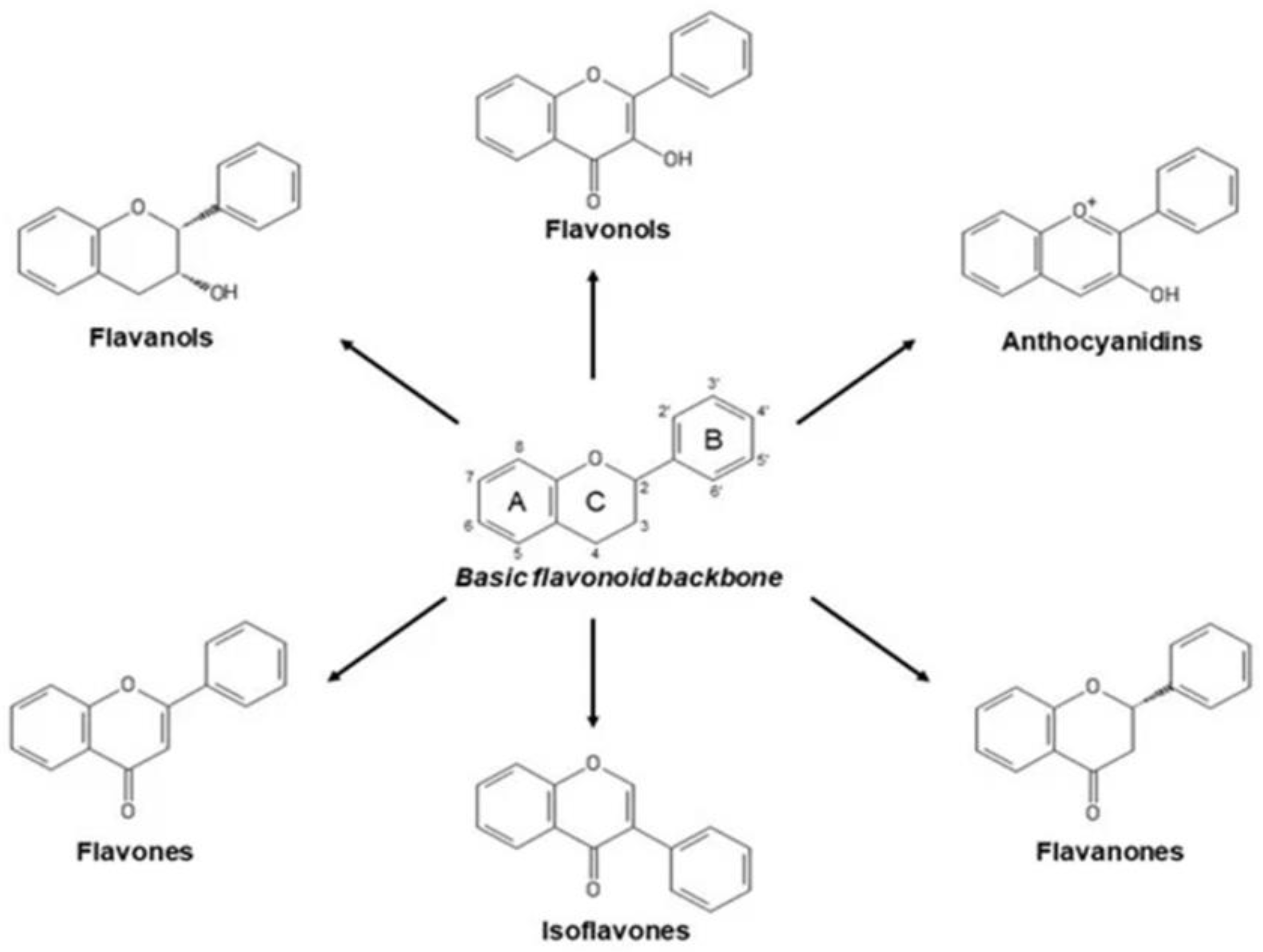
6.1. Classification
6.2. Chemical Structure, Bioavailability, and Dietary Sources
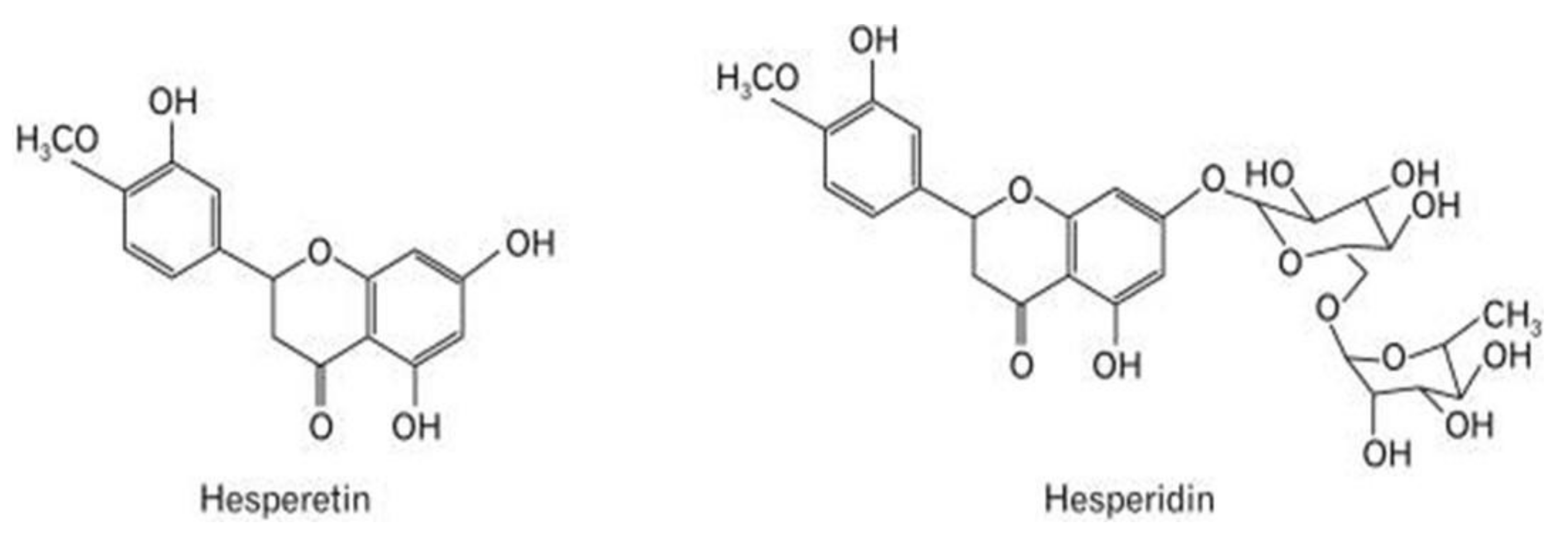
6.3. Hesperetin
6.4. Hesperetin and Microglia
6.5. Hesperetin Crossing the Blood–Brain Barrier
6.6. Hesperetin and Neuroinflammation
6.7. Hesperetin and Oxidative Stress
6.8. Hesperetin’s Effect on Canonical and Non-Canonical Mechanisms of Nrf2 Activation
6.9. Possible Effects of Hesperetin on Autophagy Modulation
6.10. The Possible Protective Effects of Hesperetin on MicroRNA
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative Diseases: An Overview of Environmental Risk Factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Ikram, M.; Hahm, J.R.; Kim, M.O. Antioxidant and Anti-Inflammatory Effects of Citrus Flavonoid Hesperetin: Special Focus on Neurological Disorders. Antioxidants 2020, 9, 609. [Google Scholar] [CrossRef] [PubMed]
- Meek, P.D.; McKeithan, K.; Schumock, G.T. Economic considerations in Alzheimer’s disease. Pharmacotherapy 1998, 18 Pt 2, 68–73; discussion 9–82. [Google Scholar] [CrossRef] [PubMed]
- Zahra, W.; Rai, S.N.; Birla, H.; Singh, S.S.; Dilnashin, H.; Rathore, A.S.; Singh, S.P. The global economic impact of neurodegenerative diseases: Opportunities and challenges. In Bioeconomy for Sustainable Development; Keswani, C., Ed.; Springer Nature: Singapore, 2020; pp. 333–345. ISBN 9789811394300. [Google Scholar]
- Cannon, J.R.; Greenamyre, J.T. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol. Sci. 2011, 124, 225–250. [Google Scholar] [CrossRef]
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef] [Green Version]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage, and neurodegenerative diseases. Neural. Regen. Res. 2013, 8, 2003–2014. [Google Scholar]
- Kang, Y.; Lee, J.-H.; Seo, Y.H.; Jang, J.-H.; Jeong, C.-H.; Lee, S.; Jeong, G.-S.; Park, B. Epicatechin Prevents Methamphetamine-Induced Neuronal Cell Death via Inhibition of ER Stress. Biomol. Ther. 2019, 27, 145–151. [Google Scholar] [CrossRef]
- De Andrade Teles, R.B.; Diniz, T.C.; Costa Pinto, T.C.; de Oliveira Júnior, R.G.; Gama e Silva, M.; de Lavor, É.M.; Fernandes, A.W.C.; de Oliveira, A.P.; de Almeida Ribeiro, F.P.R.; da Silva, A.A.M.; et al. Flavonoids as Therapeutic Agents in Alzheimer’s and Parkinson’s Diseases: A Systematic Review of Preclinical Evidence. Oxid. Med. Cell. Longev. 2018, 2018, 7043213. [Google Scholar] [CrossRef]
- Jones, Q.R.; Warford, J.; Rupasinghe, H.V.; Robertson, G.S. Target-based selection of flavonoids for neurodegenerative disorders. Trends Pharmacol. Sci. 2012, 33, 602–610. [Google Scholar] [CrossRef]
- Gonzales, G.B.; Smagghe, G.; Grootaert, C.; Zotti, M.; Raes, K.; Camp, J.V. Flavonoid interactions during digestion, absorption, distribution, and metabolism: A sequential structure-activity/property relationship-based approach in the study of bioavailability and bioactivity. Drug Metab. Rev. 2015, 47, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Parhiz, H.; Roohbakhsh, A.; Soltani, F.; Rezaee, R.; Iranshahi, M. Antioxidant, and anti-inflammatory properties of the citrus flavonoid’s hesperidin and hesperetin: An updated review of their molecular mechanisms and experimental models. Phytother. Res. 2015, 29, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-W.; Wang, W.; Sheng, H.; Bai, Y.-F.; Weng, Y.-Y.; Fan, X.-Y.; Zheng, F.; Zhu, X.T.; Xu, Z.C.; Zhang, F.; et al. hesperetin, a SIRT1 activator, inhibits hepatic inflammation via the AMPK/CREB pathway. Int. Immunopharmacol. 2020, 89, 107036. [Google Scholar] [CrossRef]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis, and Memory Impairment by Modulating TLR4/NF-κB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Qian, Y.; Chen, C.; Feng, F.; Pan, L.; Yang, L.; Wang, C. Hesperetin Exhibits Anti-Inflammatory Effects on Chondrocytes via the AMPK Pathway to Attenuate Anterior Cruciate Ligament Transection-Induced Osteoarthritis. Front. Pharmacol. 2021, 12, 735087. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A. Recent advances in our understanding of neurodegeneration. J. Neural Transm. 2009, 116, 1111–1162. [Google Scholar] [CrossRef]
- Carmona, J.J.; Michan, S. Biology of Healthy Aging and Longevity. Rev. Investig. Clin. 2016, 68, 7–16. [Google Scholar]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Azam, S.; Haque, E.; Balakrishnan, R.; Kim, I.-S.; Choi, D.-K. The Ageing Brain: Molecular and Cellular Basis of Neurodegeneration. Front. Cell Dev. Biol. 2021, 9, 683459. [Google Scholar] [CrossRef]
- Hung, C.-W.; Chen, Y.-C.; Hsieh, W.-L.; Chiou, S.-H.; Kao, C.-L. Ageing, and neurodegenerative diseases. Ageing Res. Rev. 2010, 9, S36–S46. [Google Scholar] [CrossRef] [PubMed]
- Elobeid, A.; Libard, S.; Leino, M.; Popova, S.N.; Alafuzoff, I. Altered Proteins in the Aging Brain. J. Neuropathol. Exp. Neurol. 2016, 75, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salahuddin, P.; Rabbani, G.; Khan, R.H. The role of advanced glycation end products in various types of neurodegenerative disease: A therapeutic approach. Cell. Mol. Biol. Lett. 2014, 19, 407–437. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.J. Role of RAGE in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Smith, U. Advanced glycation end products, dementia, and diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 4743–4744. [Google Scholar] [CrossRef] [Green Version]
- Van Ham, T.J.; Breitling, R.; Swertz, M.A.; Nollen, E.A. Neurodegenerative diseases: Lessons from genome-wide screens in small model organisms. EMBO Mol. Med. 2009, 1, 360–370. [Google Scholar] [CrossRef]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Small, S.A.; Tsai, W.Y.; DeLaPaz, R.; Mayeux, R.; Stern, Y. Imaging hippocampal function across the human life span: Is memory decline normal or not? Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2002, 51, 290–295. [Google Scholar] [CrossRef]
- Gómez-Isla, T.; Price, J.L.; McKeel, D.W., Jr.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar] [CrossRef]
- Rodrigue, K.M.; Raz, N. Shrinkage of the entorhinal cortex over five years predicts memory performance in healthy adults. J. Neurosci. 2004, 24, 956–963. [Google Scholar] [CrossRef] [Green Version]
- Andrews-Hanna, J.R.; Snyder, A.Z.; Vincent, J.L.; Lustig, C.; Head, D.; Raichle, M.E.; Buckner, R.L. Disruption of large-scale brain systems in advanced aging. Neuron 2007, 56, 924–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yankner, B.A.; Lu, T.; Loerch, P. The aging brain. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 41–66. [Google Scholar] [CrossRef]
- Loerch, P.M.; Lu, T.; Dakin, K.A.; Vann, J.M.; Isaacs, A.; Geula, C.; Wang, J.; Pan, Y.; Gabuzda, D.H.; Li, C.; et al. Evolution of the Aging Brain Transcriptome and Synaptic Regulation. PLoS ONE 2008, 3, e3329. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Bohl, J.R.; Müller, C.M.; Rüb, U.; de Vos, R.A.; Del Tredici, K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov. Disord. Off. J. Mov. Disord. Soc. 2006, 21, 2042–2051. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; McCann, H. The progression of pathology in Parkinson’s disease. Ann. N.Y. Acad. Sci. 2010, 1184, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, E.; Bester, J.; Page, M.; Kell, D. The Potential of LPS-Binding Protein to Reverse Amyloid Formation in Plasma Fibrin of Individuals With Alzheimer-Type Dementia. Front. Aging Neurosci. 2018, 10, 257. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Deng, J.S.; Chang, Y.S.; Huang, G.J. Ginsenoside Rh2 Ameliorates Lipopolysaccharide-Induced Acute Lung Injury by Regulating the TLR4/PI3K/Akt/mTOR, Raf-1/MEK/ERK, and Keap1/Nrf2/HO-1 Signaling Pathways in Mice. Nutrients 2018, 10, 1208. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Al-Gubory, K.H.; Garrel, C.; Faure, P.; Sugino, N. Roles of antioxidant enzymes in corpus luteum rescue from reactive oxygen species-induced oxidative stress. Reprod. Biomed. Online 2012, 25, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef]
- Ceriello, A. Possible Role of Oxidative Stress in the Pathogenesis of Hypertension. Diabetes Care 2008, 31, S181–S184. [Google Scholar] [CrossRef] [Green Version]
- Embury, C.; Dyavarshetty, B.; Lu, Y.; Wiederin, J.L.; Ciborowski, P.; E Gendelman, H.; Kiyota, T. Cathepsin B Improves ß-Amyloidosis and Learning and Memory in Models of Alzheimer’s Disease. J. Neuroimmune Pharmacol. 2016, 12, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef]
- Pacheco-Quinto, J.; Eckman, E.A. Endothelin-converting Enzymes Degrade Intracellular β-Amyloid Produced within the Endosomal/Lysosomal Pathway and Autophagosomes. J. Biol. Chem. 2013, 288, 5606–5615. [Google Scholar] [CrossRef] [Green Version]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [Green Version]
- Carrí, M.T.; Ferri, A.; Cozzolino, M.; Calabrese, L.; Rotilio, G. Neurodegeneration in amyotrophic lateral sclerosis: The role of oxidative stress and altered homeostasis of metals. Brain Res. Bull. 2003, 61, 365–374. [Google Scholar] [CrossRef]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE Signaling Pathway: Key Mediator in Oxidative Stress and Potential Therapeutic Target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef]
- Vargas, M.; Pehar, M.; Cassina, P.; Beckman, J.S.; Barbeito, L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J. Neurochem. 2006, 97, 687–696. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Hauser, D.N.; Hastings, T.G. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol. Dis. 2013, 51, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.L.; Yen, G.C. Effect of hesperetin against oxidative stress via ER- and TrkA-mediated actions in PC12 cells. J. Agric. Food Chem. 2011, 59, 5779–5785. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, O. Role of oxidative stress in Parkinson’s disease. Exp. Neurobiol. 2013, 22, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muftuoglu, M.; Elibol, B.; Dalmızrak, .; Ercan, A.; Kulaksız, G.; Ögüs, H.; Dalkara, T.; Özer, N. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov. Disord. 2003, 19, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by Protein Kinase C Regulates Antioxidant Response Element-mediated Transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2013, 79, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellezza, I.; Tucci, A.; Galli, F.; Grottelli, S.; Mierla, A.L.; Pilolli, F.; Minelli, A. Inhibition of NF-κB nuclear translocation via HO-1 activation underlies α-tocopheryl succinate toxicity. J. Nutr. Biochem. 2012, 23, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Quinti, L.; Dayalan Naidu, S.; Träger, U.; Chen, X.; Kegel-Gleason, K.; Llères, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. USA 2017, 114, e4676–e4685. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.-M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- De Vries, H.E.; Witte, M.; Hondius, D.; Rozemuller, A.J.; Drukarch, B.; Hoozemans, J.; van Horssen, J. Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free. Radic. Biol. Med. 2008, 45, 1375–1383. [Google Scholar] [CrossRef]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef]
- Lee, J.-M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related Factor-2-dependent Genes Conferring Protection against Oxidative Stress in Primary Cortical Astrocytes Using Oligonucleotide Microarray Analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef] [Green Version]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear Factor E2-Related Factor 2-Dependent Antioxidant Response Element Activation by tert-Butylhydroquinone and Sulforaphane Occurring Preferentially in Astrocytes Conditions Neurons against Oxidative Insult. J. Neurosci. 2004, 24, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Hannan, M.A.; Dash, R.; Sohag, A.A.M.; Haque, M.N.; Moon, I.S. Neuroprotection Against Oxidative Stress: Phytochemicals Targeting TrkB Signaling and the Nrf2-ARE Antioxidant System. Front. Mol. Neurosci. 2020, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. Korean Soc. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanninen, K.; Malm, T.M.; Jyrkkänen, H.-K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Ylä-Herttuala, S.; Levonen, A.-L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Gan, K.A.; Johnson, D.A.; Johnson, J.A. Increased Alzheimer’s disease-like pathology in the APP/PS1ΔE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol. Aging 2015, 36, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing Expression of Nrf2-Driven Genes Protects the Blood Brain Barrier after Brain Injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Sun, G.; Zhang, J.; Strong, R.; Dash, P.K.; Kan, Y.W.; Grotta, J.C.; Aronowski, J. Transcription Factor Nrf2 Protects the Brain From Damage Produced by Intracerebral Hemorrhage. Stroke 2007, 38, 3280–3286. [Google Scholar] [CrossRef]
- Mahomoodally, M.F.; Gurib-Fakim, A.; Subratty, A.H. Antimicrobial Activities and Phytochemical Profiles of Endemic Medicinal Plants of Mauritius. Pharm. Biol. 2005, 43, 237–242. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.; Yu, O. Metabolic engineering of flavonoids in plants and microorganisms. Appl. Microbiol. Biotechnol. 2011, 91, 949–956. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Bolwell, P.G.; Bramley, P.M.; Pridham, J.B. The Relative Antioxidant Activities of Plant-Derived Polyphenolic Flavonoids. Free Radic. Res. 1995, 22, 375–383. [Google Scholar] [CrossRef]
- Rendeiro, C.; Rhodes, J.S.; Spencer, J.P. The mechanisms of action of flavonoids in the brain: Direct versus indirect effects. Neurochem. Int. 2015, 89, 126–139. [Google Scholar] [CrossRef]
- Cosme, P.; Rodríguez, A.B.; Espino, J.; Garrido, M. Plant Phenolics: Bioavailability as a Key Determinant of Their Potential Health-Promoting Applications. Antioxidants 2020, 9, 1263. [Google Scholar] [CrossRef]
- Youdim, K.A.; Dobbie, M.S.; Kuhnle, G.; Proteggente, A.R.; Abbott, N.J.; Rice-Evans, C. Interaction between flavonoids and the blood-brain barrier: In vitro studies. J. Neurochem. 2003, 85, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Aranganathan, S.; Selvam, J.P.; Nalini, N. Effect of hesperetin, a citrus flavonoid, on bacterial enzymes and carcinogen-induced aberrant crypt foci in colon cancer rats: A dose-dependent study. J. Pharm. Pharmacol. 2010, 60, 1385–1392. [Google Scholar] [CrossRef]
- Kim, J.; Wie, M.-B.; Ahn, M.; Tanaka, A.; Matsuda, H.; Shin, T. Benefits of hesperidin in central nervous system disorders: A review. Anat. Cell Biol. 2019, 52, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, F.; Lian, Y.; Xiao, H.; Zheng, J. Biosynthesis of citrus flavonoids and their health effects. Crit. Rev. Food Sci. Nutr. 2018, 60, 566–583. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, I.L.F.; Chee, W.; Poulsen, L.; Offord-Cavin, E.; Rasmussen, S.E.; Frederiksen, H.; Enslen, M.; Barron, D.; Horcajada, M.-N.; Williamson, G. Bioavailability Is Improved by Enzymatic Modification of the Citrus Flavonoid Hesperidin in Humans: A Randomized, Double-Blind, Crossover Trial. J. Nutr. 2006, 136, 404–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlund, I.; Meririnne, E.; Alfthan, G.; Aro, A. Plasma Kinetics and Urinary Excretion of the Flavanones Naringenin and Hesperetin in Humans after Ingestion of Orange Juice and Grapefruit Juice. J. Nutr. 2001, 131, 235–241. [Google Scholar] [CrossRef]
- Hollman, P.C.H. Absorption, Bioavailability, and Metabolism of Flavonoids. Pharm. Biol. 2004, 42 (Suppl. 1), 74–83. [Google Scholar] [CrossRef]
- Jo, S.H.; Kim, M.E.; Cho, J.H.; Lee, Y.; Lee, J.; Park, Y.-D.; Lee, J.S. Hesperetin inhibits neuroinflammation on microglia by suppressing inflammatory cytokines and MAPK pathways. Arch. Pharmacal Res. 2019, 42, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Ahn, W.S. Neuroprotective effects of chronic hesperetin administration in mice. Arch. Pharmacal Res. 2008, 31, 1457–1462. [Google Scholar] [CrossRef]
- Vauzour, D.; Vafeiadou, K.; Rice-Evans, C.; Williams, R.J.; Spencer, J.P. Activation of pro-survival Akt and ERK1/2 signalling pathways underlie the anti-apoptotic effects of flavanones in cortical neurons. J. Neurochem. 2007, 103, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; De Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Hong, J.-S. Role of Microglia in Inflammation-Mediated Neurodegenerative Diseases: Mechanisms and Strategies for Therapeutic Intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boje, K.M.; Arora, P.K. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992, 587, 250–256. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Molitor, T.W.; Shaskan, E.G.; Peterson, P.K. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J. Immunol. 1992, 149, 2736–2741. [Google Scholar]
- McGuire, S.; Ling, Z.D.; Lipton, J.W.; Sortwell, C.E.; Collier, T.J.; Carvey, P.M. Tumor Necrosis Factor α Is Toxic to Embryonic Mesencephalic Dopamine Neurons. Exp. Neurol. 2001, 169, 219–230. [Google Scholar] [CrossRef]
- Teismann, P.; Schulz, J.B. Cellular pathology of Parkinson’s disease: Astrocytes, microglia, and inflammation. Cell Tissue Res. 2004, 318, 149–161. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, Y.; Pauze, M.; Fukuyama, K.; Nagai, N.; Funakoshi-Tago, M.; Sugai, T.; Tamura, H. Effect of hesperetin derivatives on the development of selenite-induced cataracts in rats. Mol. Med. Rep. 2018, 18, 1043–1050. [Google Scholar] [CrossRef] [Green Version]
- Shukitt-Hale, B. The effects of aging and oxidative stress on psychomotor and cognitive behavior. Age 1999, 22, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeter, H.; Spencer, J.P.; Rice-Evans, C.; Williams, R.J. Flavonoids protect neurons from oxidized low-density-lipoprotein-induced apoptosis involving c-Jun N-terminal kinase (JNK), c-Jun, and caspase-3. Biochem. J. 2001, 358 Pt 3, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.; Schroeter, H.; Kuhnle, G.; Srai, S.K.; Tyrrell, R.M.; Hahn, U.; Rice-Evans, C. Epicatechin and it’s in vivo metabolite, 3’-O-methyl epicatechin, protect human fibroblasts from oxidative-stress-induced cell death involving caspase-3 activation. Biochem. J. 2001, 354 Pt 3, 493–500. [Google Scholar] [CrossRef]
- Spencer, J.P.; Schroeter, H.; Crossthwaithe, A.J.; Kuhnle, G.; Williams, R.; Rice-Evans, C. Contrasting influences of glucuronidation and O -methylation of epicatechin on hydrogen peroxide-induced cell death in neurons and fibroblasts. Free Radic. Biol. Med. 2001, 31, 1139–1146. [Google Scholar] [CrossRef]
- Shimouchi, A.; Yokota, H.; Ono, S.; Matsumoto, C.; Tamai, T.; Takumi, H.; Narayanan, S.P.; Kimura, S.; Kobayashi, H.; Caldwell, R.; et al. Neuroprotective effect of water-dispersible hesperetin in retinal ischemia reperfusion injury. Jpn. J. Ophthalmol. 2015, 60, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, V.T.; de Franco, É.P.; de Araújo, M.E.; Messias, M.C.; Priviero, F.B.; Frankland Sawaya, A.C.; de Oliveira Carvalho, P. Characterization of the antioxidant activity of aglycone and glycosylated derivatives of hesperetin: An in vitro and in vivo study. J. Mol. Recognit. 2016, 29, 80–87. [Google Scholar] [CrossRef]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin Confers Neuroprotection by Regulating Nrf2/TLR4/NF-κB Signaling in an Aβ Mouse Model. Mol. Neurobiol. 2019, 56, 6293–6309. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ikram, M.; Ullah, N.; Alam, S.I.; Park, H.Y.; Badshah, H.; Choe, K.; Kim, M.O. Neurological Enhancement Effects of Melatonin against Brain Injury-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration via AMPK/CREB Signaling. Cells 2019, 8, 760. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, T.; Ali, T.; Ikram, M.; Khan, A.; Alam, S.I.; Kim, M.O. Melatonin Rescue Oxidative Stress-Mediated Neuroinflammation/Neurodegeneration and Memory Impairment in Scopolamine-Induced Amnesia Mice Model. J. Neuroimmune Pharmacol. 2019, 14, 278–294. [Google Scholar] [CrossRef]
- Zhu, C.; Dong, Y.; Liu, H.; Ren, H.; Cui, Z. Hesperetin protects against H (2)O(2)-triggered oxidative damage via upregulation of the Keap1-Nrf2/HO-1 signal pathway in ARPE-19 cells. Biomed. Pharmacother. 2017, 88, 124–133. [Google Scholar] [CrossRef]
- Haidari, F.; Keshavarz, S.A.; Rashidi, M.R.; Shahi, M.M. Orange Juice and Hesperetin Supplementation to Hyperuricemic Rats Alter Oxidative Stress Markers and Xanthine Oxidoreductase Activity. J. Clin. Biochem. Nutr. 2009, 45, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Liu, Z.; Wang, J.; Zhu, H. Antioxidative effects of hesperetin against lead acetate-induced oxidative stress in rats. Indian J. Pharmacol. 2013, 45, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.J. Antioxidative effects of hesperetin against 7,12-dimethylbenz(a)anthracene-induced oxidative stress in mice. Life Sci. 2008, 82, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Kheradmand, E.; Hajizadeh Moghaddam, A.; Zare, M. Neuroprotective effect of hesperetin and nano-hesperetin on recognition memory impairment and the elevated oxygen stress in a rat model of Alzheimer’s disease. Biomed. Pharmacother. 2018, 97, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-M.; Tsai, S.-Y.; Lin, J.-A.; Wu, C.-H.; Yen, G.-C. Cytoprotective effects of hesperetin and hesperidin against amyloid β-induced impairment of glucose transport through downregulation of neuronal autophagy. Mol. Nutr. Food Res. 2012, 56, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Islas, C.S.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The Role of Keap1 in Cellular Protective Responses. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2–ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef]
- Joshi, G.; Johnson, J.A. The Nrf2-ARE pathway: A valuable therapeutic target for the treatment of neurodegenerative diseases. Recent Pat. CNS Drug Discov. 2012, 7, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.-C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabhan, B.; Tong, K.I.; Kobayashi, A.; Yamamoto, M.; Yokoyama, S. Structural insights into the similar modes of Nrf2 transcription factor recognition by the cytoplasmic repressor Keap1. J. Synchrotron Radiat. 2008, 15, 273–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different Electrostatic Potentials Define ETGE and DLG Motifs as Hinge and Latch in Oxidative Stress Response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, S.-C.; Hannink, M. CAND1-Mediated Substrate Adaptor Recycling Is Required for Efficient Repression of Nrf2 by Keap1. Mol. Cell. Biol. 2006, 26, 1235–1244. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Llères, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yu, S.; Liu, T.; Kim, J.-H.; Blank, V.; Li, H.; Kong, A.-N.T. Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip motif. Biochim. Biophys. Acta 2008, 1783, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Chi, P.-L.; Lin, C.-C.; Chen, Y.-W.; Hsiao, L.-D.; Yang, C.-M. CO Induces Nrf2-Dependent Heme Oxygenase-1 Transcription by Cooperating with Sp1 and c-Jun in Rat Brain Astrocytes. Mol. Neurobiol. 2014, 52, 277–292. [Google Scholar] [CrossRef]
- Tanigawa, S.; Lee, C.; Lin, C.; Ku, C.; Hasegawa, H.; Qin, S.; Kawahara, A.; Korenori, Y.; Miyamori, K.; Noguchi, M.; et al. Jun dimerization protein 2 is a critical component of the Nrf2/MafK complex regulating the response to ROS homeostasis. Cell Death Dis. 2013, 4, e921. [Google Scholar] [CrossRef] [Green Version]
- Levy, S.; Forman, H.J. C-Myc is a Nrf2-interacting protein that negatively regulates phase II genes through their electrophile responsive elements. IUBMB Life 2010, 62, 237–246. [Google Scholar] [CrossRef]
- Li, W.; Yu, S.; Kong, A.-N.T. Nrf2 Possesses a Redox-sensitive Nuclear Exporting Signal in the Neh5 Transactivation Domain. J. Biol. Chem. 2006, 281, 27251–27263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.; Hao, J.; Liu, T.; Zhang, D.; Lv, H.; Song, E.; Zhu, C. Hesperetin Suppresses Inflammatory Responses in Lipopolysaccharide-Induced RAW 264.7 Cells via the Inhibition of NF-κB and Activation of Nrf2/HO-1 Pathways. Inflammation 2016, 39, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, J.; Liu, M.; Zhang, M.; Xue, Y.; Zhang, Y.; Han, X.; Jing, X. Hesperetin modulates the Sirt1/Nrf2 signaling pathway in counteracting myocardial ischemia through suppression of oxidative stress, inflammation, and apoptosis. Biomed. Pharmacother. 2021, 139, 111552. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, P.; Soliman, K.F.A. Flavonoids Activation of the Transcription Factor Nrf2 as a Hypothesis Approach for the Prevention and Modulation of SARS-CoV-2 Infection Severity. Antioxidants 2020, 9, 659. [Google Scholar] [CrossRef]
- Adinew, G.; Taka, E.; Mendonca, P.; Messeha, S.; Soliman, K. The Anticancer Effects of Flavonoids through miRNAs Modulations in Triple-Negative Breast Cancer. Nutrients 2021, 13, 1212. [Google Scholar] [CrossRef]
- Frake, R.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [Green Version]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.; Tsvetkov, A.S.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2005, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Peterhoff, C.M.; Schmidt, S.D.; Terio, N.B.; Duff, K.; Beard, M.; Mathews, P.M.; Nixon, R.A. Presenilin Mutations in Familial Alzheimer Disease and Transgenic Mouse Models Accelerate Neuronal Lysosomal Pathology. J. Neuropathol. Exp. Neurol. 2004, 63, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar]
- Rohn, T.T.; Wirawan, E.; Brown, R.J.; Harris, J.R.; Masliah, E.; Vandenabeele, P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol. Dis. 2011, 43, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Alegre-Abarrategui, J.; Christian, H.; Lufino, M.M.; Mutihac, R.; Venda, L.L.; Ansorge, O.; Wade-Martins, R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009, 18, 4022–4034. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, A.; Heimbach, A.; Gründemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.-L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Pucci, M.; Aria, F.; Premoli, M.; Maccarinelli, G.; Mastinu, A.; Bonini, S.; Memo, M.; Uberti, D.; Abate, G. Methylglyoxal affects cognitive behaviour and modulates RAGE and Presenilin-1 expression in hippocampus of aged mice. Food Chem. Toxicol. 2021, 158, 112608. [Google Scholar] [CrossRef]
- Mastrocola, R.; Nigro, D.; Cento, A.S.; Chiazza, F.; Collino, M.; Aragno, M. High-fructose intake as a risk factor for neurodegeneration: Key role for carboxymethyl lysine accumulation in mice hippocampal neurons. Neurobiol. Dis. 2016, 89, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.-S.; Yu, S.; Kong, A.N. Activation of Nrf2-antioxidant signaling attenuates NFκB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-W.; Hu, J.-J.; Fu, R.-Q.; Liu, X.; Zhang, Y.-H.; Li, J.; Liu, L.; Li, Y.-N.; Deng, Q.; Luo, Q.-S.; et al. Flavonoids inhibit cell proliferation and induce apoptosis and autophagy through downregulation of PI3Kγ mediated PI3K/AKT/mTOR/p70S6K/ULK signaling pathway in human breast cancer cells. Sci. Rep. 2018, 8, 11255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Buya, M.; Qin, W.; Sun, C.; Cai, H.; Xie, Q.; Xu, B.; Wu, Y. Anthocyanins from Chinese Bayberry Extract Activate Transcription Factor Nrf2 in β Cells and Negatively Regulate Oxidative Stress-Induced Autophagy. J. Agric. Food Chem. 2013, 61, 8765–8772. [Google Scholar] [CrossRef]
- García-Barrado, M.J.; Iglesias-Osma, M.C.; Pérez-García, E.; Carrero, S.; Blanco, E.J.; Carretero-Hernández, M.; Carretero, J. Role of Flavonoids in the Interactions among Obesity, Inflammation, and Autophagy. Pharmaceuticals 2020, 13, 342. [Google Scholar] [CrossRef]
- Hasima, N.; Ozpolat, B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis. 2014, 5, e1509. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Du, M.; Zhu, M.-J. Quercetin suppresses NLRP3 inflammasome activation in epithelial cells triggered by Escherichia coli O157:H7. Free Radic. Biol. Med. 2017, 108, 760–769. [Google Scholar] [CrossRef]
- Tsai, C.-Y.; Chen, C.-Y.; Chiou, Y.-H.; Shyu, H.-W.; Lin, K.-H.; Chou, M.-C.; Huang, M.-H.; Wang, Y.-F. Epigallocatechin-3-Gallate Suppresses Human Herpesvirus 8 Replication and Induces ROS Leading to Apoptosis and Autophagy in Primary Effusion Lymphoma Cells. Int. J. Mol. Sci. 2017, 19, 16. [Google Scholar] [CrossRef] [Green Version]
- Rezabakhsh, A.; Rahbarghazi, R.; Malekinejad, H.; Fathi, F.; Montaseri, A.; Garjani, A. Quercetin alleviates high glucose-induced damage on human umbilical vein endothelial cells by promoting autophagy. Phytomedicine 2018, 56, 183–193. [Google Scholar] [CrossRef] [PubMed]
- El-Horany, H.E.; El-Latif, R.N.; ElBatsh, M.M.; Emam, M.N. Ameliorative Effect of Quercetin on Neurochemical and Behavioral Deficits in Rotenone Rat Model of Parkinson’s Disease: Modulating Autophagy (Quercetin on Experimental Parkinson’s Disease). J. Biochem. Mol. Toxicol. 2016, 30, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Regitz, C.; Dußling, L.M.; Wenzel, U. Amyloid-beta (Aβ₁₋₄₂)-induced paralysis in Caenorhabditis elegans is inhibited by the polyphenol quercetin through activation of protein degradation pathways. Mol. Nutr. Food Res. 2014, 58, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Chesser, A.S.; Ganeshan, V.; Yang, J.; Johnson, G.V.W. Epigallocatechin-3-gallate enhances clearance of phosphorylated tau in primary neurons. Nutr. Neurosci. 2015, 19, 21–31. [Google Scholar] [CrossRef]
- Kuang, L.; Cao, X.; Lu, Z. Baicalein Protects against Rotenone-Induced Neurotoxicity through Induction of Autophagy. Biol. Pharm. Bull. 2017, 40, 1537–1543. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [Green Version]
- Maciotta, S.; Meregalli, M.; Torrente, Y. The involvement of microRNAs in neurodegenerative diseases. Front. Cell Neurosci. 2013, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.; Noble, W.; Tartaglia, G.G.; Buckley, N.J. Neurodegeneration as an RNA disorder. Prog. Neurobiol. 2012, 99, 293–315. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA Feedback Circuit in Midbrain Dopamine Neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [Green Version]
- Hebert, S.; De Strooper, B. Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci. 2009, 32, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.; Nahashon, S.; Taka, E.; Adinew, G.M.; Soliman, K.F.A. Epigallocatechin-3-Gallate (EGCG): New Therapeutic Perspectives for Neuroprotection, Aging, and Neuroinflammation for the Modern Age. Biomolecules 2022, 12, 371. [Google Scholar] [CrossRef] [PubMed]
- Benameur, T.; Soleti, R.; Porro, C. The Potential Neuroprotective Role of Free and Encapsulated Quercetin Mediated by miRNA against Neurological Diseases. Nutrients 2021, 13, 1318. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimpour, S.; Esmaeili, A.; Dehghanian, F.; Beheshti, S. Effects of quercetin-conjugated with superparamagnetic iron oxide nanoparticles on learning and memory improvement through targeting microRNAs/NF-κB pathway. Sci. Rep. 2020, 10, 15070. [Google Scholar] [CrossRef]
- Bilkei-Gorzo, A. Genetic mouse models of brain ageing and Alzheimer’s disease. Pharmacol. Ther. 2014, 142, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Yang, S.; Cai, L.; Qin, L.-Q.; Li, B.-Y.; Wan, Z. Effects of Quercetin Intervention on Cognition Function in APP/PS1 Mice was Affected by Vitamin D Status. Mol. Nutr. Food Res. 2018, 62, e1800621. [Google Scholar] [CrossRef]
- Santa-Maria, I.; Alaniz, M.E.; Renwick, N.; Cela, C.; Fulga, T.; Van Vactor, J.D.L.; Tuschl, T.; Clark, L.N.; Shelanski, M.L.; McCabe, B.; et al. Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J. Clin. Investig. 2015, 125, 681–686. [Google Scholar] [CrossRef]
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6055. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yi, P.; Yi, M.; Tong, X.; Cheng, X.; Yang, J.; Hu, Y. Protective Effect of Quercetin against H2O2-Induced Oxidative Damage in PC-12 Cells: Comprehensive Analysis of a lncRNA-Associated ceRNA Network. Oxid. Med. Cell. Longev. 2020, 2020, 6038919. [Google Scholar] [CrossRef]
- Cione, E.; La Torre, C.; Cannataro, R.; Caroleo, M.C.; Plastina, P.; Gallelli, L. Quercetin, Epigallocatechin Gallate, Curcumin, and Resveratrol: From Dietary Sources to Human MicroRNA Modulation. Molecules 2019, 25, 63. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Li, Y.; Su, B. Identification of Circulating miR-125b as a Potential Biomarker of Alzheimer’s Disease in APP/PS1 Transgenic Mouse. J. Alzheimer’s Dis. 2017, 59, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zhou, H.; Zhao, H.; Zhao, Y.; Chang, C. Hesperetin ameliorates lipopolysaccharide-induced acute lung injury via the miR-410/SOX18 axis. J. Biochem. Mol. Toxicol. 2020, 34, e22588. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evans, J.A.; Mendonca, P.; Soliman, K.F.A. Neuroprotective Effects and Therapeutic Potential of the Citrus Flavonoid Hesperetin in Neurodegenerative Diseases. Nutrients 2022, 14, 2228. https://doi.org/10.3390/nu14112228
Evans JA, Mendonca P, Soliman KFA. Neuroprotective Effects and Therapeutic Potential of the Citrus Flavonoid Hesperetin in Neurodegenerative Diseases. Nutrients. 2022; 14(11):2228. https://doi.org/10.3390/nu14112228
Chicago/Turabian StyleEvans, Jasmine A., Patricia Mendonca, and Karam F. A. Soliman. 2022. "Neuroprotective Effects and Therapeutic Potential of the Citrus Flavonoid Hesperetin in Neurodegenerative Diseases" Nutrients 14, no. 11: 2228. https://doi.org/10.3390/nu14112228
APA StyleEvans, J. A., Mendonca, P., & Soliman, K. F. A. (2022). Neuroprotective Effects and Therapeutic Potential of the Citrus Flavonoid Hesperetin in Neurodegenerative Diseases. Nutrients, 14(11), 2228. https://doi.org/10.3390/nu14112228