

Feeding the Brain: Effect of Nutrients on Cognition, Synaptic Function, and AMPA Receptors

Abstract
:1. Introduction
2. Western Diet
2.1. Cognition Studies in Animal Models
2.2. Synaptic Function and Neuroplasticity
2.3. Other Mechanisms
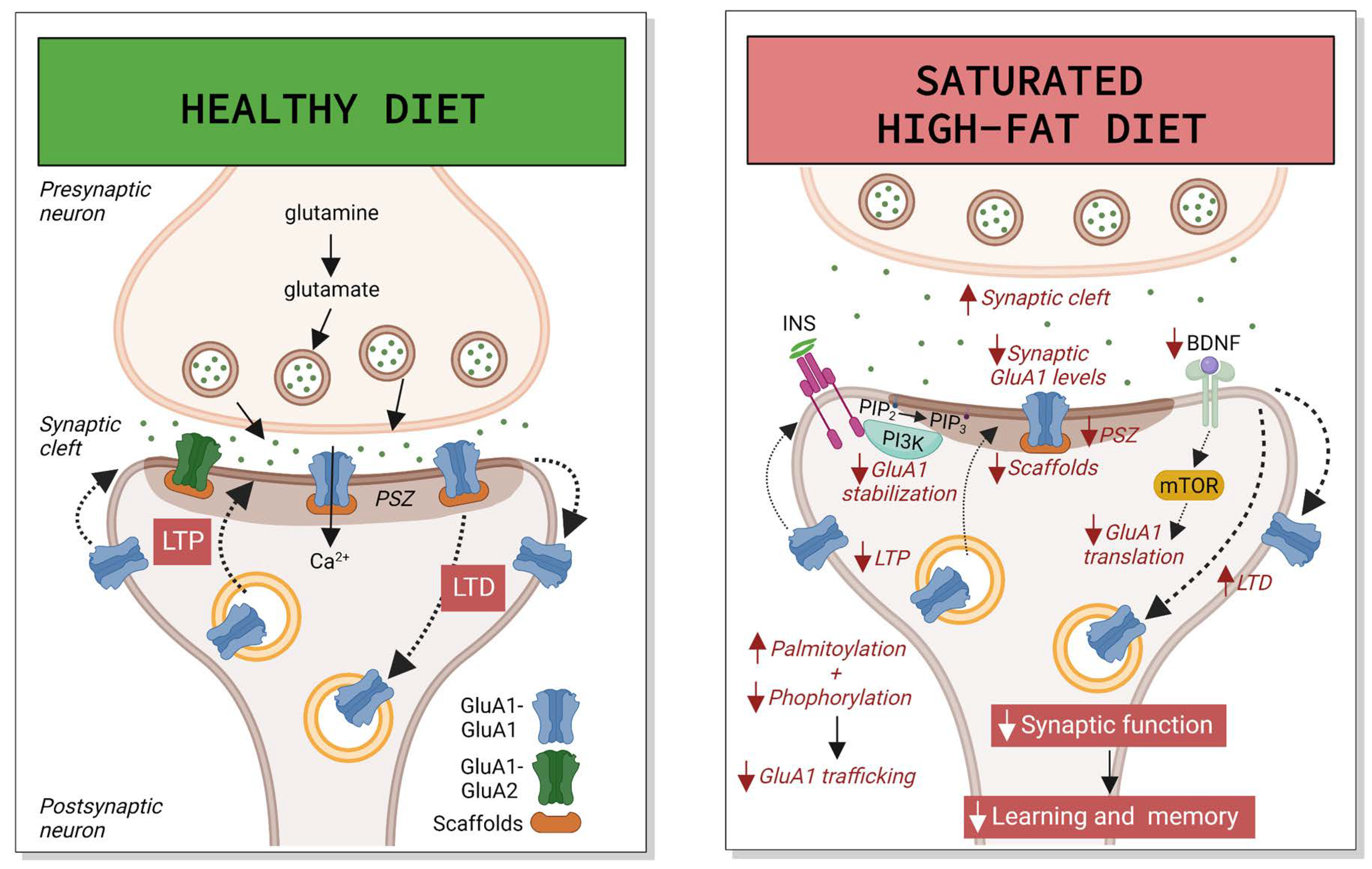
3. High-Fat Diet
3.1. Cognition Studies with Mixed and Saturated Fats
3.2. Cognition Studies with Polyunsaturated Fats
3.3. Synaptic Function and Neuroplasticity with Mixed and Saturated Fats
3.4. Synaptic Function and Neuroplasticity with Polyunsaturated Fats
3.5. Other Mecanisms of Hippocampal Damage with Mixed and Saturated Fats
4. High-Sugar Diet
4.1. Cognition Studies in Animal Models
4.2. Synaptic Function and Neuroplasticity
4.3. Insulin Resistance
4.4. Other Mechanisms
5. Ketogenic Diet
5.1. Cognition Studies in Humans
5.2. Cognition Studies in Animal Models
5.3. Synaptic Function and Neuroplasticity
5.4. Insulin Resistance
6. Paleo(lithic) Diet
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, P.J.; Blumenthal, J.A. Dietary Factors and Cognitive Decline. J. Prev. Alzheimer’s Dis. 2016, 3, 53–64. [Google Scholar] [CrossRef]
- Dye, L.; Boyle, N.B.; Champ, C.; Lawton, C. The Relationship between Obesity and Cognitive Health and Decline. Proc. Nutr. Soc. 2017, 76, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, J.C.D.; Killcross, A.S.; Jenkins, T.A. Obesity and Cognitive Decline: Role of Inflammation and Vascular Changes. Front. Neurosci. 2014, 8, 375. [Google Scholar] [CrossRef] [Green Version]
- Bramorska, A.; Zarzycka, W.; Podolecka, W.; Kuc, K.; Brzezicka, A. Age-Related Cognitive Decline May Be Moderated by Frequency of Specific Food Products Consumption. Nutrients 2021, 13, 2504. [Google Scholar] [CrossRef] [PubMed]
- WHO. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 12 September 2022).
- Loef, M.; Walach, H. Midlife Obesity and Dementia: Meta-Analysis and Adjusted Forecast of Dementia Prevalence in the United States and China. Obesity 2013, 21, E51–E56. [Google Scholar] [CrossRef]
- Singh-Manoux, A.; Dugravot, A.; Shipley, M.; Brunner, E.J.; Elbaz, A.; Sabia, S.; Kivimaki, M. Obesity Trajectories and Risk of Dementia: 28 Years of Follow-up in the Whitehall II Study. Alzheimer’s Dement. 2018, 14, 178–186. [Google Scholar] [CrossRef]
- Cournot, M.; Marquié, J.C.; Ansiau, D.; Martinaud, C.; Fonds, H.; Ferrières, J.; Ruidavets, J.B. Relation between Body Mass Index and Cognitive Function in Healthy Middle-Aged Men and Women. Neurology 2006, 67, 1208–1214. [Google Scholar] [CrossRef]
- Coppin, G.; Nolan-Poupart, S.; Jones-Gotman, M.; Small, D.M. Working Memory and Reward Association Learning Impairments in Obesity. Neuropsychologia 2014, 65, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Gunstad, J.; Paul, R.H.; Cohen, R.A.; Tate, D.F.; Spitznagel, M.B.; Gordon, E. Elevated Body Mass Index Is Associated with Executive Dysfunction in Otherwise Healthy Adults. Compr. Psychiatry 2007, 48, 57–61. [Google Scholar] [CrossRef]
- World Health Organization Diabetes. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 12 September 2022).
- Moheet, A.; Mangia, S.; Seaquist, E.R. Impact of Diabetes on Cognitive Function and Brain Structure. Ann. NY Acad. Sci. 2015, 1353, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Palta, P.; Schneider, A.L.; Biessels, G.J.; Touradji, P.; Hill-Briggs, F. Magnitude of Cognitive Dysfunction in Adults with Type 2 Diabetes: A Meta-Analysis of Six Cognitive Domains and the Most Frequently Reported Neuropsychological Tests within Domains. J. Int. Neuropsychol. Soc. 2014, 20, 278–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaven, G.M.; Thompson, L.W.; Nahum, D.; Haskins, E. Relationship between Hyperglycemia and Cognitive Function in Older NIDDM Patients. Diabetes Care 1990, 13, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamport, D.J.; Lawton, C.L.; Mansfield, M.W.; Dye, L. Impairments in Glucose Tolerance Can Have a Negative Impact on Cognitive Function: A Systematic Research Review. Neurosci. Biobehav. Rev. 2009, 33, 394–413. [Google Scholar] [CrossRef] [PubMed]
- Komulainen, P.; Lakka, T.A.; Kivipelto, M.; Hassinen, M.; Helkala, E.L.; Haapala, I.; Nissinen, A.; Rauramaa, R. Metabolic Syndrome and Cognitive Function: A Population-Based Follow-Up Study in Elderly Women. Dement. Geriatr. Cogn. Disord. 2007, 23, 29–34. [Google Scholar] [CrossRef]
- Van den Berg, E.; Biessels, G.J.; de Craen, A.J.; Gussekloo, J.; Westendorp, R.G. The Metabolic Syndrome Is Associated with Decelerated Cognitive Decline in the Oldest Old. Neurology 2007, 69, 979–985. [Google Scholar] [CrossRef]
- Yates, K.F.; Sweat, V.; Yau, P.L.; Turchiano, M.M.; Convit, A. Impact of Metabolic Syndrome on Cognition and Brain: A Selected Review of the Literature. Arter. Thromb. Vasc. Biol. 2012, 32, 2060–2067. [Google Scholar] [CrossRef] [Green Version]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [Green Version]
- Hanley, J.G. Subunit-Specific Trafficking Mechanisms Regulating the Synaptic Expression of Ca2+-Permeable AMPA Receptors. Semin. Cell Dev. Biol. 2014, 27, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Jurado, S. AMPA Receptor Trafficking in Natural and Pathological Aging. Front. Mol. Neurosci. 2018, 10, 446. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Ma, Y.Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keifer, J. Regulation of AMPAR Trafficking in Synaptic Plasticity by BDNF and the Impact of Neurodegenerative Disease. J. Neurosci. Res. 2022, 100, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, D.J.; Hogg, E.L.; Regan, P.; Piers, T.; Narayan, P.; Whitehead, G.; Winters, B.L.; Kim, D.H.; Kim, E.; St George-Hyslop, P.; et al. Intracellular Oligomeric Amyloid-Beta Rapidly Regulates GluA1 Subunit of AMPA Receptor in the Hippocampus. Sci. Rep. 2015, 5, 10934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miñano-Molina, A.J.; España, J.; Martín, E.; Barneda-Zahonero, B.; Fadó, R.; Solé, M.; Trullás, R.; Saura, C.A.; Rodríguez-Alvarez, J. Soluble Oligomers of Amyloid-β Peptide Disrupt Membrane Trafficking of α-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionic Acid Receptor Contributing to Early Synapse Dysfunction. J. Biol. Chem. 2011, 286, 27311–27321. [Google Scholar] [CrossRef] [Green Version]
- Guntupalli, S.; Widagdo, J.; Anggono, V. Amyloid-β-Induced Dysregulation of AMPA Receptor Trafficking. Neural Plast. 2016, 2016, 3204519. [Google Scholar] [CrossRef] [Green Version]
- Ammassari-Teule, M. Early-Occurring Dendritic Spines Alterations in Mouse Models of Alzheimer’s Disease Inform on Primary Causes of Neurodegeneration. Front. Synaptic Neurosci. 2020, 12, 566615. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Davidson, T.L. Western Diet Consumption and Cognitive Impairment: Links to Hippocampal Dysfunction and Obesity. Physiol. Behav. 2011, 103, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Moult, P.R.; Cross, A.; Santos, S.D.; Carvalho, A.-L.; Lindsay, Y.; Connolly, C.N.; Irving, A.J.; Leslie, N.R.; Harvey, J. Leptin Regulates AMPA Receptor Trafficking via PTEN Inhibition. J. Neurosci. 2010, 30, 4088–4101. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, L.F.; Catarino, T.; Santos, S.D.; Benoist, M.; van Leeuwen, J.F.; Esteban, J.A.; Carvalho, A.L. Ghrelin Triggers the Synaptic Incorporation of AMPA Receptors in the Hippocampus. Proc. Natl. Acad. Sci. USA 2014, 111, E149–E158. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, M.; Fusco, S.; Mainardi, M.; Scala, F.; Natale, F.; Lapenta, R.; Mattera, A.; Rinaudo, M.; Li Puma, D.D.; Ripoli, C.; et al. Brain Insulin Resistance Impairs Hippocampal Synaptic Plasticity and Memory by Increasing GluA1 Palmitoylation through FoxO3a. Nat. Commun. 2017, 8, 2009. [Google Scholar] [CrossRef]
- Ouyang, J.; Carcea, I.; Schiavo, J.K.; Jones, K.T.; Rabinowitsch, A.; Kolaric, R.; Cabeza de Vaca, S.; Froemke, R.C.; Carr, K.D. Food Restriction Induces Synaptic Incorporation of Calcium-Permeable AMPA Receptors in Nucleus Accumbens. Eur. J. Neurosci. 2017, 45, 826–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas, M.; Fadó, R.; Domínguez, J.L.; Roig, A.; Kaku, M.; Chohnan, S.; Solé, M.; Unzeta, M.; Miñano-Molina, A.J.; Rodríguez-Álvarez, J.; et al. Sensing of Nutrients by CPT1C Controls SAC1 Activity to Regulate AMPA Receptor Trafficking. J. Cell Biol. 2020, 219, e201912045. [Google Scholar] [CrossRef] [PubMed]
- Vilarnau, C.; Stracker, D.M.; Funtikov, A.; da Silva, R.; Estruch, R.; Bach-Faig, A. Worldwide Adherence to Mediterranean Diet between 1960 and 2011. Eur. J. Clin. Nutr. 2019, 72, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-García, M.I.; Martínez-González, M.A.; Razquin, C.; Fernández-Matarrubia, M.; Guillén-Grima, F.; Toledo, E. Exploratory Dietary Patterns and Cognitive Function in the “Seguimiento Universidad de Navarra” (SUN) Prospective Cohort. Eur. J. Clin. Nutr. 2022, 76, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Attuquayefio, T.; Stevenson, R.J.; Oaten, M.J.; Francis, H.M. A Four-Day Western-Style Dietary Intervention Causes Reductions in Hippocampal-Dependent Learning and Memory and Interoceptive Sensitivity. PLoS ONE 2017, 12, e0172645. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, R.J.; Francis, H.M.; Attuquayefio, T.; Gupta, D.; Yeomans, M.R.; Oaten, M.J.; Davidson, T. Hippocampal-Dependent Appetitive Control Is Impaired by Experimental Exposure to a Western-Style Diet. R. Soc. Open Sci. 2020, 7, 191338. [Google Scholar] [CrossRef] [Green Version]
- Ashby-Mitchell, K.; Peeters, A.; Anstey, K.J. Role of Dietary Pattern Analysis in Determining Cognitive Status in Elderly Australian Adults. Nutrients 2015, 7, 1052–1067. [Google Scholar] [CrossRef] [Green Version]
- Taylor, Z.B.; Stevenson, R.J.; Ehrenfeld, L.; Francis, H.M. The Impact of Saturated Fat, Added Sugar and Their Combination on Human Hippocampal Integrity and Function: A Systematic Review and Meta-Analysis. Neurosci. Biobehav. Rev. 2021, 130, 91–106. [Google Scholar] [CrossRef]
- Hargrave, S.L.; Davidson, T.L. Western Diets Induce Blood-Brain Barrier Leakage and Alter Spatial Strategies in Rats. Behav. Neurosci. 2016, 130, 123–135. [Google Scholar] [CrossRef]
- Nakajima, S.; Fukasawa, K.; Gotoh, M.; Murakami-Murofushi, K.; Kunugi, H. Saturated Fatty Acid Is a Principal Cause of Anxiety-like Behavior in Diet-Induced Obese Rats in Relation to Serum Lysophosphatidyl Choline Level. Int. J. Obes. 2019, 44, 727–738. [Google Scholar] [CrossRef]
- Dharavath, R.N.; Arora, S.; Bishnoi, M.; Kondepudi, K.K.; Chopra, K. High Fat-Low Protein Diet Induces Metabolic Alterations and Cognitive Dysfunction in Female Rats. Metab. Brain Dis. 2019, 34, 1531–1546. [Google Scholar] [CrossRef] [PubMed]
- Tsan, L.; Sun, S.; Hayes, A.M.R.; Bridi, L.; Chirala, L.S.; Noble, E.E.; Fodor, A.A.; Kanoski, S.E. Early Life Western Diet-Induced Memory Impairments and Gut Microbiome Changes in Female Rats Are Long-Lasting despite Healthy Dietary Intervention. Nutr. Neurosci. 2021, 1–17. [Google Scholar] [CrossRef]
- Sarfert, K.S.; Knabe, M.L.; Gunawansa, N.S.; Blythe, S.N. Western-Style Diet Induces Object Recognition Deficits and Alters Complexity of Dendritic Arborization in the Hippocampus and Entorhinal Cortex of Male Rats. Nutr. Neurosci. 2019, 22, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Leigh, S.J.; Kaakoush, N.O.; Westbrook, R.F.; Morris, M.J. Minocycline-Induced Microbiome Alterations Predict Cafeteria Diet-Induced Spatial Recognition Memory Impairments in Rats. Transl. Psychiatry 2020, 10, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molteni, R.; Wu, A.; Vaynman, S.; Ying, Z.; Barnard, R.J.; Gómez-Pinilla, F. Exercise Reverses the Harmful Effects of Consumption of a High-Fat Diet on Synaptic and Behavioral Plasticity Associated to the Action of Brain-Derived Neurotrophic Factor. Neuroscience 2004, 123, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, M.N.; Heo, J.W.; Schifino, A.G.; Hoffman, J.R.; Donohue, K.N.; Call, J.A.; Noble, E.E. Sexually Dimorphic Effects of a Western Diet on Brain Mitochondrial Bioenergetics and Neurocognitive Function. Nutrients 2021, 13, 4222. [Google Scholar] [CrossRef] [PubMed]
- Beilharz, J.E.; Maniam, J.; Morris, M.J. Short Exposure to a Diet Rich in Both Fat and Sugar or Sugar Alone Impairs Place, but Not Object Recognition Memory in Rats. Brain. Behav. Immun. 2014, 37, 134–141. [Google Scholar] [CrossRef]
- Beilharz, J.E.; Kaakoush, N.O.; Maniam, J.; Morris, M.J. Cafeteria Diet and Probiotic Therapy: Cross Talk among Memory, Neuroplasticity, Serotonin Receptors and Gut Microbiota in the Rat. Mol. Psychiatry 2018, 23, 351–361. [Google Scholar] [CrossRef]
- Alzoubi, K.H.; Khabour, O.F.; Salah, H.A.; Hasan, Z. Vitamin E Prevents High-Fat High-Carbohydrates Diet-Induced Memory Impairment: The Role of Oxidative Stress. Physiol. Behav. 2013, 119, 72–78. [Google Scholar] [CrossRef]
- Tran, D.M.D.; Westbrook, R.F. A High-Fat High-Sugar Diet-Induced Impairment in Place-Recognition Memory Is Reversible and Training-Dependent. Appetite 2017, 110, 61–71. [Google Scholar] [CrossRef]
- Francis, H.M.; Mirzaei, M.; Pardey, M.C.; Haynes, P.A.; Cornish, J.L. Proteomic Analysis of the Dorsal and Ventral Hippocampus of Rats Maintained on a High Fat and Refined Sugar Diet. Proteomics 2013, 13, 3076–3091. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.K.; Sheng, L.; Nguyen, M.; Di Lucente, J.; Hu, Y.; Li, Y.; Maezawa, I.; Jin, L.W.; Wan, Y.J.Y. Dysregulated Bile Acid Receptor-Mediated Signaling and IL-17A Induction Are Implicated in Diet-Associated Hepatic Health and Cognitive Function. Biomark. Res. 2020, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Paulo, S.L.; Miranda-Lourenço, C.; Belo, R.F.; Rodrigues, R.S.; Fonseca-Gomes, J.; Tanqueiro, S.R.; Geraldes, V.; Rocha, I.; Sebastião, A.M.; Xapelli, S.; et al. High Caloric Diet Induces Memory Impairment and Disrupts Synaptic Plasticity in Aged Rats. Curr. Issues Mol. Biol. 2021, 43, 2305–2319. [Google Scholar] [CrossRef] [PubMed]
- Syeda, T.; Sánchez-Tapia, M.; Orta, I.; Granados-Portillo, O.; Pérez-Jimenez, L.; Rodríguez-Callejas, J.D.D.; Toribio, S.; Silva-Lucero, M.D.C.; Rivera, A.L.; Tovar, A.R.; et al. Bioactive Foods Decrease Liver and Brain Alterations Induced by a High-Fat-Sucrose Diet through Restoration of Gut Microbiota and Antioxidant Enzymes. Nutrients 2021, 14, 22. [Google Scholar] [CrossRef]
- Mamo, J.C.L.; Lam, V.; Brook, E.; Mooranian, A.; Al-Salami, H.; Fimognari, N.; Nesbit, M.; Takechi, R. Probucol Prevents Blood-Brain Barrier Dysfunction and Cognitive Decline in Mice Maintained on pro-Diabetic Diet. Diabetes Vasc. Dis. Res. 2019, 16, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Abd-Allah, H.; Nasr, M.; Ahmed-Farid, O.A.H.; El-Marasy, S.A.; Bakeer, R.M.; Ahmed, R.F. Biological and Pharmacological Characterization of Ascorbic Acid and Nicotinamide Chitosan Nanoparticles against Insulin-Resistance-Induced Cognitive Defects: A Comparative Study. ACS Omega 2021, 6, 3587–3601. [Google Scholar] [CrossRef]
- Lama, A.; Pirozzi, C.; Annunziata, C.; Morgese, M.G.; Senzacqua, M.; Severi, I.; Calignano, A.; Trabace, L.; Giordano, A.; Meli, R.; et al. Palmitoylethanolamide Counteracts Brain Fog Improving Depressive-like Behaviour in Obese Mice: Possible Role of Synaptic Plasticity and Neurogenesis. Br. J. Pharmacol. 2021, 178, 845–859. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High Fat Diet Induces Brain Insulin Resistance and Cognitive Impairment in Mice. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef]
- Virtuoso, A.; Tveden-nyborg, P.; Schou-pedersen, A.M.V.; Lykkesfeldt, J.; Müller, H.K.; Elfving, B.; Sørensen, D.B. A Long-Term Energy-Rich Diet Increases Prefrontal BDNF in Sprague-Dawley Rats. Nutrients 2021, 14, 126. [Google Scholar] [CrossRef]
- Kanoski, S.; Zhang, Y.; Zheng, W.; Davidson, T. The Effects of a High-Energy Diet on Hippocampal-Dependent Negative Occasion Setting and Blood-Brain Barrier Integrity in the Rat. J. Alzheimer’s Dis. 2010, 21, 207–219. [Google Scholar] [CrossRef]
- Molteni, R.; Barnard, R.J.; Ying, Z.; Roberts, C.K.; Gómez-Pinilla, F. A High-Fat, Refined Sugar Diet Reduces Hippocampal Brain-Derived Neurotrophic Factor, Neuronal Plasticity, and Learning. Neuroscience 2002, 112, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Stranahan, A.M.; Norman, E.D.; Lee, K.; Cutler, R.G.; Telljohann, R.S.; Egan, J.M.; Mattson, M.P. Diet-Induced Insulin Resistance Impairs Hippocampal Synaptic Plasticity and Cognition in Middle-Aged Rats. Hippocampus 2008, 18, 1085–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Serrano, A.M.; Mohr, A.A.; Philippe, J.; Skoug, C.; Spégel, P.; Duarte, J.M.N. Cognitive Impairment and Metabolite Profile Alterations in the Hippocampus and Cortex of Male and Female Mice Exposed to a Fat and Sugar-Rich Diet Are Normalized by Diet Reversal. Aging Dis. 2022, 13, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.N.; Ipsen, D.H.; Schou-Pedersen, A.M.; Lykkesfeldt, J.; Tveden-Nyborg, P. Long Term Westernized Diet Leads to Region-Specific Changes in Brain Signaling Mechanisms. Neurosci. Lett. 2018, 676, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Marwitz, S.E.; Woodie, L.N.; Blythe, S.N. Western-Style Diet Induces Insulin Insensitivity and Hyperactivity in Adolescent Male Rats. Physiol. Behav. 2015, 151, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Z.; Li, B.; Qiang, Y.; Yuan, T.; Tan, X.; Wang, Z.; Liu, Z.; Liu, X. Lycopene Attenuates Western-Diet-Induced Cognitive Deficits via Improving Glycolipid Metabolism Dysfunction and Inflammatory Responses in Gut-Liver-Brain Axis. Int. J. Obes. 2019, 43, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Orozco, H.; Reyes-Castro, L.A.; Lomas-Soria, C.; Sandoval-Salazar, C.; Ramírez-Emiliano, J.; Díaz-Cintra, S.; Solís-Ortiz, S. High-Fat and Combined High-Fat–High-Fructose Diets Impair Episodic-like Memory and Decrease Glutamate and Glutamine in the Hippocampus of Adult Mice. Nutr. Neurosci. 2021, 1–11. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Meisel, R.L.; Mullins, A.J.; Davidson, T.L. Of the Rat. Behav. Brain Res. 2007, 182, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Chávez-Gutiérrez, E.; Fuentes-Venado, C.E.; Rodríguez-Páez, L.; Guerra-Araiza, C.; Larqué, C.; Martínez-Herrera, E.; Ocharan-Hernández, M.E.; Lomelí, J.; Loza-Mejía, M.A.; Salazar, J.R.; et al. High Fructose and High Fat Diet Impair Different Types of Memory through Oxidative Stress in a Sex- and Hormone-Dependent Manner. Metabolites 2022, 12, 341. [Google Scholar] [CrossRef]
- Takechi, R.; Lam, V.; Brook, E.; Giles, C.; Fimognari, N.; Mooranian, A.; Al-Salami, H.; Coulson, S.H.; Nesbit, M.; Mamo, J.C.L. Blood-Brain Barrier Dysfunction Precedes Cognitive Decline and Neurodegeneration in Diabetic Insulin Resistant Mouse Model: An Implication for Causal Link. Front. Aging Neurosci. 2017, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, B.; Kundu, P.; Liu, Z.; Urbanski, H.F.; Kroenke, C.D.; Kohama, S.G.; Bethea, C.L.; Raber, J. Longitudinal Effects of Immediate and Delayed Estradiol on Cognitive Performance in a Spatial Maze and Hippocampal Volume in Menopausal Macaques Under an Obesogenic Diet. Front. Neurol. 2020, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Thériault, P.; ElAli, A.; Rivest, S. High Fat Diet Exacerbates Alzheimer’s Disease-Related Pathology in APPswe/PS1 Mice. Oncotarget 2016, 7, 67808–67827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, N.; Liu, Q.; Agca, C.; Agca, Y.; Noble, E.G.; Whitehead, S.N.; Cechetto, D.F. White Matter Inflammation and Cognitive Function in a Co-Morbid Metabolic Syndrome and Prodromal Alzheimer’s Disease Rat Model. J. Neuroinflamm. 2020, 17, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, T.A. Metabolic Syndrome and Vascular-Associated Cognitive Impairment: A Focus on Preclinical Investigations. Curr. Diab. Rep. 2022, 22, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Kouvari, M.; D’cunha, N.M.; Travica, N.; Sergi, D.; Zec, M.; Marx, W.; Naumovski, N. Metabolic Syndrome, Cognitive Impairment and the Role of Diet: A Narrative Review. Nutrients 2022, 14, 333. [Google Scholar] [CrossRef]
- Sinha, K.; Sun, C.; Kamari, R.; Bettermann, K. Current Status and Future Prospects of Pathophysiology-Based Neuroprotective Drugs for the Treatment of Vascular Dementia. Drug Discov. Today 2020, 25, 793–799. [Google Scholar] [CrossRef]
- Appleton, J.P.; Scutt, P.; Sprigg, N.; Bath, P.M. Hypercholesterolaemia and Vascular Dementia. Clin. Sci. 2017, 131, 1561–1578. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Lapiscina, E.H.; Clavero, P.; Toledo, E.; Estruch, R.; Salas-Salvadó, J.; San Julián, B.; Sanchez-Tainta, A.; Ros, E.; Valls-Pedret, C.; Martinez-Gonzalez, M.Á. Mediterranean Diet Improves Cognition: The Predimed-Navarra Randomised Trial. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1318–1325. [Google Scholar] [CrossRef] [Green Version]
- Karimi, G.; Heidari, Z.; Firouzi, S.; Haghighatdoost, F. A Systematic Review and Meta-Analysis of the Association between Fish Consumption and Risk of Metabolic Syndrome. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 717–729. [Google Scholar] [CrossRef]
- Shin, J.Y.; Kim, J.Y.; Kang, H.T.; Han, K.H.; Shim, J.Y. Effect of Fruits and Vegetables on Metabolic Syndrome: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Int. J. Food Sci. Nutr. 2015, 66, 416–425. [Google Scholar] [CrossRef]
- Borroni, B.; Stanic, J.; Verpelli, C.; Mellone, M.; Bonomi, E.; Alberici, A.; Bernasconi, P.; Culotta, L.; Zianni, E.; Archetti, S.; et al. Anti-AMPA GluA3 Antibodies in Frontotemporal Dementia: A New Molecular Target. Sci. Rep. 2017, 7, 6723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortin, D.A.; Srivastava, T.; Dwarakanath, D.; Pierre, P.; Nygaard, S.; Derkach, V.A.; Soderling, T.R. Brain-Derived Neurotrophic Factor Activation of CaM-Kinase Kinase via Transient Receptor Potential Canonical Channels Induces the Translation and Synaptic Incorporation of GluA1-Containing Calcium-Permeable AMPA Receptors. J. Neurosci. 2012, 32, 8127–8137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, J.A.; Shi, Y.; Usui, H.; During, M.J.; Sakimura, K.; Nicoll, R.A. Distinct Modes of AMPA Receptor Suppression at Developing Synapses by GluN2A and GluN2B: Single-Cell NMDA Receptor Subunit Deletion in Vivo. Neuron 2011, 71, 1085–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baym, C.L.; Khan, N.A.; Monti, J.M.; Raine, L.B.; Drollette, E.S.; Moore, R.D.; Scudder, M.R.; Kramer, A.F.; Hillman, C.H.; Cohen, N.J. Dietary Lipids Are Differentially Associated with Hippocampal-Dependent Relational Memory in Prepubescent Children. Am. J. Clin. Nutr. 2014, 99, 1026–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.A.; Davidson, T.L.; McCrory, M.A. Deficits in Episodic Memory Are Related to Uncontrolled Eating in a Sample of Healthy Adults. Appetite 2018, 124, 33. [Google Scholar] [CrossRef]
- Okereke, O.I.; Rosner, B.A.; Kim, D.H.; Kang, J.H.; Cook, N.R.; Manson, J.E.; Buring, J.E.; Willett, W.C.; Grodstein, F. Dietary Fat Types and 4-Year Cognitive Change in Community-Dwelling Older Women. Ann. Neurol. 2012, 72, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Golomb, B.A.; Bui, A.K. A Fat to Forget: Trans Fat Consumption and Memory. PLoS ONE 2015, 10, e0128129. [Google Scholar] [CrossRef]
- Leigh Gibson, E.; Barr, S.; Jeanes, Y.M. Habitual Fat Intake Predicts Memory Function in Younger Women. Front. Hum. Neurosci. 2013, 7, 838. [Google Scholar] [CrossRef] [Green Version]
- Noble, E.E.; Kanoski, S.E. Early Life Exposure to Obesogenic Diets and Learning and Memory Dysfunction. Curr. Opin. Behav. Sci. 2016, 9, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Spencer, S.J.; D’Angelo, H.; Soch, A.; Watkins, L.R.; Maier, S.F.; Barrientos, R.M. High-Fat Diet and Aging Interact to Produce Neuroinflammation and Impair Hippocampal- and Amygdalar-Dependent Memory. Neurobiol. Aging 2017, 58, 88–101. [Google Scholar] [CrossRef]
- Yang, X.; Zheng, M.; Hao, S.; Shi, H.; Lin, D.; Chen, X.; Becvarovski, A.; Pan, W.; Zhang, P.; Hu, M.; et al. Curdlan Prevents the Cognitive Deficits Induced by a High-Fat Diet in Mice via the Gut-Brain Axis. Front. Neurosci. 2020, 14, 384. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Cho, J.; Lee, I.; Jin, Y.; Kang, H. Exercise Attenuates High-Fat Diet-Induced Disease Progression in 3xTg-AD Mice. Med. Sci. Sports Exerc. 2017, 49, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Freire, D.; Knable, L.; Zhao, W.; Gong, B.; Mazzola, P.; Ho, L.; Levine, S.; Pasinetti, G.M. Childhood and Adolescent Obesity and Long-Term Cognitive Consequences during Aging. J. Comp. Neurol. 2015, 523, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.A.; Paul, J.R.; Yates, S.D.; Cutts, E.J.; McMahon, L.L.; Pollock, J.S.; Pollock, D.M.; Bailey, S.M.; Gamble, K.L. Time-Restricted Feeding Rescues High-Fat-Diet-Induced Hippocampal Impairment. iScience 2021, 24, 102532. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhu, Y.; Zhou, L.; Lu, Y.; Feng, T.; Dai, M.; Liu, J.; Xu, W.; Cheng, W.; Sun, F.; et al. Parasite-Derived Excretory-Secretory Products Alleviate Gut Microbiota Dysbiosis and Improve Cognitive Impairment Induced by a High-Fat Diet. Front. Immunol. 2021, 12, 710513. [Google Scholar] [CrossRef] [PubMed]
- Hahm, J.R.; Jo, M.H.; Ullah, R.; Kim, M.W.; Kim, M.O. Metabolic Stress Alters Antioxidant Systems, Suppresses the Adiponectin Receptor 1 and Induces Alzheimer’s Like Pathology in Mice Brain. Cells 2020, 9, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Wang, Q.; Zheng, M.; Hao, S.; Lum, J.S.; Chen, X.; Huang, X.F.; Yu, Y.; Zheng, K. Supplement of Microbiota-Accessible Carbohydrates Prevents Neuroinflammation and Cognitive Decline by Improving the Gut Microbiota-Brain Axis in Diet-Induced Obese Mice. J. Neuroinflamm. 2020, 17, 77. [Google Scholar] [CrossRef]
- Beilharz, J.E.; Kaakoush, N.O.; Maniam, J.; Morris, M.J. The Effect of Short-Term Exposure to Energy-Matched Diets Enriched in Fat or Sugar on Memory, Gut Microbiota and Markers of Brain Inflammation and Plasticity. Brain. Behav. Immun. 2016, 57, 304–313. [Google Scholar] [CrossRef] [Green Version]
- Granholm, A.C.; Bimonte-Nelson, H.A.; Moore, A.B.; Nelson, M.E.; Freeman, L.R.; Sambamurti, K. Effects of a Saturated Fat and High Cholesterol Diet on Memory and Hippocampal Morphology in the Middle-Aged Rat. J. Alzheimer’s Dis. 2008, 14, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Felipe, J.; Merino, B.; Sanz-Martos, A.B.; Plaza, A.; Contreras, A.; Naranjo, V.; Morales, L.; Chowen, J.A.; Cano, V.; Ruiz-Gayo, M.; et al. Saturated and Unsaturated Fat Diets Impair Hippocampal Glutamatergic Transmission in Adolescent Mice. Psychoneuroendocrinology 2021, 133, 105429. [Google Scholar] [CrossRef]
- Khazen, T.; Hatoum, O.A.; Ferreira, G.; Maroun, M. Acute Exposure to a High-Fat Diet in Juvenile Male Rats Disrupts Hippocampal-Dependent Memory and Plasticity through Glucocorticoids. Sci. Rep. 2019, 9, 12270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, A.; Del Rio, D.; Martínez, A.; Gil, C.; Morales, L.; Ruiz-Gayo, M.; Del Olmo, N. Inhibition of Hippocampal Long-Term Potentiation by High-Fat Diets: Is It Related to an Effect of Palmitic Acid Involving Glycogen Synthase Kinase-3? Neuroreport 2017, 28, 354–359. [Google Scholar] [CrossRef] [PubMed]
- De Paula, G.C.; Brunetta, H.S.; Engel, D.F.; Gaspar, J.M.; Velloso, L.A.; Engblom, D.; de Oliveira, J.; de Bem, A.F. Hippocampal Function Is Impaired by a Short-Term High-Fat Diet in Mice: Increased Blood-Brain Barrier Permeability and Neuroinflammation as Triggering Events. Front. Neurosci. 2021, 15, 734158. [Google Scholar] [CrossRef] [PubMed]
- Chiazza, F.; Bondi, H.; Masante, I.; Ugazio, F.; Bortolotto, V.; Canonico, P.L.; Grilli, M. Short High Fat Diet Triggers Reversible and Region Specific Effects in DCX + Hippocampal Immature Neurons of Adolescent Male Mice. Sci. Rep. 2021, 11, 21499. [Google Scholar] [CrossRef]
- McLean, F.H.; Campbell, F.M.; Sergi, D.; Grant, C.; Morris, A.C.; Hay, E.A.; MacKenzie, A.; Mayer, C.D.; Langston, R.F.; Williams, L.M. Early and Reversible Changes to the Hippocampal Proteome in Mice on a High-Fat Diet. Nutr. Metab. 2019, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.J.; Cole, R.M.; Deems, N.P.; Belury, M.A.; Barrientos, R.M. Fatty Food, Fatty Acids, and Microglial Priming in the Adult and Aged Hippocampus and Amygdala. Brain. Behav. Immun. 2020, 89, 145–158. [Google Scholar] [CrossRef]
- Arcego, D.M.; Toniazzo, A.P.; Krolow, R.; Lampert, C.; Berlitz, C.; dos Santos Garcia, E.; do Couto Nicola, F.; Hoppe, J.B.; Gaelzer, M.M.; Klein, C.P.; et al. Impact of High-Fat Diet and Early Stress on Depressive-Like Behavior and Hippocampal Plasticity in Adult Male Rats. Mol. Neurobiol. 2018, 55, 2740–2753. [Google Scholar] [CrossRef]
- Mainardi, M.; Spinelli, M.; Scala, F.; Mattera, A.; Fusco, S.; D’Ascenzo, M.; Grassi, C. Loss of Leptin-Induced Modulation of Hippocampal Synaptic Trasmission and Signal Transduction in High-Fat Diet-Fed Mice. Front. Cell. Neurosci. 2017, 11, 225. [Google Scholar] [CrossRef] [Green Version]
- Vinuesa, A.; Bentivegna, M.; Calfa, G.; Filipello, F.; Pomilio, C.; Bonaventura, M.M.; Lux-Lantos, V.; Matzkin, M.E.; Gregosa, A.; Presa, J.; et al. Early Exposure to a High-Fat Diet Impacts on Hippocampal Plasticity: Implication of Microglia-Derived Exosome-like Extracellular Vesicles. Mol. Neurobiol. 2019, 56, 5075–5094. [Google Scholar] [CrossRef] [Green Version]
- Dingess, P.M.; Darling, R.A.; Kurt Dolence, E.; Culver, B.W.; Brown, T.E. Exposure to a Diet High in Fat Attenuates Dendritic Spine Density in the Medial Prefrontal Cortex. Brain Struct. Funct. 2017, 222, 1077–1085. [Google Scholar] [CrossRef]
- Spinelli, M.; Natale, F.; Rinaudo, M.; Leone, L.; Mezzogori, D.; Fusco, S.; Grassi, C. Neural Stem Cell-Derived Exosomes Revert HFD-Dependent Memory Impairment via CREB-BDNF Signalling. Int. J. Mol. Sci. 2020, 21, 8994. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Chen, J.; Zhan, L.; Lu, X.; Sun, X.; Sui, H.; Zheng, L.; Xiang, H.; Zhang, F. Endoplasmic Reticulum Stress Impairs Insulin Receptor Signaling in the Brains of Obese Rats. PLoS ONE 2015, 10, e0126384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Liu, B.-B.; Cai, M.; Li, J.-J.; Lou, S.-J. Excessive Endoplasmic Reticulum Stress and Decreased Neuroplasticity-Associated Proteins in Prefrontal Cortex of Obese Rats and the Regulatory Effects of Aerobic Exercise. Brain Res. Bull. 2018, 140, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.R.; Chen, Z.; Fang, K.; Xu, J.X.; Ge, J.F. Protective Effect of Quercetin against the Metabolic Dysfunction of Glucose and Lipids and Its Associated Learning and Memory Impairments in NAFLD Rats. Lipids Health Dis. 2021, 20, 164. [Google Scholar] [CrossRef]
- Wang, Z.; Ge, Q.; Wu, Y.; Zhang, J.; Gu, Q.; Han, J. Impairment of Long-Term Memory by a Short-Term High-Fat Diet via Hippocampal Oxidative Stress and Alterations in Synaptic Plasticity. Neuroscience 2020, 424, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Lucki, I.; Brookshire, B.R.; Carlson, G.C.; Browne, C.A.; Kazi, H.; Bang, S.; Choi, B.R.; Chen, Y.; McMullen, M.F.; et al. High Fat Diet Produces Brain Insulin Resistance, Synaptodendritic Abnormalities and Altered Behavior in Mice. Neurobiol. Dis. 2014, 67, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathan, A.R.; Gaikwad, A.B.; Viswanad, B.; Ramarao, P. Rosiglitazone Attenuates the Cognitive Deficits Induced by High Fat Diet Feeding in Rats. Eur. J. Pharmacol. 2008, 589, 176–179. [Google Scholar] [CrossRef]
- Asadbegi, M.; Yaghmaei, P.; Salehi, I.; Ebrahim-Habibi, A.; Komaki, A. Neuroprotective Effects of Metformin against Aβ-Mediated Inhibition of Long-Term Potentiation in Rats Fed a High-Fat Diet. Brain Res. Bull. 2016, 121, 178–185. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Oxidative Stress Modulates Sir2α in Rat Hippocampus and Cerebral Cortex. Eur. J. Neurosci. 2006, 23, 2573–2580. [Google Scholar] [CrossRef]
- Lee, S.; Goodson, M.L.; Vang, W.; Rutkowsky, J.; Kalanetra, K.; Bhattacharya, M.; Barile, D.; Raybould, H.E. Human Milk Oligosaccharide 2’-Fucosyllactose Supplementation Improves Gut Barrier Function and Signaling in the Vagal Afferent Pathway in Mice. Food Funct. 2021, 12, 8507–8521. [Google Scholar] [CrossRef]
- Kim, J.M.; Lee, U.; Kang, J.Y.; Park, S.K.; Kim, J.C.; Heo, H.J. Matcha Improves Metabolic Imbalance-Induced Cognitive Dysfunction. Oxid. Med. Cell. Longev. 2020, 2020, 8882763. [Google Scholar] [CrossRef] [PubMed]
- Pintana, H.; Tanajak, P.; Pratchayasakul, W.; Sa-Nguanmoo, P.; Chunchai, T.; Satjaritanun, P.; Leelarphat, L.; Chattipakorn, N.; Chattipakorn, S.C. Energy Restriction Combined with Dipeptidyl Peptidase-4 Inhibitor Exerts Neuroprotection in Obese Male Rats. Br. J. Nutr. 2016, 116, 1700–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, D.M.; Fitzgerald, D.P.; Anderson, B.M.; Lee, C.C.; Tessier, P.M.; Mcnay, E.C. Intrahippocampal administration of a domain antibody that binds aggregated amyloid-β reverses cognitive deficits produced by diet-induced obesity. Biochim. Biophys. Acta 2016, 1860, 1291–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, N.; Katsuura, G.; Yamada-Goto, N.; Novianti, E.; Inui, A.; Asakawa, A. Impaired Brain Fractalkine-CX3CR1 Signaling Is Implicated in Cognitive Dysfunction in Diet-Induced Obese Mice. BMJ Open Diabetes Res. Care 2021, 9, e001492. [Google Scholar] [CrossRef]
- Mandwie, M.; Karunia, J.; Niaz, A.; Keay, K.A.; Musumeci, G.; Rennie, C.; McGrath, K.; Al-badri, G.; Castorina, A. Metformin Treatment Attenuates Brain Inflammation and Rescues PACAP/VIP Neuropeptide Alterations in Mice Fed a High-Fat Diet. Int. J. Mol. Sci. 2021, 22, 3660. [Google Scholar] [CrossRef]
- Roy, P.; Martinelli, I.; Moruzzi, M.; Maggi, F.; Amantini, C.; Micioni Di Bonaventura, M.V.; Cifani, C.; Amenta, F.; Tayebati, S.K.; Tomassoni, D. Ion Channels Alterations in the Forebrain of High-Fat Diet Fed Rats. Eur. J. Histochem. 2021, 65, 3305. [Google Scholar] [CrossRef]
- Leyh, J.; Winter, K.; Reinicke, M.; Ceglarek, U.; Bechmann, I.; Landmann, J. Long-Term Diet-Induced Obesity Does Not Lead to Learning and Memory Impairment in Adult Mice. PLoS ONE 2021, 16, e0257921. [Google Scholar] [CrossRef]
- Wu, M.; Liao, M.; Huang, R.; Chen, C.; Tian, T.; Wang, H.; Li, J.; Li, J.; Sun, Y.; Wu, C.; et al. Hippocampal Overexpression of TREM2 Ameliorates High Fat Diet Induced Cognitive Impairment and Modulates Phenotypic Polarization of the Microglia. Genes Dis. 2020, 9, 401–414. [Google Scholar] [CrossRef]
- Xu, N.; Meng, H.; Liu, T.Y.; Feng, Y.L.; Qi, Y.; Zhang, D.H.; Wang, H.L. Sterol O-Acyltransferase 1 Deficiency Improves Defective Insulin Signaling in the Brains of Mice Fed a High-Fat Diet. Biochem. Biophys. Res. Commun. 2018, 499, 105–111. [Google Scholar] [CrossRef]
- Pathak, N.M.; Pathak, V.; Lynch, A.M.; Irwin, N.; Gault, V.A.; Flatt, P.R. Stable Oxyntomodulin Analogues Exert Positive Effects on Hippocampal Neurogenesis and Gene Expression as Well as Improving Glucose Homeostasis in High Fat Fed Mice. Mol. Cell. Endocrinol. 2015, 412, 95–103. [Google Scholar] [CrossRef]
- Choi, J.; Yin, T.; Shinozaki, K.; Lampe, J.W.; Stevens, J.F.; Becker, L.B.; Kim, J. Comprehensive Analysis of Phospholipids in the Brain, Heart, Kidney, and Liver: Brain Phospholipids Are Least Enriched with Polyunsaturated Fatty Acids. Mol. Cell. Biochem. 2018, 442, 187–201. [Google Scholar] [CrossRef]
- Canhada, S.; Castro, K.; Perry, I.S.; Luft, V.C. Omega-3 Fatty Acids’ Supplementation in Alzheimer’s Disease: A Systematic Review. Nutr. Neurosci. 2018, 21, 529–538. [Google Scholar] [CrossRef]
- Rangel-Huerta, O.D.; Gil, A. Effect of Omega-3 Fatty Acids on Cognition: An Updated Systematic Review of Randomized Clinical Trials. Nutr. Rev. 2018, 76, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Bauer, I.; Hughes, M.; Rowsell, R.; Cockerell, R.; Pipingas, A.; Crewther, S.; Crewther, D. Omega-3 Supplementation Improves Cognition and Modifies Brain Activation in Young Adults. Hum. Psychopharmacol. 2014, 29, 133–144. [Google Scholar] [CrossRef]
- Power, R.; Nolan, J.M.; Prado-Cabrero, A.; Roche, W.; Coen, R.; Power, T.; Mulcahy, R. Omega-3 Fatty Acid, Carotenoid and Vitamin E Supplementation Improves Working Memory in Older Adults: A Randomised Clinical Trial. Clin. Nutr. 2022, 41, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Külzow, N.; Witte, A.V.; Kerti, L.; Grittner, U.; Schuchardt, J.P.; Hahn, A.; Flöel, A. Impact of Omega-3 Fatty Acid Supplementation on Memory Functions in Healthy Older Adults. J. Alzheimer’s Dis. 2016, 51, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Wang, Y.; Xu, F.; Fan, W.; Zhang, Y.; Fan, K.; Wang, W.; Zhang, Y.; Zhang, C. Omega-3 Fatty Acids Ameliorate Cognitive Dysfunction in Schizophrenia Patients with Metabolic Syndrome. Brain. Behav. Immun. 2020, 88, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Rigacci, S.; Stefani, M. Nutraceutical Properties of Olive Oil Polyphenols. An Itinerary from Cultured Cells through Animal Models to Humans. Int. J. Mol. Sci. 2016, 17, 843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinciguerra, F.; Graziano, M.; Hagnäs, M.; Frittitta, L.; Tumminia, A. Influence of the Mediterranean and Ketogenic Diets on Cognitive Status and Decline: A Narrative Review. Nutrients 2020, 12, 1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutuli, D.; de Bartolo, P.; Caporali, P.; Laricchiuta, D.; Foti, F.; Ronci, M.; Rossi, C.; Neri, C.; Spalletta, G.; Caltagirone, C.; et al. N-3 Polyunsaturated Fatty Acids Supplementation Enhances Hippocampal Functionality in Aged Mice. Front. Aging Neurosci. 2014, 6, 220. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, S.; Lee, J.Y.; Yeo, Y.K.; Kim, J.S.; Lim, J. Improved Spatial Learning and Memory by Perilla Diet Is Correlated with Immunoreactivities to Neurofilament and α-Synuclein in Hilus of Dentate Gyrus. Proteome Sci. 2012, 10, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.Y.; Choi, J.M.; Lee, J.; Lee, M.H.; Lee, S.; Cho, E.J. Effects of Vegetable Oils with Different Fatty Acid Compositions on Cognition and Memory Ability in Aβ 25-35-Induced Alzheimer’s Disease Mouse Model. J. Med. Food 2016, 19, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.H.; Paris, C.; Magnien, M.; Colin, J.; Pelleïeux, S.; Coste, F.; Escanyé, M.C.; Pillot, T.; Olivier, J.L. Dietary Arachidonic Acid Increases Deleterious Effects of Amyloid-β Oligomers on Learning Abilities and Expression of AMPA Receptors: Putative Role of the ACSL4-CPLA2 Balance. Alzheimer’s Res. Ther. 2017, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Nenov, M.; Dincer, O.; Iuliano, L.; Praticò, D. Extra Virgin Olive Oil Improves Synaptic Activity, Short-Term Plasticity, Memory, and Neuropathology in a Tauopathy Model. Aging Cell 2020, 19, e13076. [Google Scholar] [CrossRef] [Green Version]
- Loehfelm, A.; Elder, M.K.; Boucsein, A.; Jones, P.P.; Williams, J.M.; Tups, A. Docosahexaenoic Acid Prevents Palmitate-Induced Insulin-Dependent Impairments of Neuronal Health. FASEB J. 2020, 34, 4635–4652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seebohm, G.; Wrobel, E.; Pusch, M.; Dicks, M.; Terhag, J.; Matschke, V.; Rothenberg, I.; Ursu, O.N.; Hertel, F.; Pott, L.; et al. Structural Basis of PI(4,5)P2-Dependent Regulation of GluA1 by Phosphatidylinositol-5-Phosphate 4-Kinase, Type II, Alpha (PIP5K2A). Pflugers Arch. 2014, 466, 1885–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, B.; Skup, M. Roles of Palmitoylation in Structural Long-Term Synaptic Plasticity. Mol. Brain 2021, 14, 8. [Google Scholar] [CrossRef]
- Mahalakshmi, B.; Maurya, N.; Da Lee, S.; Kumar, V.B. Possible Neuroprotective Mechanisms of Physical Exercise in Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 5895. [Google Scholar] [CrossRef]
- Phillips, M.C.L. Fasting as a Therapy in Neurological Disease. Nutrients 2019, 11, 2501. [Google Scholar] [CrossRef] [Green Version]
- Cansev, M.; Wurtman, R.J.; Sakamoto, T.; Ulus, I.H. Oral Administration of Circulating Precursors for Membrane Phosphatides Can Promote the Synthesis of New Brain Synapses. Alzheimer’s Dement. 2008, 4, S153–S168. [Google Scholar] [CrossRef]
- Kavraal, S.; Oncu, S.K.; Bitiktas, S.; Artis, A.S.; Dolu, N.; Gunes, T.; Suer, C. Maternal Intake of Omega-3 Essential Fatty Acids Improves Long Term Potentiation in the Dentate Gyrus and Morris Water Maze Performance in Rats. Brain Res. 2012, 1482, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Aryal, S.; Hussain, S.; Drevon, C.A.; Nagelhus, E.; Hvalby, Ø.; Jensen, V.; Walaas, S.I.; Davanger, S. Omega-3 Fatty Acids Regulate Plasticity in Distinct Hippocampal Glutamatergic Synapses. Eur. J. Neurosci. 2019, 49, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Dyall, S.C.; Michael, G.J.; Whelpton, R.; Scott, A.G.; Michael-Titus, A.T. Dietary Enrichment with Omega-3 Polyunsaturated Fatty Acids Reverses Age-Related Decreases in the GluR2 and NR2B Glutamate Receptor Subunits in Rat Forebrain. Neurobiol. Aging 2007, 28, 424–439. [Google Scholar] [CrossRef] [PubMed]
- Ettcheto, M.; Sánchez-Lopez, E.; Cano, A.; Carrasco, M.; Herrera, K.; Manzine, P.R.; Espinosa-Jimenez, T.; Busquets, O.; Verdaguer, E.; Olloquequi, J.; et al. Dexibuprofen Ameliorates Peripheral and Central Risk Factors Associated with Alzheimer’s Disease in Metabolically Stressed APPswe/PS1dE9 Mice. Cell Biosci. 2021, 11, 141. [Google Scholar] [CrossRef]
- Ye, X.; Gao, X.; Scott, T.; Tucker, K.L. Habitual Sugar Intake and Cognitive Function among Middle-Aged and Older Puerto Ricans without Diabetes. Br. J. Nutr. 2011, 106, 1423–1432. [Google Scholar] [CrossRef] [Green Version]
- Taha, A.Y.; Gao, F.; Ramadan, E.; Cheon, Y.; Rapoport, S.I.; Kim, H.W. Upregulated Expression of Brain Enzymatic Markers of Arachidonic and Docosahexaenoic Acid Metabolism in a Rat Model of the Metabolic Syndrome. BMC Neurosci. 2012, 13, 131. [Google Scholar] [CrossRef] [Green Version]
- Kendig, M.D.; Boakes, R.A.; Rooney, K.B.; Corbit, L.H. Chronic Restricted Access to 10% Sucrose Solution in Adolescent and Young Adult Rats Impairs Spatial Memory and Alters Sensitivity to Outcome Devaluation. Physiol. Behav. 2013, 120, 164–172. [Google Scholar] [CrossRef]
- Abbott, K.N.; Morris, M.J.; Westbrook, R.F.; Reichelt, A.C. Sex-Specific Effects of Daily Exposure to Sucrose on Spatial Memory Performance in Male and Female Rats, and Implications for Estrous Cycle Stage. Physiol. Behav. 2016, 162, 52–60. [Google Scholar] [CrossRef]
- Ross, A.; Barnett, N.; Faulkner, A.; Hannapel, R.; Parent, M.B. Sucrose Ingestion Induces Glutamate AMPA Receptor Phosphorylation in Dorsal Hippocampal Neurons: Increased Sucrose Experience Prevents This Effect. Behav. Brain Res. 2019, 359, 792–798. [Google Scholar] [CrossRef]
- Lemos, C.; Rial, D.; Gonçalves, F.Q.; Pires, J.; Silva, H.B.; Matheus, F.C.; da Silva, A.C.; Marques, J.M.; Rodrigues, R.J.; Jarak, I.; et al. High Sucrose Consumption Induces Memory Impairment in Rats Associated with Electrophysiological Modifications but Not with Metabolic Changes in the Hippocampus. Neuroscience 2016, 315, 196–205. [Google Scholar] [CrossRef]
- Soares, E.; Prediger, R.D.; Nunes, S.; Castro, A.A.; Viana, S.D.; Lemos, C.; De Souza, C.M.; Agostinho, P.; Cunha, R.A.; Carvalho, E.; et al. Spatial Memory Impairments in a Prediabetic Rat Model. Neuroscience 2013, 250, 565–577. [Google Scholar] [CrossRef]
- Li, J.M.; Yu, R.; Zhang, L.P.; Wen, S.Y.; Wang, S.J.; Zhang, X.Y.; Xu, Q.; Kong, L.D. Dietary Fructose-Induced Gut Dysbiosis Promotes Mouse Hippocampal Neuroinflammation: A Benefit of Short-Chain Fatty Acids. Microbiome 2019, 7, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.C.; Wu, C.W.; Tain, Y.L.; Fu, M.H.; Hung, C.Y.; Chen, I.C.; Chen, L.W.; Wu, K.L.H. Oral Pioglitazone Ameliorates Fructose-Induced Peripheral Insulin Resistance and Hippocampal Gliosis but Not Restores Inhibited Hippocampal Adult Neurogenesis. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Fierros-Campuzano, J.; Ballesteros-Zebadúa, P.; Manjarrez-Marmolejo, J.; Aguilera, P.; Méndez-Diaz, M.; Prospero-García, O.; Franco-Pérez, J. Irreversible Hippocampal Changes Induced by High Fructose Diet in Rats. Nutr. Neurosci. 2020, 25, 1325–1337. [Google Scholar] [CrossRef]
- Agrawal, R.; Noble, E.; Vergnes, L.; Ying, Z.; Reue, K.; Gomez-Pinilla, F. Dietary Fructose Aggravates the Pathobiology of Traumatic Brain Injury by Influencing Energy Homeostasis and Plasticity. J. Cereb. Blood Flow Metab. 2015, 36, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Djordjevic, A.; Bursać, B.; Veličković, N.; Vasiljević, A.; Matić, G. The Impact of Different Fructose Loads on Insulin Sensitivity, Inflammation, and PSA-NCAM-Mediated Plasticity in the Hippocampus of Fructose-Fed Male Rats. Nutr. Neurosci. 2015, 18, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Gomez-Pinilla, F. “Metabolic Syndrome” in the Brain: Deficiency in Omega-3 Fatty Acid Exacerbates Dysfunctions in Insulin Receptor Signalling and Cognition. J. Physiol. 2012, 590, 2485–2499. [Google Scholar] [CrossRef]
- Dos Santos, B.; Schmitz, A.E.; de Almeida, G.R.L.; de Souza, L.F.; Szczepanik, J.C.; Nunes, E.A.; Brunetta, H.S.; Mack, J.M.; Prediger, R.D.; Cunha, M.P.; et al. Fructose Intake Impairs Cortical Antioxidant Defenses Allied to Hyperlocomotion in Middle-Aged C57BL/6 Female Mice. Neurochem. Res. 2020, 45, 2868–2883. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.M.; Konanur, V.R.; Taing, L.; Usui, R.; Kayser, B.D.; Goran, M.I.; Kanoski, S.E. Effects of Sucrose and High Fructose Corn Syrup Consumption on Spatial Memory Function and Hippocampal Neuroinflammation in Adolescent Rats. Hippocampus 2015, 25, 227–239. [Google Scholar] [CrossRef]
- Nuthikattu, S.; Milenkovic, D.; Norman, J.E.; Rutledge, J.; Villablanca, A. Inhibition of Soluble Epoxide Hydrolase Is Protective against the Multiomic Effects of a High Glycemic Diet on Brain Microvascular Inflammation and Cognitive Dysfunction. Nutrients 2021, 13, 3913. [Google Scholar] [CrossRef]
- Orr, M.E.; Salinas, A.; Buffenstein, R.; Oddo, S. Mammalian Target of Rapamycin Hyperactivity Mediates the Detrimental Effects of a High Sucrose Diet on Alzheimer’s Disease Pathology. Neurobiol. Aging 2014, 35, 1233–1242. [Google Scholar] [CrossRef] [Green Version]
- Iacovides, S.; Goble, D.; Paterson, B.; Meiring, R.M. Three Consecutive Weeks of Nutritional Ketosis Has No Effect on Cognitive Function, Sleep, and Mood Compared with a High-Carbohydrate, Low-Fat Diet in Healthy Individuals: A Randomized, Crossover, Controlled Trial. Am. J. Clin. Nutr. 2019, 110, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary Ketosis Enhances Memory in Mild Cognitive Impairment. Neurobiol. Aging 2012, 33, 425.e19–425.e27. [Google Scholar] [CrossRef] [Green Version]
- Ródenas-González, F.; Blanco-Gandía, M.C.; Miñarro, J.; Rodríguez-Arias, M. Cognitive Profile of Male Mice Exposed to a Ketogenic Diet. Physiol. Behav. 2022, 254, 113883. [Google Scholar] [CrossRef] [PubMed]
- Miles, K.N.; Skelton, M.R. Male Mice Placed on a Ketogenic Diet from Postnatal Day (P) 21 through Adulthood Have Reduced Growth, Are Hypoactive, Show Increased Freezing in a Conditioned Fear Paradigm, and Have Spatial Learning Deficits. Brain Res. 2020, 1734, 146697. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Stafstrom, C.E.; Fu, D.D.; Hu, Y.; Holmes, G.L. Detrimental Effects of the Ketogenic Diet on Cognitive Function in Rats. Pediatr. Res. 2004, 55, 498–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaise, J.H.; Ruskin, D.N.; Koranda, J.L.; Masino, S.A. Effects of a Ketogenic Diet on Hippocampal Plasticity in Freely Moving Juvenile Rats. Physiol. Rep. 2015, 3, e12411. [Google Scholar] [CrossRef] [PubMed]
- Thio, L.L.; Rensing, N.; Maloney, S.; Wozniak, D.F.; Xiong, C.; Yamada, K.A. A Ketogenic Diet Does Not Impair Rat Behavior or Long-Term Potentiation: BRIEF COMMUNICATION. Epilepsia 2010, 51, 1619–1623. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.C.; Rocha, J.; Pires, C.S.; Ribeiro, L.C.; Brolese, G.; Leite, M.C.; Almeida, L.M.V.; Tramontina, F.; Ziegler, D.R.; Gonçalves, C.A. Transitory Gliosis in the CA3 Hippocampal Region in Rats Fed on a Ketogenic Diet. Nutr. Neurosci. 2005, 8, 259–264. [Google Scholar] [CrossRef]
- Fukushima, A.; Ogura, Y.; Furuta, M.; Kakehashi, C.; Funabashi, T.; Akema, T. Ketogenic Diet Does Not Impair Spatial Ability Controlled by the Hippocampus in Male Rats. Brain Res. 2015, 1622, 36–42. [Google Scholar] [CrossRef]
- Huang, J.; Li, Y.Q.; Wu, C.H.; Zhang, Y.L.; Zhao, S.T.; Chen, Y.J.; Deng, Y.H.; Xuan, A.; Sun, X.D. The Effect of Ketogenic Diet on Behaviors and Synaptic Functions of Naive Mice. Brain Behav. 2019, 9, e01246. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.R.; Hernandez, C.M.; Campos, K.; Truckenbrod, L.; Federico, Q.; Moon, B.; McQuail, J.A.; Maurer, A.P.; Bizon, J.L.; Burke, S.N. A Ketogenic Diet Improves Cognition and Has Biochemical Effects in Prefrontal Cortex That Are Dissociable From Hippocampus. Front. Aging Neurosci. 2018, 10, 391. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.R.; Hernandez, C.M.; Truckenbrod, L.M.; Campos, K.T.; McQuail, J.A.; Bizon, J.L.; Burke, S.N. Age and Ketogenic Diet Have Dissociable Effects on Synapse-Related Gene Expression Between Hippocampal Subregions. Front. Aging Neurosci. 2019, 11, 239. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab. 2017, 26, 547–557.e8. [Google Scholar] [CrossRef] [Green Version]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic Diet Improves Motor Performance but Not Cognition in Two Mouse Models of Alzheimer’s Pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Jiang, C.; Wu, J.; Liu, P.; Deng, X.; Zhang, Y.; Peng, B.; Zhu, Y. Ketogenic Diet Ameliorates Cognitive Impairment and Neuroinflammation in a Mouse Model of Alzheimer’s Disease. CNS Neurosci. Ther. 2021, 28, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Page, K.A.; Williamson, A.; Yu, N.; McNay, E.C.; Dzuira, J.; McCrimmon, R.J.; Sherwin, R.S. Medium-Chain Fatty Acids Improve Cognitive Function in Intensively Treated Type 1 Diabetic Patients and Support in Vitro Synaptic Transmission during Acute Hypoglycemia. Diabetes 2009, 58, 1237–1244. [Google Scholar] [CrossRef] [Green Version]
- Jensen, N.J.; Nilsson, M.; Ingerslev, J.S.; Olsen, D.A.; Fenger, M.; Svart, M.; Møller, N.; Zander, M.; Miskowiak, K.W.; Rungby, J. Effects of β-Hydroxybutyrate on Cognition in Patients with Type 2 Diabetes. Eur. J. Endocrinol. 2020, 182, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Yomogida, Y.; Matsuo, J.; Ishida, I.; Ota, M.; Nakamura, K.; Ashida, K.; Kunugi, H. An FMRI Investigation into the Effects of Ketogenic Medium-Chain Triglycerides on Cognitive Function in Elderly Adults: A Pilot Study. Nutrients 2021, 13, 2134. [Google Scholar] [CrossRef]
- Fortier, M.; Castellano, C.A.; St-Pierre, V.; Myette-Côté, É.; Langlois, F.; Roy, M.; Morin, M.C.; Bocti, C.; Fulop, T.; Godin, J.P.; et al. A Ketogenic Drink Improves Cognition in Mild Cognitive Impairment: Results of a 6-Month RCT. Alzheimer’s Dement. 2021, 17, 543–552. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a Medium-Chain Triglyceride-Based Ketogenic Formula on Cognitive Function in Patients with Mild-to-Moderate Alzheimer’s Disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of β-Hydroxybutyrate on Cognition in Memory-Impaired Adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Hu, E.; Du, H.; Zhu, X.; Wang, L.; Shang, S.; Wu, X.; Lu, H.; Lu, X. Beta-Hydroxybutyrate Promotes the Expression of BDNF in Hippocampal Neurons under Adequate Glucose Supply. Neuroscience 2018, 386, 315–325. [Google Scholar] [CrossRef]
- Wang, D.; Mitchell, E.S. Cognition and Synaptic-Plasticity Related Changes in Aged Rats Supplemented with 8- and 10-Carbon Medium Chain Triglycerides. PLoS ONE 2016, 11, e0160159. [Google Scholar] [CrossRef] [Green Version]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. β-Hydroxybutyrate Inhibits Inflammasome Activation to Attenuate Alzheimer’s Disease Pathology. J. Neuroinflamm. 2020, 17, 280. [Google Scholar] [CrossRef]
- Yin, J.X.; Maalouf, M.; Han, P.; Zhao, M.; Gao, M.; Dharshaun, T.; Ryan, C.; Whitelegge, J.; Wu, J.; Eisenberg, D.; et al. Ketones Block Amyloid Entry and Improve Cognition in an Alzheimer’s Model. Neurobiol. Aging 2016, 39, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Kashiwaya, Y.; Pawlosky, R.; Markis, W.; King, M.T.; Bergman, C.; Srivastava, S.; Murray, A.; Clarke, K.; Veech, R.L. A Ketone Ester Diet Increases Brain Malonyl-CoA and Uncoupling Proteins 4 and 5 While Decreasing Food Intake in the Normal Wistar Rat. J. Biol. Chem. 2010, 285, 25950–25956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cao, Q.; Li, S.; Lu, X.; Zhao, Y.; Guan, J.S.; Chen, J.C.; Wu, Q.; Chen, G.Q. 3-Hydroxybutyrate Methyl Ester as a Potential Drug against Alzheimer’s Disease via Mitochondria Protection Mechanism. Biomaterials 2013, 34, 7552–7562. [Google Scholar] [CrossRef] [PubMed]
- Bostock, E.C.S.; Kirkby, K.C.; Taylor, B.V.; Hawrelak, J.A. Consumer Reports of “Keto Flu” Associated With the Ketogenic Diet. Front. Nutr. 2020, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Hori, A.; Tandon, P.; Holmes, G.L.; Stafstrom, C.E. Ketogenic Diet: Effects on Expression of Kindled Seizures and Behavior in Adult Rats. Epilepsia 1997, 38, 750–758. [Google Scholar] [CrossRef]
- Thio, L.L.; Wong, M.; Yamada, K.A. Ketone Bodies Do Not Directly Alter Excitatory or Inhibitory Hippocampal Synaptic Transmission. Neurology 2000, 54, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Youssef, F.F. Ketone Bodies Attenuate Excitotoxic Cell Injury in the Rat Hippocampal Slice under Conditions of Reduced Glucose Availability. Neurol. Res. 2015, 37, 211–216. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, S.S.; Rensing, N.R.; Thio, L.L.; Yamada, K.A.; Wong, M. The Ketogenic Diet Inhibits the Mammalian Target of Rapamycin (MTOR) Pathway. Epilepsia 2011, 52, e7–e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.; Augustin, K.; Boddum, K.; Williams, S.; Sun, M.; Terschak, J.A.; Hardege, J.D.; Chen, P.E.; Walker, M.C.; Williams, R.S.B. Seizure Control by Decanoic Acid through Direct AMPA Receptor Inhibition. Brain 2016, 139, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of Action for the Medium-Chain Triglyceride Ketogenic Diet in Neurological and Metabolic Disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone Bodies: From Enemy to Friend and Guardian Angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Craft, S.; Newcomer, J.; Kanne, S.; Dagogo-Jack, S.; Cryer, P.; Sheline, Y.; Luby, J.; Dagogo-Jack, A.; Alderson, A. Memory Improvement Following Induced Hyperinsulinemia in Alzheimer’s Disease. Neurobiol. Aging 1996, 17, 123–130. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-Degrading Enzyme Regulates the Levels of Insulin, Amyloid β-Protein, and the β-Amyloid Precursor Protein Intracellular Domain in Vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Alldred, M.J.; Ginsberg, S.D.; Ohno, M. Mechanisms Underlying Insulin Deficiency-Induced Acceleration of β-Amyloidosis in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2012, 7, e32792. [Google Scholar] [CrossRef] [Green Version]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-Beta-Hydroxybutyrate Protects Neurons in Models of Alzheimer’s and Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef] [PubMed]
- Koppel, S.J.; Pei, D.; Wilkins, H.M.; Weidling, I.W.; Wang, X.; Menta, B.W.; Perez-Ortiz, J.; Kalani, A.; Manley, S.; Novikova, L.; et al. A Ketogenic Diet Differentially Affects Neuron And Astrocyte Transcription. J. Neurochem. 2021, 157, 1930. [Google Scholar] [CrossRef] [PubMed]
- Mujica-Parodi, L.R.; Amgalan, A.; Sultan, S.F.; Antal, B.; Sun, X.; Skiena, S.; Lithen, A.; Adra, N.; Ratai, E.M.; Weistuch, C.; et al. Diet Modulates Brain Network Stability, a Biomarker for Brain Aging, in Young Adults. Proc. Natl. Acad. Sci. USA 2020, 117, 6170–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la O, V.; Zazpe, I.; Martínez, J.A.; Santiago, S.; Carlos, S.; Zulet, M.Á.; Ruiz-Canela, M. Scoping Review of Paleolithic Dietary Patterns: A Definition Proposal. Nutr. Res. Rev. 2020, 34, 78–106. [Google Scholar] [CrossRef]
- Du, K.; Markus, E.; Fecych, M.; Rhodes, J.S.; Beverly, J.L. Satiety and Memory Enhancing Effects of a High-Protein Meal Depend on the Source of Protein. Nutr. Neurosci. 2018, 21, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Méndez-López, M.; Méndez, M.; Arias, J.; Arias, J.L. Effects of a High Protein Diet on Cognition and Brain Metabolism in Cirrhotic Rats. Physiol. Behav. 2015, 149, 220–228. [Google Scholar] [CrossRef]
- Su, Z.W.; Liao, J.Y.; Zhang, H.; Zhang, T.; Wu, F.; Tian, X.H.; Zhang, F.T.; Sun, W.W.; Cui, Q.L. Postnatal High-Protein Diet Improves Learning and Memory in Premature Rats via Activation of MTOR Signaling. Brain Res. 2015, 1611, 1–7. [Google Scholar] [CrossRef]
- Boraxbekk, C.J.; Stomby, A.; Ryberg, M.; Lindahl, B.; Larsson, C.; Nyberg, L.; Olsson, T. Diet-Induced Weight Loss Alters Functional Brain Responses during an Episodic Memory Task. Obes. Facts 2015, 8, 261–272. [Google Scholar] [CrossRef]
- Stomby, A.; Otten, J.; Ryberg, M.; Nyberg, L.; Olsson, T.; Boraxbekk, C.J. A Paleolithic Diet with and without Combined Aerobic and Resistance Exercise Increases Functional Brain Responses and Hippocampal Volume in Subjects with Type 2 Diabetes. Front. Aging Neurosci. 2017, 9, 391. [Google Scholar] [CrossRef] [Green Version]
- Gyorkos, A.; Baker, M.H.; Miutz, L.N.; Lown, D.A.; Jones, M.A.; Houghton-Rahrig, L.D. Carbohydrate-Restricted Diet and Exercise Increase Brain-Derived Neurotrophic Factor and Cognitive Function: A Randomized Crossover Trial. Cureus 2019, 11, e5604. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Exp. Approach | Species, Sex, Age | Model | Learning/Memory | Synaptic Function, Neuroplasticity | Other Pathways | Refs. |
|---|---|---|---|---|---|---|
| Short periods (≤2 months) | ||||||
| Young animals | Rat (♀; 3 w) | CAFD (F/sucrose: 45%) & HFru solution (11%) or HFru solution alone vs. CD (F: 13.4% & C: 56.7%) for 5 w. Reversion: 5 w with CD | Impaired memory (novel object in context) at 5 w for CAFD and HFru solution. No reversion in CAFD group | - | Gut dysbiosis (before and after reversion) | [44] |
| Rat (♂; 6 w) | HFD-HDextrose (F: 41.7% & C: 36.7%) vs. CD (F: 13.5% & C: 58%) for 11 w | Impaired memory (NOR; no changes with MWM) | ↓ dendritic arborization in HPC neurons and ↑ in entorhinal cortex neurons | ↑ TNFα levels in blood | [45] | |
| Rat (♂; 6 w) | CAFD (cakes, biscuits & a protein source) & HSu solution (10%) vs. CD for 6 w | Impaired memory (NOR and NLR) | No changes in BDNF, TrkB and synapsin in HPC | ↑ inflammation and gut dysbiosis | [46] | |
| Rat (♀; 2 m) | HFD (F:39%) & refined sugar (40%) vs. CD (F: 13% & complex C: 59%) for 2 m | Impaired memory (MWM) | ↓ BDNF, phosphosynapsin I and phosphoCREB | - | [47] | |
| Adult animals | Rat (♂ and ♀; 9–10 w) | HFD (F: 60% & C: 20%) HFru solution (11%) vs. CD (F: 13% & C: 62%) for 6 w | Impaired hippocampal-dependent memory (NLR) in ♂ (no changes in ♀) | - | - | [48] |
| Rat (♂; adult) | CAFD (F: 45% & C: 50%) & HSu solution (10%) vs. CD (F: 15%, & C: 59%) for 5, 11 & 20 days | Impaired hippocampal-dependent memory (NLR; no changes in NOR) | No changes in BDNF | ↑ inflammation in HPC at 20 d | [49] | |
| Rat (♂) | CAFD (CD supplemented with cakes, biscuits & protein source) & HSu solution (10%) vs. CD for 5 w | Impaired memory (NLR) | - | Gut dysbiosis | [50] | |
| Rat (♂; adult) | HFD-HC (F: 25%, C: 44% & P: 18%) vs. CD (F: 5%, C: 62% & P: 18) for 6 w | Impaired short-term and long-term memory (RAWM) | No changes in BDNF | ↑ oxidative stress in HPC | [51] | |
| Rat (♂) | HFD (F: 40%, P: 5% & C: 15%) & HSu solution (40%) vs. CD (F: 15%, P: 25% & C: 55%) for 6 w. Reversion: 3 w with HFD-HSu and 3 w with CD and training | Impaired memory (NLR). Reversed by CD and training | - | - | [52] | |
| Rat (♂) | HFD (SFAs & MUFAs; 38%) & refined sugar (38%) vs. CD (F: 6% & sugar: 4.1%) for 8 w | Impaired memory (NOR) and learning (MWM) | ↓ GluA3 levels in dorsal HPC and altered levels in synaptic plasticity markers | Altered levels in energy metabolism markers (by proteomic analysis) | [53] | |
| Long periods (>2 months) | ||||||
| Young animals | Rat (♂; 21 days) | HFD (F: 29%, sucrose: 34% & cholesterol 1.25%) & HGlucose-HFru solution (55%/45%) vs. CD (F: 6% & C: 44%) for 8 m | - | ↓ PSD95 and BDNF, ↓ LTP in HPC | Gut dysbiosis, ↑ inflammation and microglia activation in HPC | [54] |
| Rat (♀; 1 m) | HFD (30% lard & 66% sucrose) & HSu solution (30%) vs. CD (F: 3%, C: 61% & P: 19%) for 24 m | Impaired memory (NOR) | ↓ TrKB, ↓ LTP in HPC | No changes in neurogenesis | [55] | |
| Rat (♂; 5–7 w) | HFD & HSu solution (5%) vs. CD for 4 m. Reversion: 3 m with bioactive food | Impaired spatial and working memory (T-maze and NOR). Reversed by bioactive foods | - | Gut dysbiosis. Reversed by bioactive foods | [56] | |
| Rat (♂; 6 w) | HFD-HFru (F: 30% & fructose: 15%) vs. CD for 6 m | Impaired learning (MWM) | - | ↑ BBB permeability, neurodegeneration and microglia activation | [57] | |
| Rat (♂; 6 w) | HFD-HFru (saturated F: 45% & fructose: 20%) vs. CD for 11 w | Impaired memory (NOR) | - | ↓ IGF1 and ↑ oxidative stress | [58] | |
| Mouse (♂; 6 w) | HFD-HSu (F: 60% & sucrose: 7%) vs. CD (F: 17%) for 13 w | Impaired memory (NOR) | ↓ GluA1, BDNF, phosphoCREB, TrkB in HPC and PFC | ↓ neurogenesis | [59] | |
| Mouse (♂; 7 w) | HFD (F: 40% & C: 20%) & HSu solution vs. CD (F: 12% & C: 67%) for 14 w | - | ↓ PSD95 and ↑ phosphoTau in brain (no changes in synaptophysin) | ↓ GLUT1/3, ↑ ER stress and inflammation responses and INS resistance in brain | [60] | |
| Rat (♂; 7 w) | CAFD (CD with cookies, cakes & biscuits) vs. CD once per day, 5 days per week for 5 m | - | ↑ BDNF and TrkB and ↓ phosphoTrkB in PFC. No changes in HPC | Redox imbalance | [61] | |
| Rat (♀; 8–10 w) | HFD (F: 40%, C: 45% & P 15%) & HFru solution (15%) vs. CD (F: 6%, C: 64% & P: 25%) for 12, 16 & 24 w | Impaired memory (MWM) at 16 and 24 w (no changes at 12 w) | - | ↑ oxidative stress and reduced antioxidant levels in HPC and CTX | [43] | |
| Rat (♂; 2 m) | HFD-HGlucose (F: 40%) vs. CD (F: 13%) for 3 m | Impaired learning (nonspatial discrimination learning problem) | - | ↑ BBB permeability in HPC | [62] | |
| Rat (♀; 2 m) | HFD (SFAs & MUFAs: 39%) & refined sugar (40%) vs. CD (F: 13% & C: 59%) for 1, 2 & 6 m or 2 y | Impaired learning and memory (MWM) at 1 and 2 m | ↓ BDNF and synapsin I in HPC | - | [63] | |
| Rat (♂; 2 m) | HFD (high-lard or high-olive oil) & HSu solution vs. CD for 10 w | No changes in spatial memory (Y-maze) | ↓ GluN2A in high-lard-HSu (no changes in high-olive oil-HSu) in CTX | - | [42] | |
| Rat (♂; 2 m) | HFD-HSu & HFru corn syrup solution (20%) vs. CD for 8 m | Impaired learning (MWM) | ↓ dendritic spine density, ↓ LTP and ↓ BDNF levels in CA1-HPC | - | [64] | |
| Mouse (♀ and ♂; 10 w) | HFD (F: 60%) & HSu solution (20%) vs. CD (F: 10%) for 4 or 6 m. Reversion: 8 w with CD | Impaired memory (NOR and NLR. No change in working memory (Y-maze). Recovered after 8 w of CD | ↑ microglia activation (no inflammation and neuronal loss). Recovered after 8 w of CD | [65] | ||
| Guinea pigs (♀; 10 w) | HFD-HSu (F: 20% & sucrose: 15%) vs. CD (F: 4% & sucrose: 0%) for 7 m | - | ↓ BDNF levels in HPC | - | [66] | |
| Rat (♂; adolescent) | HFD-HDextrose (SFAs: 41.7%) vs. CD (F: 13.4%) for 10 w | Impaired memory (NOR) | - | - | [67] | |
| Adult animals | Mouse (♂; 3 m) | HFD (F: 45%) & HFru solution (10%) vs. CD for 10 w | Impaired memory (MWM) | ↓ PSD95 and SNAP25 in HPC | INS resistance, ↑ microglia activation and inflammation in brain | [68] |
| Rat (♂) | HFD-HDextrose (F: 38%, P: 24%, C: 18% & dextrose: 20%) vs. CD (F: 18%, P: 24%, C: 58%) for 10, 40 & 90 days | Impaired learning at 10 and 90 d, no changes at 40 d (Y-shaped maze) | - | ↑ BBB permeability in HPC (only in obese rats) | [41] | |
| Mouse (♂; 3 m) | HFD-HFru (F: 48%, fructose: 33% & P: 19%) or HFD (F: 48%, C: 33% & P: 19%) vs. CD for 14 w | Impaired memory (NOR and NLR). HF-HFru more affected than HFD | ↓ glutamate and glutamine in HPC (no changes in GABA) | - | [69] | |
| Rat (♂) | HFD-HDextrose (F: 40%, P: 21% & C: 38%) or HFD-HSu (F: 40%, P: 21% & C: 38%) vs. CD (F: 12%, P: 28% & C: 59%) for 3 m | Impaired learning (nonspatial Pavlovian discrimination and reversal learning problem) | ↓ BDNF in prefrontal CTX and HPC with HF-HDextrose (no changes with HF-HSu) | - | [70] | |
| Rat (♂ and ♀) | HFD-HFru (CD: 60%, fructose: 30% & pork fat: 10%) vs. CD for 12 w | Impaired learning (MWM and passive avoidance test). ♂ more affected than ♀ | - | ↑ oxidative stress | [71] | |
| Short vs. long periods | ||||||
| Young animals | Mouse (♂; 6 w) | HFD-HFru (30% lard, 0.5% cholesterol and 15% fructose, all in weight/weight) vs. CD for 4 or 24 w | Impaired learning (MWM) at 14 w (no changes at 4 w) | - | ↑ BBB permeability and astrocytocis | [72] |
| Exp. Approach | Species, Sex, Age | Model | Learning/Memory | Synaptic Function, Neuroplasticity | Other Pathways | Refs. |
|---|---|---|---|---|---|---|
| Days (≤2 weeks) | ||||||
| Young animals | Rat (♂; 3 w & 2 m) | Saturated HFD (F: 60% & C: 20%) vs. CD (C: 35%) for 7 days | Impaired long-term memory (NLR) in young animals but improved in old ones. No changes in short-term memory | ↓ LTP in CA1-HPC in young animals, but ↑ in old ones | ↑ glucocorticoids release in young animals | [103] |
| Mouse (♂; 5 w) | HFD (F: 45% & C: 35%) vs. CD (F: 18% & C: 58%) for 2 days | - | ↓ LTP in HPC. No changes in LTD | - | [104] | |
| Mouse (♂; 6 w) | HFD (F: 60% & C: 27%) vs. CD (F: 10% & C: 70%) for 1 to 7 days | Impaired memory (NOR) at 3–7 days | ↓ synaptophysin at 7 days in HPC. No changes at 3 days | ↑ BBB permeability at 1–3 days (no changes at 4–6 days) and inflammation at 2 days (no changes at 1 or 3–7 days) | [105] | |
| Mouse (♂; 6 w) | HFD (F: 60% & C: 21%) vs. CD (F: 13% & C: 67%) for 7 days | - | ↓ BDNF and dendritic tree in HPC | - | [106] | |
| Adult animals | Rat (♂) | Saturated HFD (F: 48% & C: 37%) or PUFA HFD (F: 46% & C: 37%) vs. CD (F: 21% & C: 56%) for 2 w | Impaired memory (NLR) with saturated. No changes with PUFA | No changes in BDNF in HPC | Gut dysbiosis. No changes in inflammation in HPC | [100] |
| Mouse (♂; 10 w) | HFD (F: 30% by weight) vs. CD (F: 5% by weight) for 7 days | Impaired memory (NOR, Y-maze & temporal order memory test) | ↓ PSD95, BDNF, thickened of post synaptic density, & ↑ width of synaptic cleft in HPC & PFC | Gut dysbiosis, ↑ microglia activation, & inflammation in HPC & PFC | [93] | |
| Mouse (♂; 12 w) | Saturated HFD (F: 60%) vs. CD (F: 10%) in animals for 3 days, 1 or 2 w | - | - | Alterations in metabolism, cell stress, inflammation, cell signaling & cytoskeleton | [107] | |
| Young vs. aged animals | Rat (♂; 3 m & 24 m) | Mixed HFD (F: 60.3% & C: 21.3%) vs. CD (F:17% & C: 54%) for 3 days | Impaired long-term memory (FC & MWM) in aged animals. No changes in short-term memory or in young animals | - | ↑ microglia activation & inflammation in aged animals in HPC & amygdala | [92] |
| Rat (♂; 3 m & 24 m) | Mixed HFD (F: 60.3% & C: 21.3%) vs. CD (F:17% & C: 54%) for 3 days | - | - | ↑ inflammation in microglia of young & old animals in HPC & amygdala | [108] | |
| Short periods (>2 weeks and ≤2 months) | ||||||
| Young animals | Rat (♂; 3 w) | Mixed HFD (F: 42% & C: 25%) or CD (F: 4% & C: 50%) for 7 w | - | ↓ GluA2, PSD95, synaptophysin, & TrKB receptor in HPC. No changes in BDNF | ↓ glucocorticoid receptor in HPC | [109] |
| Mouse (♂; 3 w) | HFD (F: 60%) vs. CD (F: 6.55%) for 8 w | - | ↓ response to leptin induction of AMPAR-mediated synaptic transmission in HPC neurons | - | [110] | |
| Mouse (♂; 3 w) | HFD (F: 21.2% & C: 22.5% by weight) & CD (F:3.6% & C: 28.8% by weight) for 6 w | Impaired memory (NOR) | Synaptic loss in CA1-HPC neurons | ↓ neurogenesis and ↑ inflammation in HPC | [111] | |
| Mouse (♂; 8 w) | HFD (F: 60% & C: 20%) calorically matched or ad libitum vs. CD (F: 13% & C: 58%) for 3 w. | - | ↓ spine density in PFC in both HFD protocols | - | [112] | |
| Mouse (♂; 4 w) | Saturated HFD (F: 60%) vs. CD (F: 6.55%) for 6–7 w | Impaired memory (MWM) | ↓ LTP in HPC. ↓ GluA1 phosphorylation & ↑ palmitoylation in HPC | INS resistance in HPC | [32] | |
| Mouse (♂; 4 w) | Saturated HFD (F: 60%) vs. CD (F: 6.55%) for 6–7 w | Impaired memory (NOR & NLR) | ↓ BDNF, phosphoTRKB, phosphoCREB & glutamate metabotropic receptors in HPC | - | [113] | |
| Mouse (♂; 5 & 8 w) | SOLF (60% CD + 40% saturated oil-enriched food) or UOLF (60% CD + 40% unsaturated oil-enriched food) for 8 w | Impaired memory (Y-maze) by SOLF | ↓ GluN2A & 2B by SOLF/UOLF in young animals (no changes in old). ↓ LTP & ↑ LTD by SOLF in young animals | - | [102] | |
| Rat (♂; 6 w) | HFD (F: 40%) vs. CD for 6 w | Impaired learning & memory (MWM) | - | ↑ ER stress & INS resistance in HPC | [114] | |
| Rat (♂; 7 w) | HFD (F: 40% & C: 40%) vs. CD (F: 12.5% & C: 62.9%) for 8 w | - | ↓ BDNF in PFC | ↑ ER stress in PFC | [115] | |
| Rat (♂; 2 m) | HFD (5000 kcal/kg) vs. CD (3600 kcal/kg) for 8 w | Impaired learning & memory (MWM) | ↓ synaptotagmin1 & synapsin 1 | - | [116] | |
| Mouse (♂; 6–8 w) | HFD (F: 21.2% & C: 61.3% by weight) vs. CD (F: 7.5% & C: 75.1% by weight) for 4 or 7 w | Impaired long-term memory (FC). No changes in short-term memory (NLR) | ↓ LTP in CA1-HPC. ↓ cfos, synaptophysin, CaMKII & IV, calcineurin A in HPC. No changes in BDNF | ↑ oxidative stress | [117] | |
| Mouse (♂; 8 w) | HFD (F: 45%) vs. CD (F: 10%) for 2 m | Impaired short-term memory (T-maze) | - | INS resistance in brain | [118] | |
| Adult animals | Rat (♂; adult) | HFD (F: 58% & C: 17%) vs. CD for 5 w | Impaired learning & memory (MWM) | - | - | [119] |
| Rat (♂) | HFD (F: 20% & C: 48% by weight) vs. CD (F: 5% & C: 47% by weight) for 8 w | - | No changes in LTP in DG-HPC | - | [120] | |
| Rat | Mixed HFD (F: 39%) vs. CD (F: 13%) for 2 m | - | - | ↑ oxidative stress | [121] | |
| Rat (♂; 16 m) | Saturated HFD (2 % cholesterol + 10 % trans coconut oil) vs. soybean oil (12%) for 8 w | Impaired memory (water radial arm maze) | Dendritic loss in HPC | ↑ microglia activation & inflammation in HPC | [101] | |
| Mouse (♂) | Mixed HFD (F: 45% & C: 45%) vs. CD (F: 10% & C: 70%) for 8 w | No changes in memory (Y-maze & NOR) | - | - | [122] | |
| Long periods (>2 months) | ||||||
| Young animals | Mouse (♂; 4 w) | Mixed HFD (F: 60% & C: 20%) vs. SD (F: 10% & C: 70%) for 14 w | Impaired memory (Y-maze & MWM) | ↓ BDNF in HPC & CTX | Altered antioxidant defense, ↑ oxidative stress, inflammation & INS resistance in HPC & CTX | [123] |
| Rat (♂; 5–6 w) | HFD (F: 59.28%) vs. CD (F: 19.77%) for 16 w | Impaired memory (MWM) | ↓ spine density & LTP in HPC | INS resistance & ↓ mitochondrial function in brain | [124] | |
| Rat (♂; 6 w) | Mixed HFD (F: 60% & C: 20%) vs. CD (F: 13% & C: 58%) for 6 m | Impaired learning (FC) | ↑ surface GluA1 in HPC. No changes in total levels or GluN2B | - | [125] | |
| Mouse (♂; 6 w) | Mixed HFD (F: 60%) vs. CD (F: 12.6%) for 16 w | Impaired memory (Y-maze) | ↓ GluN1/2A, GluA1, PSD95 & synaptophysin. No changes in GluN2B or GluA2 | - | [126] | |
| Mouse (♂; 6 w) | HFD (F: 22% by weight) vs. CD (F: 6% by weight) for 16 w | - | - | ↑ inflammation in HPC & CTX & gliosis in CTX | [127] | |
| Rat (♂; 7 w) | Mixed HFD (F: 45%) vs. CD (F: 6%) for 17 w | - | ↓ PSD95. No changes in synaptophysin | - | [128] | |
| Mouse (♂; 8 w) | Saturated HFD (F: 59% & C: 26%) vs. CD (F: 11% & C: 59%) for 8, 16, 24 & 28 w | No changes in short-term (Y-maze), long-term & learning (MWM). Impaired cognitive flexibility | - | No changes in microglia activation in HPC & CTX | [129] | |
| Mouse (♂; 8 w) | Mixed HFD (F: 60% & C: 20%) vs. CD (F: 10% & C 70%) for 46 w | Impaired memory (MWM, NOR & Y-maze) | ↓ branching, spine density, PSD95, spinophilin, & synaptophysin in HPC | ↑ microglia activation, inflammation & iNOS in HPC | [130] | |
| Mouse (♂; 8 w) | Mixed HFD (F: 60%) vs. CD for 16 w | Impaired learning & memory (MWM) | - | ↑ inflammation & INS resistance in HPC & CTX | [131] | |
| Mouse (♂; 8–10 w) | Mixed HFD (F: 45%) vs. CD (F: 10% & C: 60%) for 13 w | - | ↓ synaptophysin in HPC | ↑ oxidative stress & INS resistance, & ↓ neurogenesis in HPC | [132] | |
| Mouse (♀; 2 m) | Mixed HFD (F: 60%) vs. CD (F: 10%) for 4 m. Intervention: 16 m with CD | Impaired learning & memory (MWM & FC) | ↓ BDNF in HPC | No changes in inflammation or mitochondrial function in HPC | [95] | |
| Adult animals | Mouse (♂) | HFD (F: 45%) vs. CD (F: 10%) for 17 w | Impaired memory (T-maze) | ↓ LTP in HPC | - | [96] |
| Mouse (♂; 9 w) | HFD (F: 60% by weight) vs. CD (F: 5% by weight) for 13–15 w | Impaired memory (NOR & NLR) | ↓ BDNF, synaptophysin, & PSD95 in HPC & PFC | Gut dysbiosis, ↑ inflammation, & microglia activation in HPC & PFC | [97] | |
| Mouse (♂; 9 w) | Mixed HFD (F: 60% & C: 20%) vs. CD (F: 10% & C: 70%) for 24 w | Impaired memory (MWM & Y-maze) | ↓ PSD95 & SNAP23 in HPC & CTX | ↑ inflammation, microglia activation, oxidative stress, INS resistance & Aβ in HPC & CTX | [98] | |
| Mouse (♂; 12 w) | HFD (F: 55%) vs. CD (F: 13%) for 15 w | Impaired memory (NOR & NLR) | ↓ PSD95, synaptophysin, thickened of post synaptic density, & ↑ width of synaptic cleft in CA1-HPC | Gut dysbiosis, ↑ inflammation, microglia activation, & INS resistance in CA1-HPC | [99] | |
| Exp. Approach | Species, Sex, Age | Diet Model | Learning/Memory | Synaptic Function, Neuroplasticity | Other Pathways | Refs. |
|---|---|---|---|---|---|---|
| High sucrose | ||||||
| Young animals | Rat (♂; 3 w) | HSu diet vs. CD for 8 w | - | ↓ BDNF and synaptophysin in brain | - | [158] |
| Rat (♂; 3 and 8 w) | HSu solution (10%) 2 h/day vs. 0.1% sodium saccharin solution for 4 w | Impaired learning and memory (MWM) | - | - | [159] | |
| Rat (♂ and ♀; 4 w) | HSu solution (10%) 2 h/day vs. water for 2 & 4 w | Impaired memory (NLR; more deficits in ♂) | - | - | [160] | |
| Rat (♂; 8 w) | HSu solution (32%) vs. water for 3, 5 or 10 days | - | ↑ GluA1 phosphorylation in dorsal HPC at 3 days and ↓ at 5 and 10 days | - | [161] | |
| Rat (♂; 12 w) | HSu solution (35%) vs. water for 9 w | Impaired memory (NLR and NOR) | ↓ LTD in CA1-HPC (no changes in LTP) | No changes in metabolic profile in HPC | [162] | |
| Adult animals | Rat (♂; 4 m) | HSu solution (35%) vs. water for 9 w | Impaired memory (MWM and Y-maze) | ↑ GluA1 and GluN1 protein levels in HPC | No changes in oxidative stress or inflammation in HPC | [163] |
| High fructose | ||||||
| Young animals | Mouse (♂; 5 w) | HFru (35%) vs. CD for 8 w | - | - | Microglia activation, ↑ inflammation, ↓ neurogenesis, and neuronal loss in HPC | [164] |
| Rat (♂; 6 w) | HFru (60%) vs. CD for 12 w | - | ↓ BDNF and PSD95 in HPC | INS resistance and microglia activation, and ↓ neurogenesis in HPC | [165] | |
| Rat (♂; 6 w) | HFru solution (10%) vs. water for 12 w | Impaired spatial memory (Barnes maze) | - | Astrocytosis, ↓ neurogenesis, ↑ inflammation, and in HPC and PFC | [166] | |
| Rat (♂; 2 m) | HFru solution (15%) vs. water for 8 w | Impaired memory (Barnes maze) | ↓ phosphoTrKB and synaptophysin. No changes in BDNF in HPC | Alterations in metabolism, mitochondrial function and INS resistance in HPC | [167] | |
| Rat (♂; 12 w) | HFru solution (10 % or 60%) vs. water for 9 w | - | - | INS resistance and ↓ inflammation in HPC with 10% of HFru. No changes in Ins and ↑ inflammation with 60% of HFru | [168] | |
| Adult animals | Rat (♂) | HFru solution (15%) vs. water for 6 w | Impaired memory (Barnes maze) | - | INS resistance in HPC | [169] |
| Mouse (♀; 9 m) | HFru solution (10%) vs. water for 12 w | No changes in spatial memory (Y maze) | - | ↓ antioxidant defense in PFC | [170] | |
| High sucrose vs. high fructose | ||||||
| Young animals | Rat (♂; 4 and 9 w) | HSu or HFru solution (11%) vs. water for 30 days | Impaired memory in young animals with HFru (Barnes maze). No changes in adults or with HSu | - | ↑ inflammation in young animals with HFru. No changes in adults or with HSu | [171] |
| Simple carbohydrates | ||||||
| Adult animals | Rat (♂) | HC (simple C: 30%) vs. CD (simple C: 16%) for 8 days | Impaired memory (NLR) | No changes in BDNF in HPC | Gut dysbiosis. No changes in inflammation in HPC | [100] |
| Mouse (♂; 22 w) | HC (simple C: 36% weight) vs. CD (simple C: 12%) for 10 w | - | - | ↑ expression of neurodegeneration genes, inflammation, mitochondrial function, and oxidation in HPC | [172] | |
| Exp. Approach | Species, Sex, Age | Diet Model | Learning/Memory | Synaptic Function, Neuroplasticity | Other Pathways | Refs. |
|---|---|---|---|---|---|---|
| Ketogenic diet | ||||||
| Humans | Human (♂ and ♀; 18–40 y) | KD (F: 60%, C: 15% & P: 25%) vs. CD (F: 20%, C: 55% & P 25%) for 3 w | No changes in cognitive performance | - | - | [174] |
| Human (♂ and ♀; ~70 y) | KD (C: 5–10%) vs. CD (C: 50%) for 6 w | Improved paired associate learning | - | - | [175] | |
| Young animals | Mouse (♂; 3 w) | KD (F: 90.5%, C: 0.3% & P: 9.1%) vs. CD (F: 13%, C: 67% & P: 20%) for 2 w | No differences in learning (Hebb Williams Maze) or memory (passive avoidance test) | - | - | [176] |
| Mouse (♂; 3 w) | KD (F: 75.1%, C: 3.2% & P: 8.6%) vs. CD (F: 7.1%, C: 63.2% & P: 18.3%) for 5.5 w | Impaired learning (MWM) but no differences in memory (NOR) | - | - | [177] | |
| Rat (♂; 3 w) | KD (F: 78.8%, C: 0.8% & P: 9.5%) vs. CD (F: 10%, C: 49% & P: 23.4%) for 1 m | Impaired learning and memory (MWM) | - | - | [178] | |
| Rat (♂; 3 w) | KD (F: 87% & C + P: 13%) vs. CD for 3 w | - | No changes in short-term plasticity but ↓ LTP magnitude | - | [179] | |
| Rat (3 w) | KD (F: 92%, C: 3% & P: 5%) vs. CD (F: 12%, C: 65% & P: 24%) for 2–3 w | No changes in memory (FC) | No changes in short-term or long-term plasticity | - | [180] | |
| Rat (♂; 4 w) | KD (F: 69%, P: 24% & C: 0%) vs. CD (F: 12%, P: 23% & C: 54%) for 6 w | No changes in learning (MWM) | - | ↑ transitory glia activation in CA3-HPC at 1 w (no changes at 6 w) | [181] | |
| Rat (♂; 7–8 w) | KD (F: 90%, P: 10% & C: 0%) vs. CD (F: 10%, P 10% & C: 80%) for 3 w | Improved short-term memory (Y-maze) but no changes in long-term memory (MWM) | ↑ GluA1 levels in HPC (no changes in GluA2) | - | [182] | |
| Adult animals | Mouse (♂; 3 m) | KD (F: 90%, C: 0% & P 10%) vs. CD (F: 10%, C: 80% & P 10%) for 3 m | No changes in learning (MWM) and memory (MWM and Y-maze) | No changes in LTP | - | [183] |
| Rat (86% ♂; 4 and 20 m) | KD (F: 75.9%, C: 3.9% & P 20.1%) vs. CD (F: 16.4%, C: 64.9% & P 18.8%) for 12 w | Improved memory (WM/BAT) | - | ↓ GLUT1 in PFC | [184] | |
| Rat (♂; 4 and 20 m) | KD (F: 75.9%, C: 3.9% & P 20.1%) vs. CD (F: 16.4%, C: 64.9% & P 18.8%) for 12 w | - | ↓ expression of gria1, 2 and 4, and other postsynaptic proteins in DG-HPC (no changes in CA3) | ↓ presynaptic proteins in DG-HPC (no changes in CA3) | [185] | |
| Mouse (♂; 12 m) | Cyclic KD: KD (F: 90% & P: 10%) & CD (F: 13%, P: 10% & C: 77%) alternate weekly; vs. CD (F: 14%, P: 24% & C: 62%) for 12 m | Improved memory (place avoidance test and NOR) | - | - | [186] | |
| Diseased animals | Mouse (5 m; AD model and WT) | KD (F: 77.1%, C: 0.5% & P: 22.4%) vs. CD (F: 14%, C: 62.2% & P: 23.8%) for 3 m | No changes in memory (RAWM) but improved motor function (AD or WT animals) | No changes in neural loss | - | [187] |
| Mouse (7 m; AD model and WT) | KD (F: 76%, C: 3% & P: 16%) vs. CD (F: 12%, C: 65% & P: 23%) for 2 & 4 m | Improved learning and memory at 4 m (no changes at 2 m; T and Barnes maze tests) | - | ↓ Aβ, microgliosis, inflammation and ↑ number of spines and neurons in HPC | [188] | |
| Standard diet supplemented with ketone bodies | ||||||
| Humans | Human (♂ and ♀; ~15 y; T1D) | KB solution (40 g of MCT) vs. placebo drink in a single session of 1 h under hypoglycemic conditions (tests performed 1 h before & 1 h latter) | Improved hypoglycemia-mediated memory deficits | - | - | [189] |
| Human (♂ and ♀; 35–70 y; T2D) | BHB (0.9% weight/volume) i.v., infusion vs. placebo (tests performed 120 min latter) | Improved working memory (no changes in global cognition) | - | - | [190] | |
| Human (♂ and ♀; 60–74 y) | Ketogenic formula (20 g of MCT in 36 g of total F) or placebo 90 min before tested | Improved working memory | - | - | [191] | |
| Human (♂ and ♀; ≥55 y; MCI) | Ketogenic solution (MCT) daily or placebo 6 m | Improved executive function, memory, and language | - | - | [192] | |
| Human (♂ and ♀; ~73 y; mild-to moderate AD) | Ketogenic formula (20 g of MCT in 35.9 g of total F) daily or placebo for 12 w vs. baseline | Improved in immediate and delayed logical memory | - | - | [193] | |
| Human (~74.7 y; mild-to moderate AD) | Ketogenic solution (MCT) vs. placebo solution (long chain triglycerides; tests performed 120 min latter) | Improved cognitive performance only in ApoEε4—subjects | - | - | [194] | |
| Young animals | Mouse (♂ and ♀; 8 w) | BHB supplementation (60 mg/kg) for 2 days (twice/day) via intragastric gavage | - | ↑ BDNF levels in HPC | [195] | |
| Aged animals | Rat (♂; 21 m) | MCT8 formula (5 % of octanoic triglyceride) or MCT10 formula (5 % of decanoic triglyceride) vs. CD (5 % sunflower oil) for 8 w | Improved memory (NOR) with MCT10 (no changes with MCT8) | No changes in PSD95 and synaptophysin in brain. ↓ expression of plasticity-related genes | ↑ INS signaling in brain | [196] |
| Diseased animals | Mouse (♂ and ♀; 8 w; AD model) | BHB solution (0.019 g/mL) daily for 8 w | - | - | ↓ Aβ and inflammation in brain | [197] |
| Mouse (4 m; AD model and WT) | BHB & ACA solution via subcutaneous injection (600 mg/kg/day & 150 mg/kg/day) daily for 2 m | Improved learning and memory (MWM and NOR; no changes in WT) | ↑ LTP (no changes in WT) | ↓ Aβ and oxidative stress in brain | [198] | |
| Mouse (8.5 m; AD model) | KE + BHB supplementation (ketone esters: 21.5%; F: 8.2% C: 43.5% & P: 23.9%) vs. CD (F: 8.2% C: 64.9% & P: 23.9%) for 4 & 7 m | Improved memory (MWM and FC) | - | ↓ Aβ and phosphorylated tau in CA1 and CA3-HPC, amygdala and CTX | [199] | |
| Mouse (♂; 7.5 m; AD model) | HBME (10, 40 & 80 mg/kg/d) daily intragastric administration vs. water for 2.5 m | Improved learning and memory (MWM) | - | ↓ Aβ in HPC and CTX. ↓ ROS and apoptosis under glucose deprivation in cultured neurons | [200] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fadó, R.; Molins, A.; Rojas, R.; Casals, N. Feeding the Brain: Effect of Nutrients on Cognition, Synaptic Function, and AMPA Receptors. Nutrients 2022, 14, 4137. https://doi.org/10.3390/nu14194137
Fadó R, Molins A, Rojas R, Casals N. Feeding the Brain: Effect of Nutrients on Cognition, Synaptic Function, and AMPA Receptors. Nutrients. 2022; 14(19):4137. https://doi.org/10.3390/nu14194137
Chicago/Turabian StyleFadó, Rut, Anna Molins, Rocío Rojas, and Núria Casals. 2022. "Feeding the Brain: Effect of Nutrients on Cognition, Synaptic Function, and AMPA Receptors" Nutrients 14, no. 19: 4137. https://doi.org/10.3390/nu14194137
APA StyleFadó, R., Molins, A., Rojas, R., & Casals, N. (2022). Feeding the Brain: Effect of Nutrients on Cognition, Synaptic Function, and AMPA Receptors. Nutrients, 14(19), 4137. https://doi.org/10.3390/nu14194137