
Arachidonic Acid as Mechanotransducer of Renin Cell Baroreceptor

Abstract
:1. Introduction
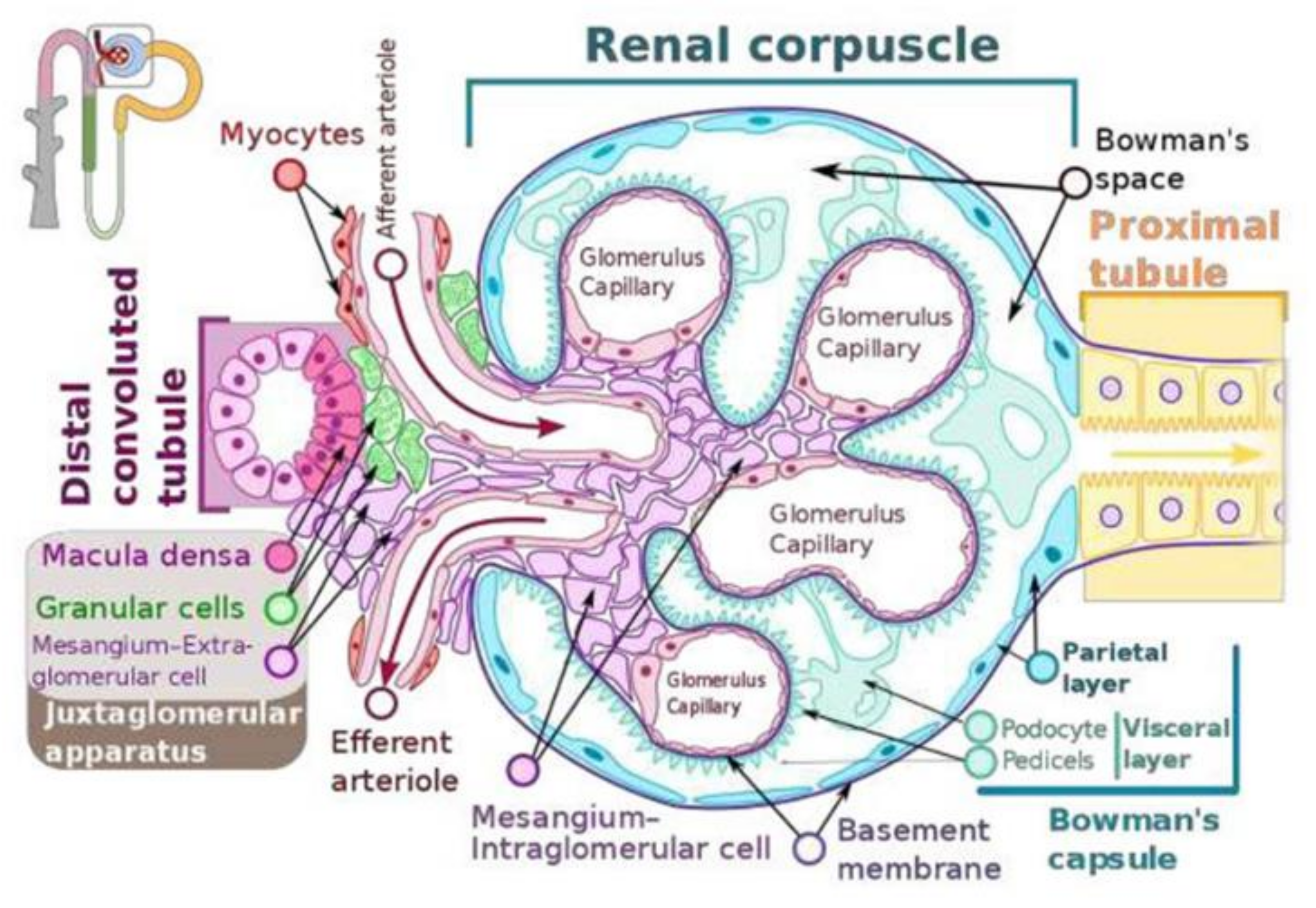
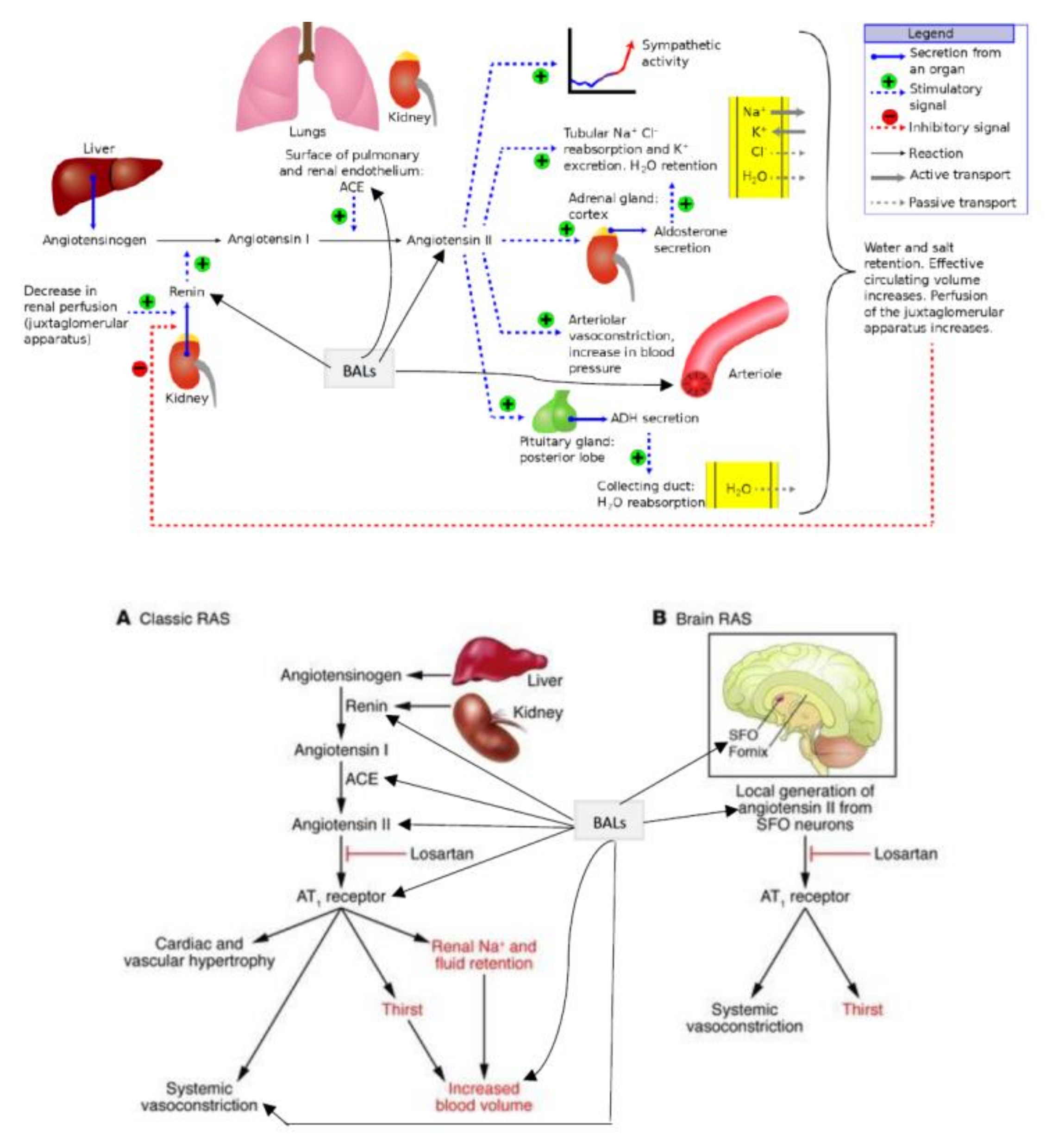
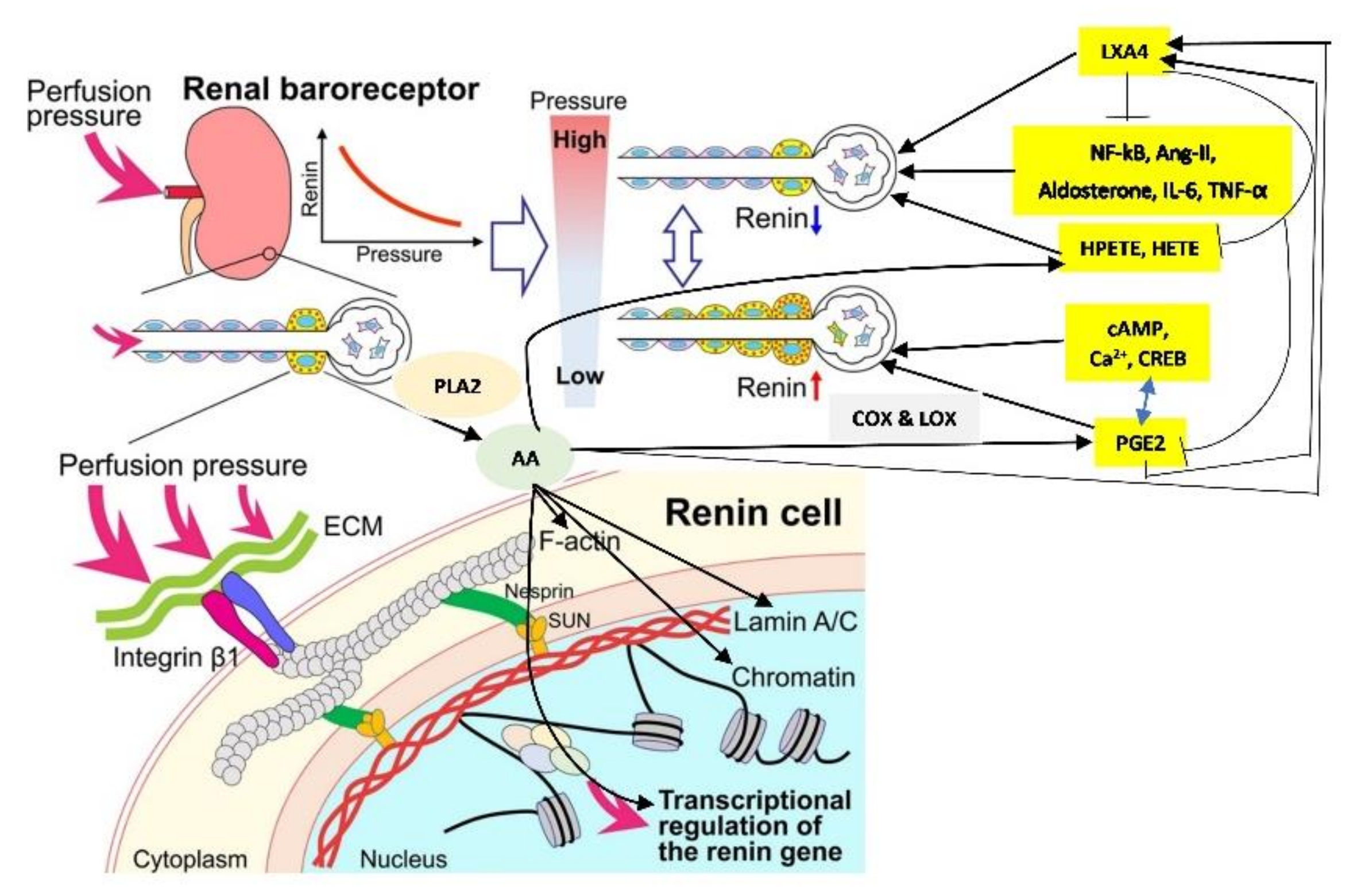
2. JGA (Juxta Glomerular Apparatus) Functions as a Baroreceptor
3. Arteriole Myogenic Mechanism
4. Tubuloglomerular Feedback
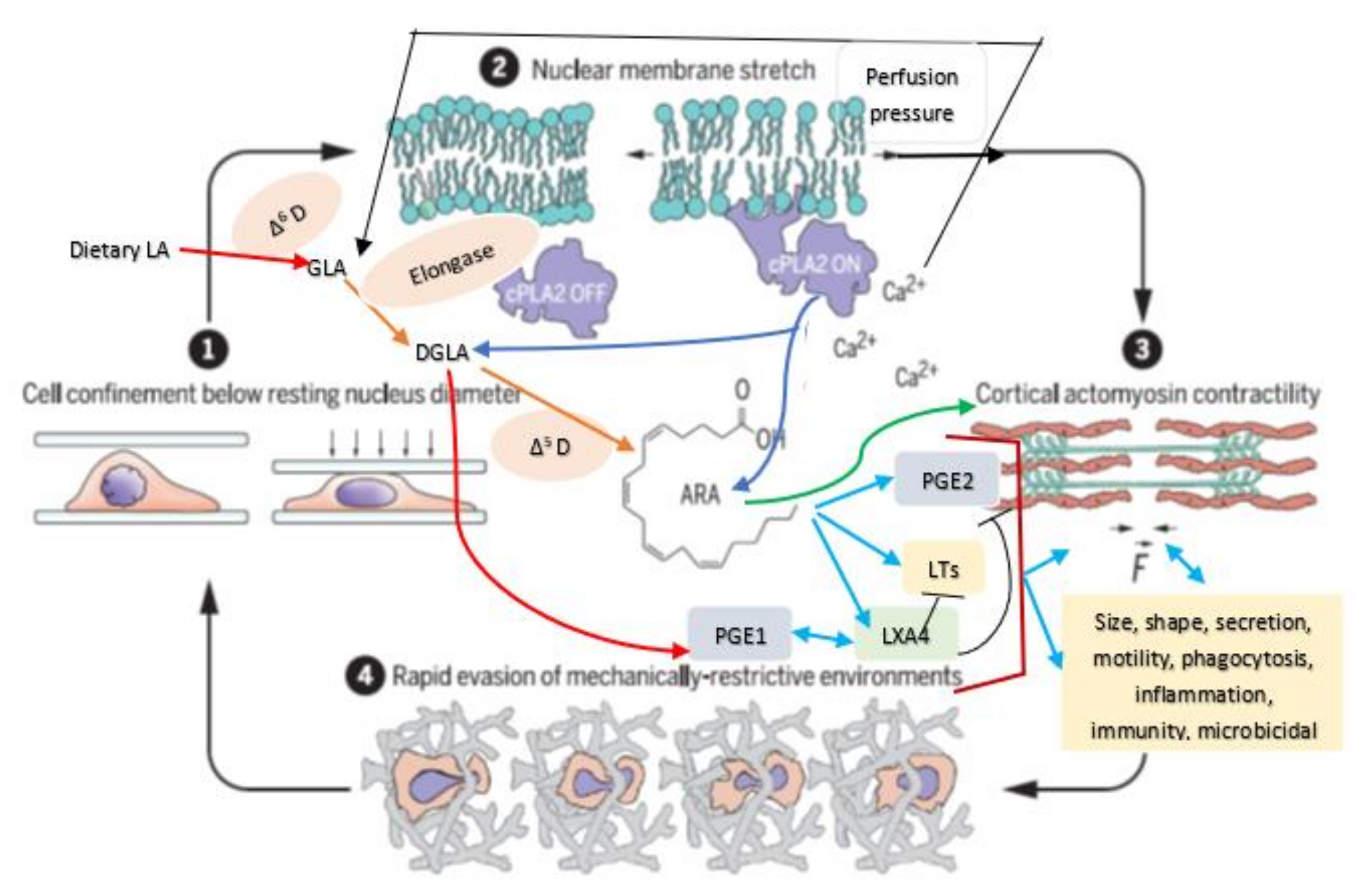
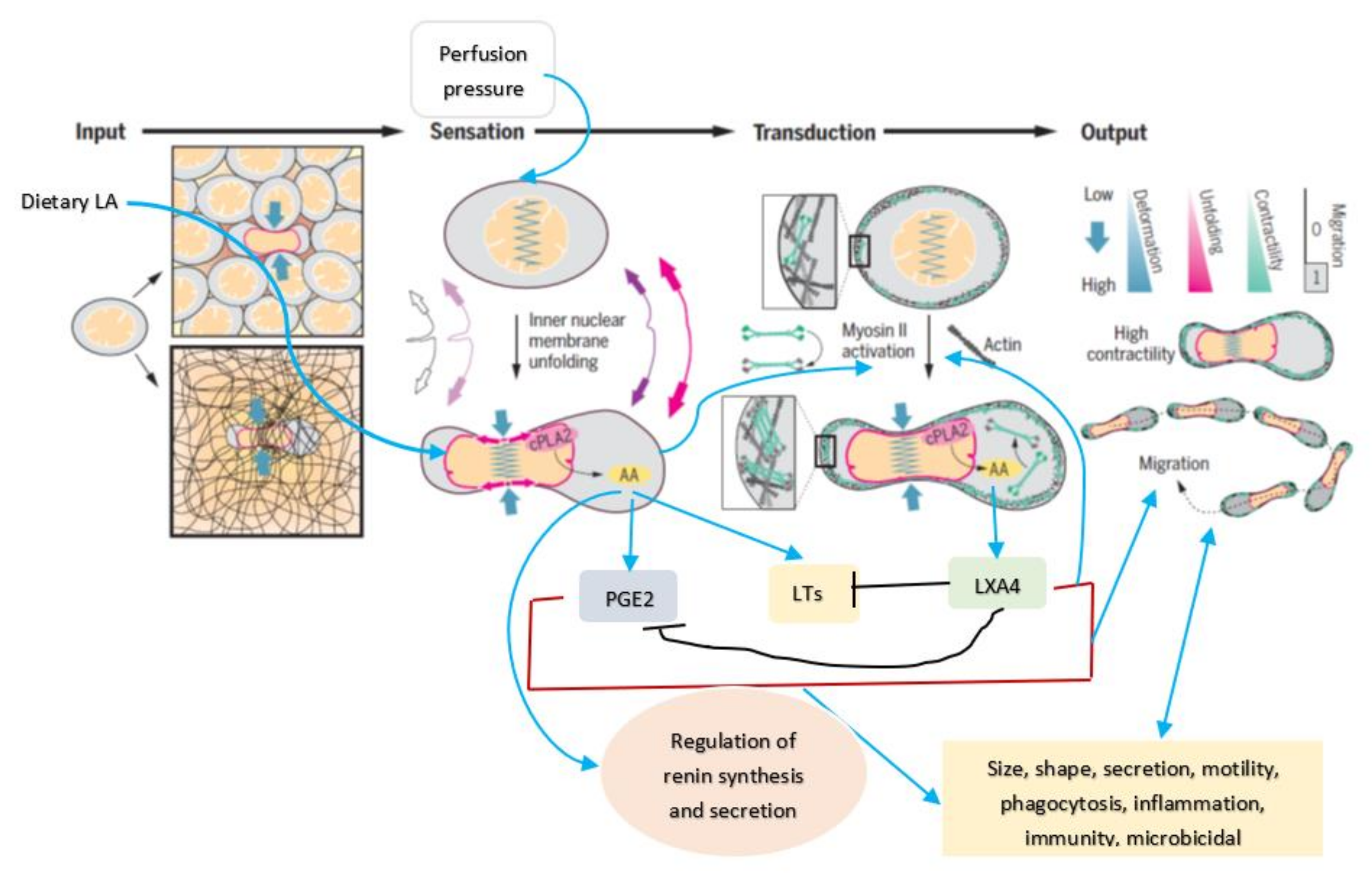
5. Mechanotransduction from Cell Membrane to the Nucleus
6. AA Functions as a Mechanotransducer



7. AA and Its Metabolites Regulate Gene(s) Expression and Renin Release and Action
8. AA and Its Metabolites in Renin Synthesis, Secretion, and Action
9. Renin Secretion at the Cellular Level
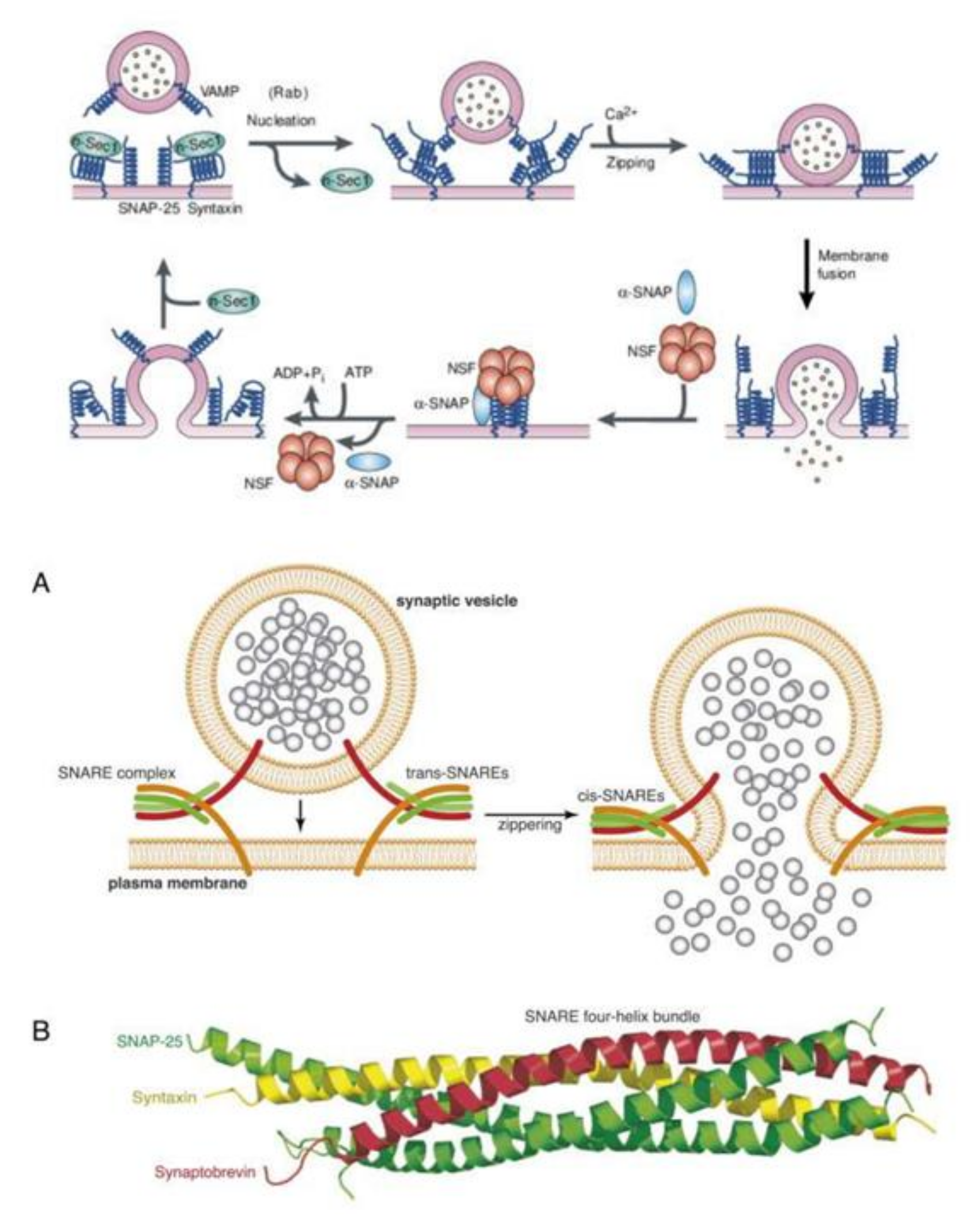
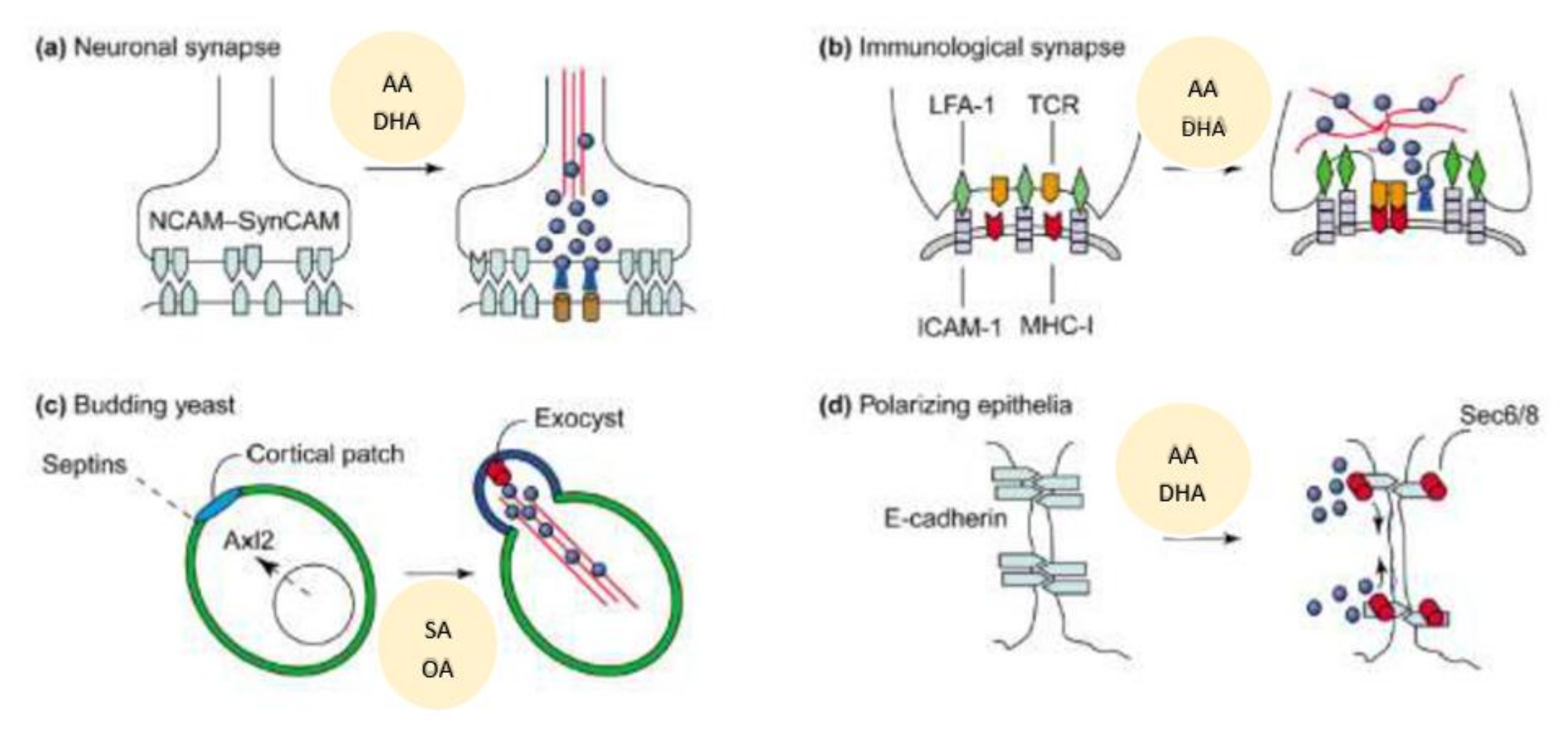
10. AA Interacts with Syntaxin to Regulate Exocytosis/Renin Secretion
11. Conclusions and Therapeutic Implications
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| EPA | Eicosapentaenoic acid |
| DHA | Docosahexaenoic acid |
| LXA4 | Lipoxin A4 |
| RAS | Renin-angiotensin aldosterone system |
| And-II | Angiotensin-II |
| JGA | Juxtaglomerular apparatus |
| PLA2 | Phospholipase A2 |
| TRPV1 | Transient receptor potential vanilloid type |
| 1 ATP | Adenosine triphosphate |
| LA | Cis-linoleic acid |
| ALA | Alpha-linolenic acid |
| GLA | Gamma-linolenic acid |
| COX-2 | Cyclooxygenase-2 |
| PGE2 | Prostaglandin E2 |
| PGs | Prostaglandins |
| LTs | Leukotrienes |
| TXs | Thromboxanes |
| mPGES1 and PGES2 | Microsomal PGE synthases 1 and 2 EP2 and EP4 = PGE2 receptors |
| LOX | Lipoxygenase |
| MSCs | Mesenchymal stem cells |
| CREB | cAMP-binding protein |
| PPAR-γ | Peroxisome proliferator-activated receptor |
| γ cAMP | Cyclic adenosine monophosphate |
| HPETE | Hydroperoxyeicosatetraenoic acid |
| HETE | Hydroxyeicosatetraenoic acid |
| PGI2 | Prostacyclin |
| cGMP | Cyclic guanosine monophosphate |
| NO | Nitric oxide |
| SNAREs | Soluble N-ethylmaleimide-sensitive factor attachment proteins |
| NSF | N-ethylmaleimide-sensitive factor |
| SNAP | Synaptosome-associated protein |
| APC | Antigen-presenting cell |
| VAMP | Vesicle-associated membrane protein |
| STX3 | Syntaxin 3 |
| PUFAs | Polyunsaturated fatty acids |
| EFAs | Essential fatty acids |
| NF-kB | Nuclear factor-kappa B |
| IkB | Inhibitory kappa B |
| IL-6 | Interleukin-6 |
| TNF | Tumor necrosis factor |
References
- Watanabe, H.; Belyea, B.C.; Paxton, R.L.; Li, M.; Dzamba, B.J.; DeSimone, D.W.; Gomez, R.A.; Sequeira-Lopez, M.L.S. Renin Cell Baroreceptor, a Nuclear Mechanotransducer Central for Homeostasis. Circ. Res. 2021, 129, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Heesch, C.M. Reflexes that Control Cardiovascular Function. Am. J. Physiol. Content 1999, 277, S234. [Google Scholar] [CrossRef] [PubMed]
- Das, U. “Cell Membrane Theory of Senescence” and the Role of Bioactive Lipids in Aging, and Aging Associated Diseases and their Therapeutic Implications. Biomolecules 2021, 11, 241. [Google Scholar] [CrossRef]
- Lomakin, A.J.; Cattin, C.J.; Cuvelier, D.; Alraies, Z.; Molina, M.; Nader, G.P.F.; Srivastava, N.; Sáez, P.J.; Garcia-Arcos, J.M.; Zhitnyak, I.Y.; et al. The Nucleus Acts as a Ruler Tailoring Cell Responses to Spatial Constraints. Science 2020, 370, eaba2894. [Google Scholar] [CrossRef]
- Venturini, V.; Pezzano, F.; Castro, F.C.; Häkkinen, H.-M.; Jiménez-Delgado, S.; Colomer-Rosell, M.; Marro, M.; Tolosa-Ramon, Q.; Paz-López, S.; Valverde, M.A.; et al. The Nucleus Measures Shape Changes for Cellular Proprioception to Control Dynamic Cell Behavior. Science 2020, 370, eaba2644. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Wang, D.H. Inhibition of Renin Release by Arachidonic Acid Metabolites, 12(s)-HPETE and 12-HETE: Role of TRPV1 Channels. Endocrinology 2011, 152, 3811–3819. [Google Scholar] [CrossRef] [Green Version]
- Nasrallah, R.; Hébert, R.L. Prostacyclin Signaling in the Kidney: Implications for Health and Disease. Am. J. Physiol. Physiol. 2005, 289, F235–F246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonipillai, I.; Nadler, J.L.; Robin, E.C.; Horton, R. The Inhibitory Role of 12- and 15-Lipoxygenase Products on Renin Release. Hypertension 1987, 10, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerkens, J.; Williams, A.; Branch, R. Effect of Precursors of the 1, 2, and 3 Series Prostaglandins on Renin Release and Renal Blood Flow in the Dog. Prostaglandins 1981, 22, 513–520. [Google Scholar] [CrossRef]
- Weber, P.C. Renal Prostaglandins in the Control of Renin1. Contr. Nephrol. 1978, 12, 92–105. [Google Scholar] [CrossRef]
- Facemire, C.S.; Nguyen, M.; Jania, L.; Beierwaltes, W.H.; Kim, H.-S.; Koller, B.H.; Coffman, T.M. A Major Role for the EP4 Receptor in Regulation of Renin. Am. J. Physiol. Renal Physiol. 2011, 301, F1035–F1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzdorf, C.; Kurtz, A.; Höcherl, K. COX-2 Activity Determines the Level of Renin Expression but Is Dispensable for Acute Upregulation of Renin Expression in Rat Kidneys. Am. J. Physiol. Renal Physiol. 2007, 292, F1782–F1790. [Google Scholar] [CrossRef] [PubMed]
- Schweda, F.; Klar, J.; Narumiya, S.; Nüsing, R.M.; Kurtz, A. Stimulation of Renin Release by Prostaglandin E2 Is Mediated by EP2 and EP4 Receptors in Mouse Kidneys. Am. J. Physiol. Renal Physiol. 2004, 287, F427–F433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breyer, M.D.; Breyer, R. Prostaglandin E Receptors and the Kidney. Am. J. Physiol. Renal Physiol. 2000, 279, F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Vasandan, A.B.; Jahnavi, S.; Shashank, C.; Prasad, P.; Kumar, A.; Prasanna, S.J. Human Mesenchymal Stem Cells Program Macrophage Plasticity by Altering their Metabolic Status via a PGE2-Dependent Mechanism. Sci. Rep. 2016, 6, 38308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenberg, A.; Rezk, A.; Boivin, M.-N.; Darlington, P.J.; Nyirenda, M.; Li, R.; Jalili, F.; Winer, R.; Artsy, E.A.; Uccelli, A.; et al. Human Mesenchymal Stem Cells Impact Th17 and Th1 Responses Through a Prostaglandin E2 and Myeloid-Dependent Mechanism. Stem Cells Transl. Med. 2016, 5, 1506–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Németh, K.; Leelahavanichkul, A.; Yuen, P.S.; Mayer, B.; Parmelee, A.; Doi, K.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.M.; et al. Bone Marrow Stromal Cells Attenuate Sepsis via Prostaglandin E2–Dependent Reprogramming of Host Macrophages to Increase their Interleukin-10 Production. Nat. Med. 2008, 15, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ylöstalo, J.H.; Bartosh, T.J.; Coble, K.; Prockop, D.J. Human Mesenchymal Stem/Stromal Cells Cultured as Spheroids are Self-activated to Produce Prostaglandin E2 that Directs Stimulated Macrophages into an Anti-inflammatory Phenotype. Stem Cells 2012, 30, 2283–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poloso, N.J.; Urquhart, P.; Nicolaou, A.; Wang, J.; Woodward, D.F. PGE2 Differentially Regulates Monocyte-Derived Dendritic Cell Cytokine Responses Depending on Receptor Usage (EP2/EP4). Mol. Immunol. 2013, 54, 284–295. [Google Scholar] [CrossRef]
- Kalim, K.W.; Groettrup, M. Prostaglandin E2 inhibits IL-23 and IL-12 Production by Human Monocytes through Down-Regulation of their Common p40 Subunit. Mol. Immunol. 2013, 53, 274–282. [Google Scholar] [CrossRef]
- Loynes, C.A.; Lee, J.A.; Robertson, A.L.; Steel, M.J.; Ellett, F.; Feng, Y.; Levy, B.D.; Whyte, M.K.; Renshaw, S.A. PGE 2 Production at Sites of Tissue Injury Promotes an Anti-Inflammatory Neutrophil Phenotype and Determines the Outcome of Inflammation Resolution in Vivo. Sci. Adv. 2018, 4, eaar8320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
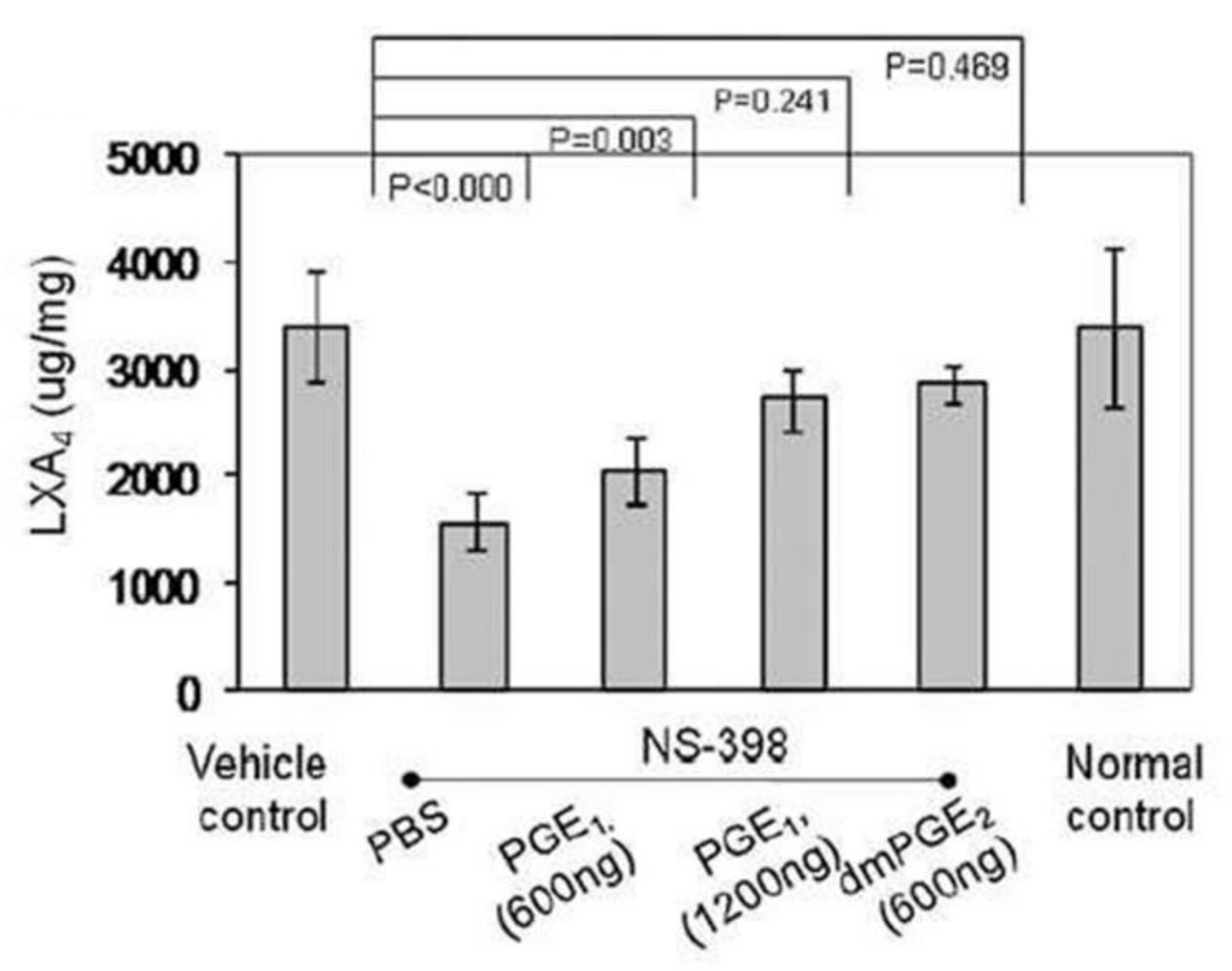
- Chan, M.M.-Y.; Moore, A.R. Resolution of Inflammation in Murine Autoimmune Arthritis Is Disrupted by Cyclooxygenase-2 Inhibition and Restored by Prostaglandin E2-Mediated Lipoxin A4 Production. J. Immunol. 2010, 184, 6418–6426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Kim, J.; Saima, F.T.; Rhee, K.-J.; Hwang, S.; Kim, M.Y.; Baik, S.K.; Eom, Y.W.; Kim, H.-S. Adipose-Derived Stem Cells Ameliorate Colitis by Suppression of Inflammasome Formation and Regulation of M1-Macrophage Population through Prostaglandin E2. Biochem. Biophys. Res. Commun. 2018, 498, 988–995. [Google Scholar] [CrossRef]
- An, J.-H.; Song, W.-J.; Li, Q.; Kim, S.-M.; Yang, J.-I.; Ryu, M.-O.; Nam, A.R.; Bhang, D.H.; Jung, Y.-C.; Youn, H.-Y. Prostaglandin E2 Secreted from Feline Adipose Tissue-Derived Mesenchymal Stem Cells Alleviate DSS-Induced Colitis by Increasing Regulatory T Cells in Mice. BMC Veter.-Res. 2018, 14, 354. [Google Scholar] [CrossRef] [PubMed]
- Terraza-Aguirre, C.; Campos-Mora, M.; Elizondo-Vega, R.; Contreras-López, R.; Luz-Crawford, P.; Jorgensen, C.; Djouad, F. Mechanisms behind the Immunoregulatory Dialogue between Mesenchymal Stem Cells and Th17 Cells. Cells 2020, 9, 1660. [Google Scholar] [CrossRef]
- Chiossone, L.; Conte, R.; Spaggiari, G.M.; Serra, M.; Romei, C.; Bellora, F.; Becchetti, F.; Andaloro, A.; Moretta, L.; Bottino, C. Mesenchymal Stromal Cells Induce Peculiar Alternatively Activated Macrophages Capable of Dampening Both Innate and Adaptive Immune Responses. Stem Cells 2016, 34, 1909–1921. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Qu, X.; Chen, Y.; Liao, L.; Cheng, K.; Shao, C.; Zenke, M.; Keating, A.; Zhao, R.C.H. Mesenchymal Stem/Stromal Cells Induce the Generation of Novel IL-10–Dependent Regulatory Dendritic Cells by SOCS3 Activation. J. Immunol. 2012, 189, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Yamane, H.; Sugimoto, Y.; Tanaka, S.; Ichikawa, A. Prostaglandin E(2) Receptors, EP2 and EP4, Differentially Modulate TNF-Alpha and IL-6 Production Induced by Lipopolysaccharide in Mouse Peritoneal Neutrophils. Biochem. Biophys. Res. Commun. 2000, 278, 224–228. [Google Scholar] [CrossRef]
- Das, U.N. Bioactive Lipids in Age-Related Disorders. Rev. New Drug Targets Age-Related Disord. 2020, 1260, 33–83. [Google Scholar] [CrossRef]
- Das, U.N. Essential Fatty Acids, Lipid Peroxidation and Apoptosis. Prostaglandins Leukot Essen Fat. Acids 1999, 61, 157–164. [Google Scholar] [CrossRef]
- Gilroy, D.; Colvillenash, P.R.; Willis, D.K.; Chivers, J.; Paulclark, M.J.; Willoughby, D. Inducible Cyclooxygenase May Have Anti-Inflammatory Properties. Nat. Med. 1999, 5, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Gundala, N.K.V.; Naidu, V.G.M.; Das, U.N. Arachidonic Acid and LipoxinA4 Attenuate Streptozotocin-Induced Cytotoxicity to RIN5 F Cells in Vitro and Type 1 and Type 2 Diabetes Mellitus in Vivo. Nutrition 2017, 35, 61–80. [Google Scholar] [CrossRef]
- Todorov, V.T.; Völkl, S.; Müller, M.; Bohla, A.; Klar, J.; Kunz-Schughart, L.A.; Hehlgans, T.; Kurtz, A. Tumor Necrosis Factor-Alpha Activates NFkappaB to Inhibit Renin Transcription by Targeting cAMP-Responsive Element. J. Biol. Chem. 2004, 279, 1458–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorov, V.T.; Völkl, S.; Friedrich, J.; Kunz-Schughart, L.A.; Hehlgans, T.; Vermeulen, L.; Haegeman, G.; Schmitz, M.L.; Kurtz, A. Role of CREB1 and NF {κ}B-p65 in the Down-Regulation of Renin Gene Expression by Tumor Necrosis Factor {α}. J. Biol. Chem. 2005, 280, 24356–24362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friis, U.G.; Jensen, B.L.; Sethi, S.; Andreasen, D.; Hansen, P.B.; Skøtt, O. Control of Renin Secretion from Rat Juxtaglomerular Cells by cAMP-Specific Phosphodiesterases. Circ Res. 2002, 90, 996–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, A. Renin Release: Sites, Mechanisms, and Control. Annu. Rev Physiol. 2011, 73, 377–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beierwaltes, W.H. The Role of Calcium in the Regulation of Renin Secretion. Am. J. Physiol. Physiol. 2010, 298, F1–F11. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, A.; Penner, R. Angiotensin II Induces Oscillations of Intracellular Calcium and Blocks Anomalous Inward Rectifying Potassium Current in Mouse Renal Juxtaglomerular Cells. Proc. Natl. Acad. Sci. USA 1989, 86, 3423–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritthaler, T.; Scholz, H.; Ackermann, M.; Riegger, G.; Kurtz, A.; Krämer, B.K. Effects of Endothelins on Renin Secretion from Isolated Mouse Renal Juxtaglomerular Cells. Am. J. Physiol. Content 1995, 268, F39–F45. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.Y.; Wang, Y.; Persson, A.E.G.; Manning, R.D., Jr.; Liu, R. Pressure Induces Intracellular Calcium Changes in Juxtaglomerular Cells in Perfused Afferent Arterioles. Hypertens. Res. 2011, 34, 942–948. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, A.; Della Bruna, R.; Pfeilschifter, J.; Taugner, R.; Bauer, C. Atrial Natriuretic Peptide Inhibits Renin Release from Juxtaglomerular Cells by a cGMP-Mediated Process. Proc. Natl. Acad. Sci. USA 1986, 83, 4769–4773. [Google Scholar] [CrossRef] [Green Version]
- Vandongen, R.; Peart, W.S.; Boyd, G.W. Adrenergic Stimulation of Renin Secretion in the Isolated Perfused Rat Kidney. Circ. Res. 1973, 32, 290–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbs, J.-L.; Schmidt, M.; Schwartz, J. Effect of Dopamine on Renin Secretion in the Anesthetized Dog. Eur. J. Pharmacol. 1975, 33, 151–157. [Google Scholar] [CrossRef]
- Holdaas, H.; Langaard, O.; Eide, I.; Kiil, F. Conditions for Enhancement of Renin Release by Isoproterenol, Dopamine, and Glucagon. Am. J. Physiol. Content 1982, 242, F267–F273. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, A.; Muff, R.; Born, W.; Lundberg, J.M.; I Millberg, B.; Gnädinger, M.P.; E Uehlinger, D.; Weidmann, P.; Hökfelt, T.; A Fischer, J. Calcitonin Gene-Related Peptide Is a Stimulator of Renin Secretion. J. Clin. Investig. 1988, 82, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, J.P.; Ganong, W.F. Vasoactive Intestinal Peptide and Renin Secretion. Ann. N. Y. Acad. Sci. 1988, 527, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Hautmann, M.; Friis, U.G.; Desch, M.; Todorov, V.; Castrop, H.; Segerer, F.; Otto, C.; Schutz, G.; Schweda, F. Pituitary Adenylate Cyclase–Activating Polypeptide Stimulates Renin Secretion via Activation of PAC1 Receptors. J. Am. Soc. Nephrol. 2007, 18, 1150–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackenthal, E.; Aktories, K.; Jakobs, K.H.; E Lang, R. Neuropeptide Y Inhibits Renin Release by a Pertussis Toxin-Sensitive Mechanism. Am. J. Physiol. Content 1987, 252, F543–F550. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, A.; Michel, M.C. Renal effects of neuropeptide Y. Pflügers Archiv 1998, 435, 443–453. [Google Scholar] [CrossRef]
- Boivin, V.; Jahns, R.; Gambaryan, S.; Ness, W.; Boege, F.; Lohse, M.J. Immunofluorescent Imaging of Beta 1- and Beta 2-Adrenergic Receptors in Rat Kidney. Kidney Int. 2001, 59, 515–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinos, P.T.; Dimitriadis, K. Radiance-Htn Trio: How the Saga of Renal Denervation Revisits Hypertensiontherapy. Cardiovasc. Res. 2021, 117, e141–e143. [Google Scholar]
- Castrop, H.; Höcherl, K.; Kurtz, A.; Schweda, F.; Todorov, V.; Wagner, C. Physiology of Kidney Renin. Physiol. Rev. 2010, 90, 607–673. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.L.; Schmid, C.; Kurtz, A. Prostaglandins Stimulate Renin Secretion and Renin mRNA in Mouse Renal Juxtaglomerular Cells. Am. J. Physiol. Physiol. 1996, 271, F659–F669. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, A.; Götz, K.-H.; Hamann, M.; Wagner, C. Stimulation of Renin Secretion by Nitric Oxide Is Mediated by Phosphodiesterase 3. Proc. Natl. Acad. Sci. USA 1998, 95, 4743–4747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez, M.; Gross, K.W.; Glenn, S.T.; Garvin, J.L.; Carretero, O.A. Vesicle-Associated Membrane Protein-2 (VAMP2) Mediates cAMP-Stimulated Renin Release in Mouse Juxtaglomerular Cells. J. Biol. Chem. 2011, 286, 28608–28618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez, M.; Gaisano, H.Y. Role of the SNARE Protein SNAP23 on cAMP-Stimulated Renin Release in Mouse Juxtaglomerular Cells. Am. J. Physiol. Ren. Physiol. 2013, 304, F498–F504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goda, Y. SNAREs and Regulated Vesicle Exocytosis. Proc. Natl. Acad. Sci. USA 1997, 94, 769–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, A. Control of Renin Synthesis and Secretion. Am. J. Hypertens. 2012, 25, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez, M. Renin Release: Role of SNAREs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R484–R486. [Google Scholar] [CrossRef] [PubMed]
- Latham, C.F.; Osborne, S.L.; Cryle, M.J.; Meunier, F.A. Arachidonic Acid Potentiates Exocytosis and Allows Neuronal SNARE Complex to Interact with Munc18a. J. Neurochem. 2006, 100, 1543–1554. [Google Scholar] [CrossRef]
- Chen, Y.A.; Scheller, R.H. SNARE-Mediated Membrane Fusion. Nat. Rev. Mol. Cell Biol. 2001, 2, 98–106. [Google Scholar] [CrossRef]
- Lang, T.; Bruns, D.; Wenzel, D.; Riedel, D.; Holroyd, P.; Thiele, C.; Jahn, R. SNAREs are Concentrated in Cholesterol-Dependent Clusters that Define Docking and Fusion Sites for Exocytosis. EMBO J. 2001, 20, 2202–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owicki, J.C.; McConnell, H.M. Theory of Protein-Lipid and Protein-Protein Interactions in Bilayer Membranes. Proc. Natl. Acad. Sci. USA 1979, 76, 4750–4754. [Google Scholar] [CrossRef] [Green Version]
- Weber, T.; Zemelman, B.; McNew, J.; Westermann, B.; Gmachl, M.; Parlati, F.; Söllner, T.H.; Rothman, J.E. SNAREpins: Minimal Machinery for Membrane Fusion. Cell 1998, 92, 759–772. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, B.L. SNARES in Neurons–Beyond Synaptic Vesicle Exocytosis. Mol Membr Biol. 2006, 23, 377–384. [Google Scholar] [CrossRef]
- Söllner, T.; Whiteheart, S.; Brunner, M.; Erdjument-Bromage, H.; Geromanos, S.; Tempst, P.; Rothman, J.E. SNAP Receptors Implicated in Vesicle Targeting and Fusion. Nature 1993, 362, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.J.; Ungermann, C.; Pelham, H.R.B.; Wickner, W.T.; Haas, A. Homotypic Vacuolar Fusion Mediated by t- and v-SNAREs. Nature 1997, 387, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Jahn, R.; Scheller, R.H. SNARES–Engines for Membrane Fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.Y.H.; Wang, Y.; Tang, B.L. The Syntaxins. Genome Biol. 2001, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Connell, E.; Darios, F.; Broersen, K.; Gatsby, N.; Peak-Chew, S.; Rickman, C.; Davletov, B. Mechanism of Arachidonic Acid Action on Syntaxin–Munc18. EMBO Rep. 2007, 8, 414–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darios, F.; Davletov, B. Omega-3 and Omega-6 Fatty Acids Stimulate Cell Membrane Expansion by Acting on Syntaxin 3. Nature 2006, 440, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Connell, E.; Davletov, B. Phospholipases and Fatty Acid Signalling in Exocytosis. J. Physiol. 2007, 585, 699–704. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Sun, P.; Wang, P.; Abonyo, B.; Cross, N.L.; Liu, L. Fusion of Lamellar Body with Plasma Membrane Is Driven by the Dual Action of Annexin II Tetramer and Arachidonic Acid. J. Biol. Chem. 2003, 278, 39675–39683. [Google Scholar] [CrossRef] [Green Version]
- Darios, F.; Ruipérez, V.; López, I.; Villanueva, J.; Gutierrez, L.M.; Davletov, B. Alpha-Synuclein Sequesters Arachidonic Acid to Modulate SNARE-Mediated Exocytosis. EMBO Rep. 2010, 11, 528–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickman, C.; Davletov, B. Arachidonic Acid Allows SNARE Complex Formation in the Presence of Munc18. Chem. Biol. 2005, 12, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Gerber, S.H.; Suüdhof, T.C. Molecular Determinants of Regulated Exocytosis. Diabetes 2002, 51, S3–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizo, J.; Südhof, T.C. Snares and munc18 in Synaptic Vesicle Fusion. Nat. Rev. Neurosci. 2002, 3, 641–653. [Google Scholar] [CrossRef]
- Bossi, G.; Trambas, C.; Booth, S.; Clark, R.; Stinchcombe, J.; Griffiths, G.M. The Secretory Synapse: The Secrets of a Serial Killer. Immunol. Rev. 2002, 189, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Sheetz, M.P. Cell Control by Membrane–Cytoskeleton Adhesion. Nat. Rev. Mol. Cell Biol. 2001, 2, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Finger, F.P.; White, J.G. Fusion and Fission: Membrane Trafficking in Animal Cytokinesis. Cell 2002, 108, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Bednarek, S.Y.; Falbel, T.G. Membrane Trafficking during Plant Cytokinesis. Traffic 2002, 3, 621–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiliotis, E.T.; Nelson, W.J. Spatial Control of Exocytosis. Curr. Opin. Cell Biol. 2003, 15, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Drubin, D.G.; Nelson, W. Origins of Cell Polarity. Cell 1996, 84, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Juhl, K.; Høy, M.; Olsen, H.L.; Bokvist, K.; Efanov, A.M.; Hoffmann, E.K.; Gromada, J. cPLA2alpha-Evoked Formation of Arachidonic Acid and Lysophospholipids Is Required for Exocytosis in Mouse Pancreatic Beta-Cells. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E73–E81. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.M.; Persaud, S.J. Arachidonic Acid as a Second Messenger in Glucose-Induced Insulin Secretion from Pancreatic Beta-Cells. J. Endocrinol. 1993, 137, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Persaud, S.J.; Muller, D.; Belin, V.D.; Kitsou-Mylona, I.; Asare-Anane, H.; Papadimitriou, A.; Burns, C.J.; Huang, G.C.; Amiel, S.A.; Jones, P.M. The Role of Arachidonic Acid and Its Metabolites in Insulin Secretion from Human Islets of Langerhans. Diabetes 2007, 56, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Ahrén, B.; Magrum, L.J.; Havel, P.J.; Greene, S.F.; Phinney, S.D.; Johnson, P.R.; Stern, J.S. Augmented Insulinotropic Action of Arachidonic Acid through the Lipoxygenase Pathway in the Obese Zucker Rat. Obes. Res. 2000, 8, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Jayaraja, S.; Dakhama, A.; Yun, B.; Ghosh, M.; Lee, H.; Redente, E.F.; Uhlson, C.L.; Murphy, R.C.; Leslie, C.C. Cytosolic Phospholipase A2 Contributes to Innate Immune Defense against Candida Albicans Lung Infection. BMC Immunol. 2016, 17, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suram, S.; Silveira, L.J.; Mahaffey, S.; Brown, G.D.; Bonventre, J.V.; Williams, D.L.; Gow, N.A.R.; Bratton, D.L.; Murphy, R.C.; Leslie, C.C. Cytosolic Phospholipase A2α and Eicosanoids Regulate Expression of Genes in Macrophages Involved in Host Defense and Inflammation. PLoS ONE 2013, 8, e69002. [Google Scholar] [CrossRef] [Green Version]
- Clancy, R.M.; Dahinden, C.A.; Hugli, T.E. Complement-Mediated Arachidonate Metabolism. Prog. Biochem. Pharmacol. 1985, 20, 120–131. [Google Scholar]
- Duvall, M.G.; Fuhlbrigge, M.E.; Reilly, R.B.; Walker, K.H.; Kılıç, A.; Levy, B.D. Human NK Cell Cytoskeletal Dynamics and Cytotoxicity Are Regulated by LIM Kinase. J. Immunol. 2020, 205, 801–810. [Google Scholar] [CrossRef]
- Putta, P.; Smith, A.H.; Chaudhuri, P.; Guardia-Wolff, R.; Rosenbaum, M.A.; Graham, L.M. Activation of the Cytosolic Calcium Independent Phospholipase A2 β Isoform Contributes to TRPC6 Externalization via Release of Arachidonic Acid. J. Biol. Chem. 2021, 297, 101180. [Google Scholar] [CrossRef] [PubMed]
- Sheppe, A.E.F.; Edelmann, M.J. Roles of Eicosanoids in Regulating Inflammation and Neutrophil Migration as an Innate Host Response to Bacterial Infections. Infect. Immun. 2021, 89, e00095-21. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wang, Z.; Ma, R.; Chen, Y.; Yan, Y.; Miao, S.; Jiao, J.; Cheng, X.; Kong, L.; Ye, D. Lipoxin A4 Protects against Lipopolysaccharide-Induced Sepsis by Promoting Innate Response Activator B Cells Generation. Int. Immunopharmacol. 2016, 39, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Inflammatory bowel disease as a disorder of an imbalance between pro- and anti-inflammatory molecules and deficiency of resolution bioactive lipids. Lipids Health Dis. 2016, 15, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, B.S.; Kantarci, A.; Zarrough, A.; Hasturk, H.; Leung, K.P.; Van Dyke, T.E. LXA4 Actions Direct Fibroblast Function and Wound Closure. Biochem. Biophys. Res. Commun. 2015, 464, 1072–1077. [Google Scholar] [CrossRef] [Green Version]
- Babbin, B.A.; Laukoetter, M.G.; Nava, P.; Koch, S.; Lee, W.Y.; Capaldo, C.T.; Peatman, E.; Severson, E.A.; Flower, R.J.; Perretti, M.; et al. Annexin A1 Regulates Intestinal Mucosal Injury, Inflammation, and Repair. J. Immunol. 2008, 181, 5035–5044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, U.N. Bioactive Lipids as Modulators of Immune Check Point Inhibitors. Med. Hypotheses 2020, 135, 109473. [Google Scholar] [CrossRef]
- Das, U.N. Essential Fatty Acids in Health and Disease. J. Assoc. Physicians India 1999, 47, 906–911. [Google Scholar]
- Poorani, R.; Bhatt, A.N.; Dwarakanath, B.; Das, U.N. COX-2, Aspirin and Metabolism of Arachidonic, Eicosapentaenoic and Docosahexaenoic Acids and their Physiological and Clinical Significance. Eur. J. Pharmacol. 2016, 785, 116–132. [Google Scholar] [CrossRef]
- Morise, T.; Miyamori, I.; Ikeda, M.; Takeda, Y.; Koshida, H.; Yasuhara, S.; Takeda, R. The Role of Prostacyclin (PGI2) in the Regulation of Blood Pressure and Aldosterone Response to Angiotensin II. Folia Endocrinol. Jpn. 1983, 59, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Schölkens, B.A. Antihypertensive Effect of Prostacyclin (PGI2) in Experimental Hypertension and its Influence on Plasma Renin Activity in Rats. Prostaglandins Med. 1978, 1, 359–372. [Google Scholar] [CrossRef]
- Frölich, J.C. Prostacyclin in Hypertension. Int. Soc. Hypertens. 1990, 8, S73–S78. [Google Scholar]
- Das, U.N. Essential Fatty Acids and their Metabolites Could Function as Endogenous HMG-CoA Reductase and ACE Enzyme Inhibitors, Anti-Arrhythmic, Anti-Hypertensive, Anti-Atherosclerotic, Anti-Inflammatory, Cytoprotective, and Cardioprotective Molecules. Lipids Health Dis. 2008, 7, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graciano, M.F.R.; Leonelli, M.; Curi, R.; Carpinelli, A. Omega-3 Fatty Acids Control Productions of Superoxide and Nitrogen Oxide and Insulin Content in INS-1E Cells. J. Physiol. Biochem. 2016, 72, 699–710. [Google Scholar] [CrossRef]
- Tanaka, S.; Ito, S.; Shimamoto, C.; Matsumura, H.; Inui, T.; Marunaka, Y.; Nakahari, T. Nitric Oxide Synthesis Stimulated by Arachidonic Acid Accumulation via PPARα in Acetylcholine-Stimulated Gastric Mucous Cells. Exp. Physiol. 2021, 106, 1939–1949. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic Acid Evokes an Increase in Intracellular Ca2+ Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef] [Green Version]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of Membrane/Lipid Rafts with the Cytoskeleton: Impact on Signaling and Function. Biochim. Biophys. Acta Biomembr. 2013, 1838, 532–545. [Google Scholar] [CrossRef] [Green Version]
- Head, B.P.; Patel, H.; Roth, D.M.; Murray, F.; Swaney, J.S.; Niesman, I.R.; Farquhar, M.G.; Insel, P.A. Microtubules and Actin Microfilaments Regulate Lipid Raft/Caveolae Localization of Adenylyl Cyclase Signaling Components. J. Biol. Chem. 2006, 281, 26391–26399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Persson, S.; Zhang, Y. The Connection of Cytoskeletal Network with Plasma Membrane and the Cell Wall. J. Integr. Plant Biol. 2015, 57, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Luna, E.J.; Hitt, A.L. Cytoskeleton--Plasma Membrane Interactions. Science 1992, 258, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Di Giaimo, R.; Penna, E.; Pizzella, A.; Cirillo, R.; Perrone-Capano, C.; Crispino, M. Cross Talk at the Cytoskeleton-Plasma Membrane Interface: Impact on Neuronal Morphology and Functions. Int. J. Mol. Sci. 2020, 21, 9133. [Google Scholar] [CrossRef]
- Saarikangas, J.; Zhao, H.; Lappalainen, P. Regulation of the Actin Cytoskeleton-Plasma Membrane Interplay by Phosphoinositides. Physiol. Rev. 2010, 90, 259–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raucher, D.; Stauffer, T.; Chen, W.; Shen, K.; Guo, S.; York, J.D.; Sheetz, M.P.; Meyer, T. Phosphatidylinositol 4,5-Bisphosphate Functions as a Second Messenger that Regulates Cytoskeleton-Plasma Membrane Adhesion. Cell 2000, 100, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Ammar, M.R.; Kassas, N.; Chasserot-Golaz, S.; Bader, M.-F.; Vitale, N. Lipids in Regulated Exocytosis: What are They Doing? Front. Endocrinol. 2013, 4, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, U.N.; Ramesh, G.; Kumar, G.S.; Madhavi, N.; Kumar, K.V.; Sagar, P.S.; Koratkar, R.; Padma, M. Free Radicals, Lipid Peroxidation and Essential Fatty Acids in Patients with Pneumonia, Septicemia and Collagen Vascular Diseases. J. Nutr. Med. 1992, 3, 117–127. [Google Scholar] [CrossRef]
- Das, U. Essential Fatty Acid Metabolism in Patients with Essential Hypertension, Diabetes Mellitus and Coronary Heart Disease. Prostaglandins Leukot. Essent. Fat. Acids 1995, 52, 387–391. [Google Scholar] [CrossRef]
- Das, U.N. Bioactive Lipids and Vascular Disease. Eur. J. Clin. Nutr. 2021, 75, 1528–1531. [Google Scholar] [CrossRef]
- Das, U. Cross Talk Among Leukocytes, Platelets, and Endothelial Cells and its Relevance to Atherosclerosis and Coronary Heart Disease. Curr. Nutr. Food Sci. 2009, 5, 75–93. [Google Scholar] [CrossRef]
- Cornwell, D.G.; Panganamala, R.V. Atherosclerosis: An Intracellular Deficiency in Essential Fatty Acids. Prog. Lipid Res. 1981, 20, 365–376. [Google Scholar] [CrossRef]
- Das, U.N. Essential Fatty Acids -A Review. Curr. Pharm. Biotechnol. 2006, 7, 467–482. [Google Scholar] [CrossRef]
- Das, U.N. Essential fatty Acids Enhance Free Radical Generation and Lipid Peroxidation to Induce Apoptosis of Tumor Cells. Clin. Lipidol. 2011, 6, 463–489. [Google Scholar] [CrossRef]
- Tateishi, N.; Kakutani, S.; Kawashima, H.; Shibata, H.; Morita, I. Dietary Supplementation of Arachidonic Acid Increases Arachidonic Acid and Lipoxin A4 Contents in Colon, but Does not Affect Severity or Prostaglandin E2 Content in Murine Colitis Model. Lipids Health Dis. 2014, 13, 30. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, N.; Kaneda, Y.; Kakutani, S.; Kawashima, H.; Shibata, H.; Morita, I. Dietary Supplementation with Arachidonic Acid Increases Arachidonic Acid Content in Paw, but Does not Affect Arthritis Severity or Prostaglandin E2 Content in Rat Adjuvant-Induced Arthritis Model. Lipids Health Dis. 2015, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Kakutani, S.; Ishikura, Y.; Tateishi, N.; Horikawa, C.; Tokuda, H.; Kontani, M.; Kawashima, H.; Sakakibara, Y.; Kiso, Y.; Shibata, H.; et al. Supplementation of Arachidonic Acid-Enriched Oil Increases Arachidonic Acid Contents in Plasma Phospholipids, but Does not Increase their Metabolites and Clinical Parameters in Japanese Healthy Elderly Individuals: A Randomized Controlled Study. Lipids Health Dis. 2011, 10, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathina, S.; Das, U. Resolvin D1 Decreases Severity of Streptozotocin-Induced Type 1 Diabetes Mellitus by Enhancing BDNF Levels, Reducing Oxidative Stress, and Suppressing Inflammation. Int. J. Mol. Sci. 2021, 22, 1516. [Google Scholar] [CrossRef] [PubMed]
- Haworth, O.; Cernadas, M.; Yang, R.; Serhan, C.N.; Levy, B.D. Resolvin E1 Regulates Interleukin 23, Interferon-Gamma and Lipoxin A4 to Promote the Resolution of Allergic Airway Inflammation. Nat. Immunol. 2008, 9, 873–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathina, S.; Gundala, N.K.; Rhenghachar, P.; Polavarapu, S.; Hari, A.D.; Sadananda, M.; Das, U.N. Resolvin D1 Ameliorates Nicotinamide-Streptozotocin-Induced Type 2 Diabetes Mellitus by its Anti-inflammatory Action and Modulating PI3K/Akt/mTOR Pathway in the Brain. Arch. Med. Res. 2020, 51, 492–503. [Google Scholar] [CrossRef]
- Bathina, S.; Das, U.N. PUFAs, BDNF and Lipoxin A4 Inhibit Chemical-Induced Cytotoxicity of RIN5F Cells in Vitro and Streptozotocin-Induced Type 2 Diabetes Mellitus in Vivo. Lipids Health Dis. 2019, 18, 214. [Google Scholar] [CrossRef] [Green Version]
- Gundala, N.K.V.; Naidu, V.G.M.; Das, U.N. Arachidonic Acid and Lipoxin A4 Attenuate Alloxan-Induced Cytotoxicity to RIN5F Cells in Vitro and Type 1 Diabetes Mellitus in Vivo. Biofactors 2017, 43, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.S.; Mozaffarian, D.; Rimm, E.; Kris-Etherton, P.; Rudel, L.L.; Appel, L.J.; Engler, M.M.; Engler, M.B.; Sacks, F. Omega-6 Fatty Acids and Risk for Cardiovascular Disease: A Science Advisory from the American Heart Association Nutrition SubCommittee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Cardiovascular Nursing; and Council on Epidemiology and Prevention. Circulation 2009, 119, 902–907. [Google Scholar]
- Nelson, G.J.; Schmidt, P.C.; Bartolini, G.; Kelley, D.S.; Kyle, D. The Effect of Dietary Arachidonic Acid on Platelet Function, Platelet Fatty Acid Composition, and Blood Coagulation in Humans. Lipids 1997, 32, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Wilborn, C.M.; Roberts, C.; Kerksick, M.I.; Taylor, L.; Campbell, B.; Kreider, R. Changes in Whole Blood and Clinical Safety Markers over 50 Days of Concomitant Arachidonic Acid Supplementation and Resistance Training. In Proceedings of the International Society of Sports Nutrition Archived, Clearwater Beach, FL, USA, 9–11 June 2006. [Google Scholar]
- De Souza, E.O.; Lowery, R.P.; Wilson, J.M.; Sharp, M.H.; Mobley, C.B.; Fox, C.D.; Lopez, H.L.; Shields, K.A.; Rauch, J.T.; Healy, J.C.; et al. Effects of Arachidonic Acid Supplementation on Acute Anabolic Signaling and Chronic Functional Performance and Body Composition Adaptations. PLoS ONE 2016, 11, e0155153. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fatty Acid | Control | HTN | CHD | Type | 2 DM | Diabetic Nephropathy |
|---|---|---|---|---|---|---|
| 16:0 | 25.9 ± 3.0 | 29.3 ± 2.7 * | 27.8 ± 3.5 | 26.6 | ± 5.2 | 26.8 ± 2.7 |
| 18:0 | 20.9 ± 3.6 | 23.2 ± 4.9 * | 18:0 ± 10.7 | 14.6 | ± 4.1 | 11.6 ± 3.6 * |
| 18:1 n-9 | 13.0 ± 2.3 | 12.1 ± 1.5 | 11.5 ± 3.1 | 12.0 | ± 2.6 | 14.5 ± 3.1 |
| 18:2 n-6 (LA) | 18.6 ± 3.1 | 14.5 ± 3.1 * | 17.8 ± 5.0 | 13.9 | ± 5.3 | 15.1 ± 3.1 |
| 18:3 n-6 (GLA) | 0.14 ± 0.1 | 0.4 ± 0.3 * | 0.1 ± 0.1 * | 0.2 ± 0.3 | 0.1 ± 0.2 | |
| 20:3 n-6 (DGLA) | 3.4 ± 1.0 | 3.1 ± 0.9 | 2.7 ± 1.1 | 1.7 ± 1.0 * | 2.0 ± 0.8 * | |
| 20:4 n-6 (AA) | 9.4 ± 1.8 | 7.8 ± 2.0 * | 7.0 ± 2.1 * | 4.6 ± 1.8 * | 6.6 ± 2.6 * | |
| 22:5 n-6 | 0.7 ± 0.4 | 0.4 ± 0.4 * | 1.0 ± 0.9 | 2.1 ± 0.6 * | 1.3 ± 0.5 * | |
| 18:3 n-6/18:2 n-6 | 0.008 | 0.026 | 0.005 | 0.017 | 0.008 | |
| 20:4 n-6/18:2 n-6 | 0.51 | 0.54 | 0.39 | 0.33 | 0.43 | |
| 20:4 n-6/20:3 -6 | 2.8 | 2.53 | 2,59 | 2.8 | 3.3 | |
| 18:3 n-3 (ALA) | 0.2 ± 0.1 | 0.4 ± 0.2 * | 0.3 ± 0.5 | 0.1 ± 0.2 * | 0.1 ± 0.1 * | |
| 20:5 n-3 (EPA) | 0.4 ± 0.4 | 0.6 ± 0.6 | 0.1 ± 0.2 * | 0.3 ± 0.3 | 0.2 ± 0.3 | |
| 22:5 n-3 | 0.5 ± 0.2 | 0.4 ± 0.5 | 0.3 ± 0.3 * | 1.6 ± 1.3 | 1.7 ± 1.1 | |
| 22:6 n-3 (DHA) | 1.4 ± 0.5 | 1.2 ± 0.6 | 0.8 ± 0.4 * | 0.5 ± 0.4 * | 0.5 ± 0.3 * | |
| 20:5 n-3/18:3 n-3 | 1.8 | 1.39 | 0.41 | 3.2 | 4.0 | |
| Control (n = 10) | Pneumonia (n = 12) | Septicemia (n = 14) | RA (n = 12) | SLE (Lupus) (n = 5) | |
|---|---|---|---|---|---|
| 16:0 | 24.8 ± 3.4 | 32.5 ± 3.6 | 26.95 ± 4.1 | 30.2 ± 3.0 | 32.0 ± 3.75 |
| 18:0 | 23.3 ± 4.1 | 21.4 ± 7.1 | 24.58 ± 6.0 | 19.0 ±6.1 | 14.6 ± 5.82 |
| 18:1 n-9 | 13.1 ± 2.3 | 15.6 ± 3.2 | 16.5 ± 3.3 * | 14.8 ± 2.1 | 16.0 ± 2.78 |
| 18:2 n-6 | 17.7 ± 3.1 | 14.2 ± 0.3 * | 16.3 ± 2.4 | 17.5 ± 2.7 | 20.8 ± 2.2 |
| 18:3 n-6 | 0.13 ± 0.09 | 0.13 ± 0.08 | 0.04 ± 0.05 * | 0.02 ± 0.04 ** | 0.01 ± 0.01 ** |
| 20:3 n-6 | 3.2 ± 0.79 | 1.5 ± 0.4 * | 0.46 ± 0.54 * | 2.5 ± 0.58 | 2.12 ± 0.52 |
| 20:4 n-6 | 8.8± 2.0 | 5.1 ± 0.4 * | 5.8 ± 1.6 * | 9.5 ± 2.2 | 8.93 ± 2.0 |
| 22:4 n-6 | 0.42 ± 0.23 | 0.8 ± 0.9 | 0.34 ± 0.28 | 0.26 ± 0.37 ** | 0.18 ± 0.18 ** |
| 22:5 n-6 | 0.73 ± 0.55 | 0.45 ± 0.63 | 1.5 ± 1.02 * | 0.6 ± 0.7 | 0.8 ± 1.0 |
| 18:3 n-3 | 0.27 ± 0.12 | 0.09 ± 0.04 * | 0.16 ± 0.11 * | 0.12 ± 0.16 * | 0.1 ± 0.1 * |
| 20:5 n-3 | 0.25 ± 0.26 | 0.23 ± 0.24 | 0.01 ± 0.01 * | 0.05 ± 0.14 ** | 0.04 ± 0.04 ** |
| 22:6 n-3 | 1.43 ± 0.43 | 0.54 ± 0.43 * | 1.2 ± 1.14 | 0.62 ± 0.56 * | 0.88 ± 0.75 * |
| Measurement (Fatty Acid) | Normal Intact Liver | Intact Yoshida Cells | Normal Liver Microsomes | Yoshida Microsomes |
|---|---|---|---|---|
| 16:0 | 18.5 ± 0.2 | 18.7 ± 2.0 | 18.9 ± 1.1 | 18.5 ±0.5 |
| 18:0 | 17.5 ± 0.5 | 13.3 ± 1.1 | 22.0 ± 3.0 | 13.7 ± 0.2 |
| 18:1, n-9 (oleic acid) | 12.1 ± 1.0 | 21.5 ± 0.8 | 8.6 ± 1.0 | 18.1 ± 0.3 |
| 20:4 (AA) | 16.7 ± 2.4 | 8.7 ± 0.7 | 19.1 ± 2.4 | 9.6 ± 0.8 |
| 22:5 | - | 2.9 ± 0.1 | - | 2.4 ± 0.3 |
| 22:6 (DHA) | 6.3 ± 0.2 | 5.2 ± 0.6 | 6.1 ± 0.3 | 5.3 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, U.N. Arachidonic Acid as Mechanotransducer of Renin Cell Baroreceptor. Nutrients 2022, 14, 749. https://doi.org/10.3390/nu14040749
Das UN. Arachidonic Acid as Mechanotransducer of Renin Cell Baroreceptor. Nutrients. 2022; 14(4):749. https://doi.org/10.3390/nu14040749
Chicago/Turabian StyleDas, Undurti N. 2022. "Arachidonic Acid as Mechanotransducer of Renin Cell Baroreceptor" Nutrients 14, no. 4: 749. https://doi.org/10.3390/nu14040749
APA StyleDas, U. N. (2022). Arachidonic Acid as Mechanotransducer of Renin Cell Baroreceptor. Nutrients, 14(4), 749. https://doi.org/10.3390/nu14040749