
Sex, Nutrition, and NAFLD: Relevance of Environmental Pollution



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nutrition, Sex Differences, and NAFLD
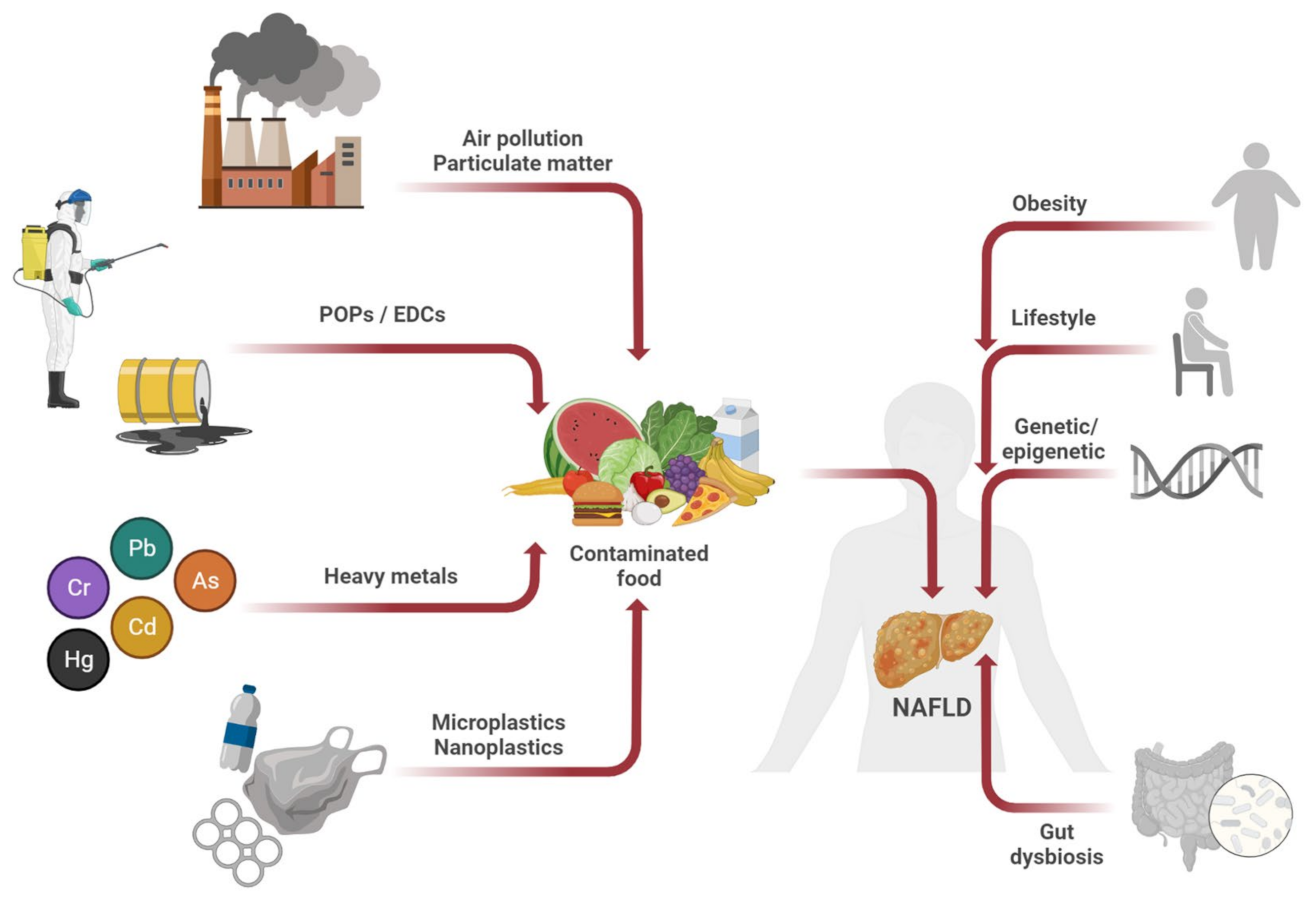
3. Nutrition, Environmental Pollutants, and NAFLD
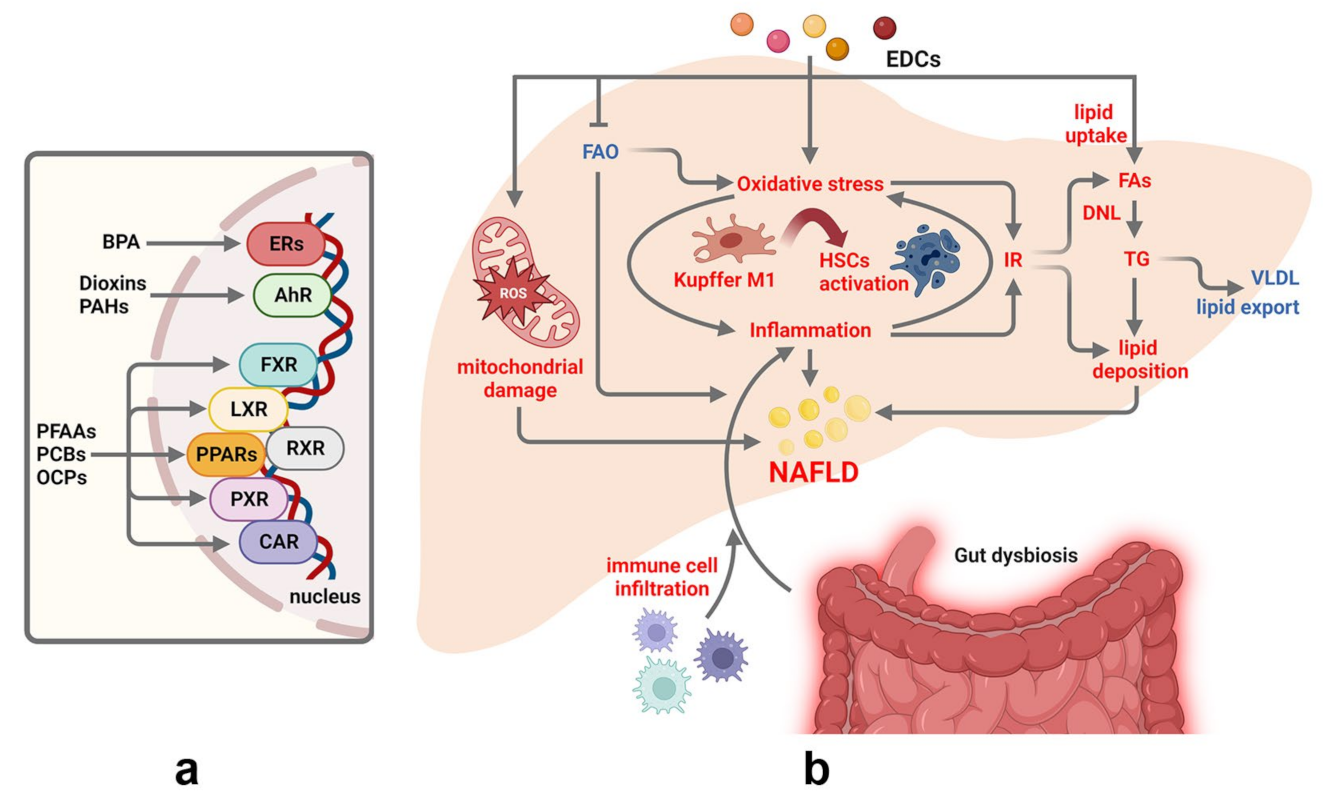
3.1. Persistent Endocrine Disrupting Chemicals
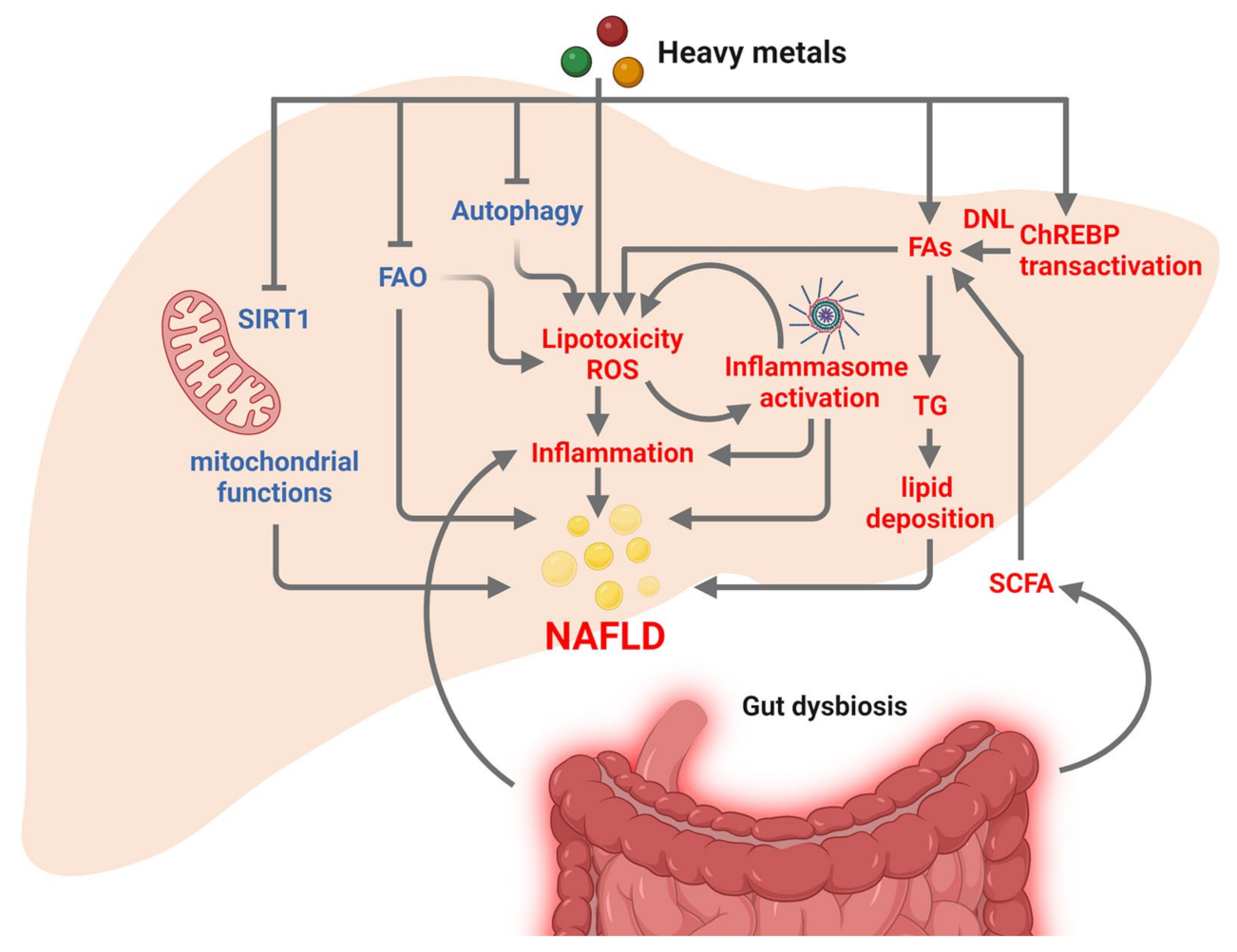
3.2. Heavy Metals
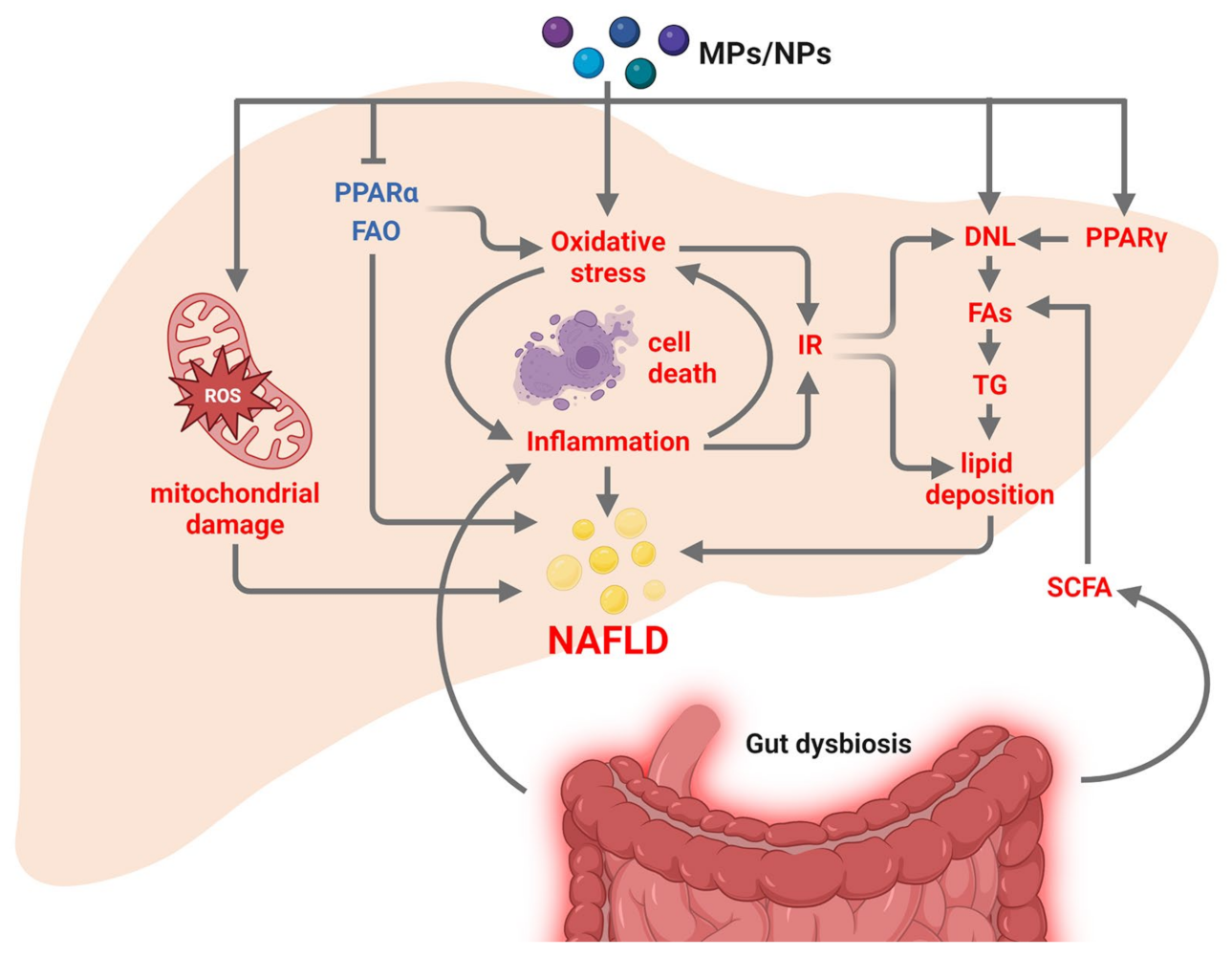
3.3. Microplastics and Nanoplastics
3.4. Air Particulate Matter
4. Climate Change, Food Insecurity, and NAFLD
5. Dietary Intake of Environmental Pollutants, Female Subfertility, and NAFLD
6. Maternal Exposure to Pollutants and Developmental Origins of NAFLD
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The Global Epidemiology of Nonalcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH): A Systematic Review. Hepatol. Baltim. Md 2023, 77, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Corey, K.E.; Byrne, C.D.; Roden, M. The Complex Link between NAFLD and Type 2 Diabetes Mellitus—Mechanisms and Treatments. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Scorletti, E.; Mosca, A.; Alisi, A.; Byrne, C.D.; Targher, G. Complications, Morbidity and Mortality of Nonalcoholic Fatty Liver Disease. Metabolism 2020, 111, 154170. [Google Scholar] [CrossRef]
- Adams, L.A.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-Alcoholic Fatty Liver Disease and Its Relationship with Cardiovascular Disease and Other Extrahepatic Diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.-S.; Rinella, M. Non-Alcoholic Fatty Liver Disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef] [PubMed]
- Monserrat-Mesquida, M.; Quetglas-Llabrés, M.; Abbate, M.; Montemayor, S.; Mascaró, C.M.; Casares, M.; Tejada, S.; Abete, I.; Zulet, M.A.; Tur, J.A.; et al. Oxidative Stress and Pro-Inflammatory Status in Patients with Non-Alcoholic Fatty Liver Disease. Antioxidants 2020, 9, 759. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic Reticulum Stress Signalling and the Pathogenesis of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and Resolution of Inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The Role of Macrophages in Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Luci, C.; Bourinet, M.; Leclère, P.S.; Anty, R.; Gual, P. Chronic Inflammation in Non-Alcoholic Steatohepatitis: Molecular Mechanisms and Therapeutic Strategies. Front. Endocrinol. 2020, 11, 597648. [Google Scholar] [CrossRef] [PubMed]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.-A. The Prevalence and Incidence of NAFLD Worldwide: A Systematic Review and Meta-Analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.; Wang, S.; Friedman, S.L. Ten Thousand Points of Light: Heterogeneity Among the Stars of NASH Fibrosis. Hepatology 2021, 74, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Jonas, W.; Schürmann, A. Genetic and Epigenetic Factors Determining NAFLD Risk. Mol. Metab. 2021, 50, 101111. [Google Scholar] [CrossRef]
- Choudhary, N.S.; Duseja, A. Genetic and Epigenetic Disease Modifiers: Non-Alcoholic Fatty Liver Disease (NAFLD) and Alcoholic Liver Disease (ALD). Transl. Gastroenterol. Hepatol. 2021, 6, 2. [Google Scholar] [CrossRef]
- Berná, G.; Romero-Gomez, M. The Role of Nutrition in Non-Alcoholic Fatty Liver Disease: Pathophysiology and Management. Liver Int. Off. J. Int. Assoc. Study Liver 2020, 40 (Suppl. 1), 102–108. [Google Scholar] [CrossRef]
- Yki-Järvinen, H.; Luukkonen, P.K.; Hodson, L.; Moore, J.B. Dietary Carbohydrates and Fats in Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 770–786. [Google Scholar] [CrossRef]
- Hallsworth, K.; Adams, L.A. Lifestyle Modification in NAFLD/NASH: Facts and Figures. JHEP Rep. Innov. Hepatol. 2019, 1, 468–479. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut Microbiota and Human NAFLD: Disentangling Microbial Signatures from Metabolic Disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The Role of the Microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin Resistance Drives Hepatic de Novo Lipogenesis in Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and Sugar: A Major Mediator of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S. Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology. Cells 2021, 10, 2502. [Google Scholar] [CrossRef]
- Della Torre, S. Non-Alcoholic Fatty Liver Disease as a Canonical Example of Metabolic Inflammatory-Based Liver Disease Showing a Sex-Specific Prevalence: Relevance of Estrogen Signaling. Front. Endocrinol. 2020, 11, 572490. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Della Torre, S.; Stell, A.; Cook, J.; Brown, M.; Maggi, A. Tetradian Oscillation of Estrogen Receptor Is Necessary to Prevent Liver Lipid Deposition. Proc. Natl. Acad. Sci. USA 2012, 109, 11806–11811. [Google Scholar] [CrossRef]
- Della Torre, S.; Mitro, N.; Fontana, R.; Gomaraschi, M.; Favari, E.; Recordati, C.; Lolli, F.; Quagliarini, F.; Meda, C.; Ohlsson, C.; et al. An Essential Role for Liver ERα in Coupling Hepatic Metabolism to the Reproductive Cycle. Cell Rep. 2016, 15, 360–371. [Google Scholar] [CrossRef]
- Della Torre, S.; Benedusi, V.; Pepe, G.; Meda, C.; Rizzi, N.; Uhlenhaut, N.H.; Maggi, A. Dietary Essential Amino Acids Restore Liver Metabolism in Ovariectomized Mice via Hepatic Estrogen Receptor α. Nat. Commun. 2021, 12, 6883. [Google Scholar] [CrossRef]
- Della Torre, S.; Mitro, N.; Meda, C.; Lolli, F.; Pedretti, S.; Barcella, M.; Ottobrini, L.; Metzger, D.; Caruso, D.; Maggi, A. Short-Term Fasting Reveals Amino Acid Metabolism as a Major Sex-Discriminating Factor in the Liver. Cell Metab. 2018, 28, 256–267. [Google Scholar] [CrossRef]
- Meda, C.; Barone, M.; Mitro, N.; Lolli, F.; Pedretti, S.; Caruso, D.; Maggi, A.; Della Torre, S. Hepatic ERα Accounts for Sex Differences in the Ability to Cope with an Excess of Dietary Lipids. Mol. Metab. 2020, 32, 97–108. [Google Scholar] [CrossRef]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Della Latta, V.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered Amino Acid Concentrations in NAFLD: Impact of Obesity and Insulin Resistance. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.G.; Reily, M.D.; Lehman-McKeeman, L.D.; Vaillancourt, R.R.; Cherrington, N.J. Branched Chain Amino Acid Metabolism Profiles in Progressive Human Nonalcoholic Fatty Liver Disease. Amino Acids 2015, 47, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Grzych, G.; Vonghia, L.; Bout, M.-A.; Weyler, J.; Verrijken, A.; Dirinck, E.; Chevalier Curt, M.J.; Van Gaal, L.; Paumelle, R.; Francque, S.; et al. Plasma BCAA Changes in Patients With NAFLD Are Sex Dependent. J. Clin. Endocrinol. Metab. 2020, 105, dgaa175. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Ishigami, M.; Luo, F.; Lingyun, M.; Ishizu, Y.; Kuzuya, T.; Hayashi, K.; Nakano, I.; Ishikawa, T.; Feng, G.-G.; et al. Branched-Chain Amino Acids Alleviate Hepatic Steatosis and Liver Injury in Choline-Deficient High-Fat Diet Induced NASH Mice. Metabolism 2017, 69, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S.; Maggi, A. Sex Differences: A Resultant of an Evolutionary Pressure? Cell Metab. 2017, 25, 499–505. [Google Scholar] [CrossRef]
- Maggi, A.; Della Torre, S. Sex, Metabolism and Health. Mol. Metab. 2018, 15, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Erkan, G.; Yilmaz, G.; Konca Degertekin, C.; Akyol, G.; Ozenirler, S. Presence and Extent of Estrogen Receptor-Alpha Expression in Patients with Simple Steatosis and NASH. Pathol. Res. Pract. 2013, 209, 429–432. [Google Scholar] [CrossRef]
- Beulens, J.W.J.; Pinho, M.G.M.; Abreu, T.C.; den Braver, N.R.; Lam, T.M.; Huss, A.; Vlaanderen, J.; Sonnenschein, T.; Siddiqui, N.Z.; Yuan, Z.; et al. Environmental Risk Factors of Type 2 Diabetes—An Exposome Approach. Diabetologia 2022, 65, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, A.; Dachraoui, M.; Baumert, T.F. Perturbation of the Circadian Clock and Pathogenesis of NAFLD. Metabolism 2020, 111S, 154337. [Google Scholar] [CrossRef]
- Guo, W.; Pan, B.; Sakkiah, S.; Yavas, G.; Ge, W.; Zou, W.; Tong, W.; Hong, H. Persistent Organic Pollutants in Food: Contamination Sources, Health Effects and Detection Methods. Int. J. Environ. Res. Public. Health 2019, 16, 4361. [Google Scholar] [CrossRef]
- Myrmel, L.S.; Fjære, E.; Midtbø, L.K.; Bernhard, A.; Petersen, R.K.; Sonne, S.B.; Mortensen, A.; Hao, Q.; Brattelid, T.; Liaset, B.; et al. Macronutrient Composition Determines Accumulation of Persistent Organic Pollutants from Dietary Exposure in Adipose Tissue of Mice. J. Nutr. Biochem. 2016, 27, 307–316. [Google Scholar] [CrossRef]
- Reina-Pérez, I.; Artacho-Cordón, F.; Mustieles, V.; Castellano-Castillo, D.; Cardona, F.; Jiménez-Díaz, I.; López-Medina, J.A.; Alcaide, J.; Ocaña-Wilhelmi, L.; Iribarne-Durán, L.M.; et al. Cross-Sectional Associations of Persistent Organic Pollutants Measured in Adipose Tissue and Metabolic Syndrome in Clinically Diagnosed Middle-Aged Adults. Environ. Res. 2023, 222, 115350. [Google Scholar] [CrossRef]
- Rolle-Kampczyk, U.; Gebauer, S.; Haange, S.-B.; Schubert, K.; Kern, M.; Moulla, Y.; Dietrich, A.; Schön, M.R.; Klöting, N.; von Bergen, M.; et al. Accumulation of Distinct Persistent Organic Pollutants Is Associated with Adipose Tissue Inflammation. Sci. Total Environ. 2020, 748, 142458. [Google Scholar] [CrossRef]
- Moriceau, M.-A.; Cano-Sancho, G.; Kim, M.; Coumoul, X.; Emond, C.; Arrebola, J.-P.; Antignac, J.-P.; Audouze, K.; Rousselle, C. Partitioning of Persistent Organic Pollutants between Adipose Tissue and Serum in Human Studies. Toxics 2022, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Lind, L.; Salihovic, S.; van Bavel, B.; Ingelsson, E.; Lind, P.M. Persistent Organic Pollutants and Liver Dysfunction Biomarkers in a Population-Based Human Sample of Men and Women. Environ. Res. 2014, 134, 251–256. [Google Scholar] [CrossRef]
- Deierlein, A.L.; Rock, S.; Park, S. Persistent Endocrine-Disrupting Chemicals and Fatty Liver Disease. Curr. Environ. Health Rep. 2017, 4, 439–449. [Google Scholar] [CrossRef]
- Cano, R.; Pérez, J.L.; Dávila, L.A.; Ortega, Á.; Gómez, Y.; Valero-Cedeño, N.J.; Parra, H.; Manzano, A.; Véliz Castro, T.I.; Albornoz, M.P.D.; et al. Role of Endocrine-Disrupting Chemicals in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 4807. [Google Scholar] [CrossRef] [PubMed]
- La Merrill, M.A.; Vandenberg, L.N.; Smith, M.T.; Goodson, W.; Browne, P.; Patisaul, H.B.; Guyton, K.Z.; Kortenkamp, A.; Cogliano, V.J.; Woodruff, T.J.; et al. Consensus on the Key Characteristics of Endocrine-Disrupting Chemicals as a Basis for Hazard Identification. Nat. Rev. Endocrinol. 2020, 16, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Casals-Casas, C.; Desvergne, B. Endocrine Disruptors: From Endocrine to Metabolic Disruption. Annu. Rev. Physiol. 2011, 73, 135–162. [Google Scholar] [CrossRef]
- Foulds, C.E.; Treviño, L.S.; York, B.; Walker, C.L. Endocrine-Disrupting Chemicals and Fatty Liver Disease. Nat. Rev. Endocrinol. 2017, 13, 445–457. [Google Scholar] [CrossRef]
- Lv, Q.; Gao, R.; Peng, C.; Yi, J.; Liu, L.; Yang, S.; Li, D.; Hu, J.; Luo, T.; Mei, M.; et al. Bisphenol A Promotes Hepatic Lipid Deposition Involving Kupffer Cells M1 Polarization in Male Mice. J. Endocrinol. 2017, 234, 143–154. [Google Scholar] [CrossRef]
- Manzoor, M.F.; Tariq, T.; Fatima, B.; Sahar, A.; Tariq, F.; Munir, S.; Khan, S.; Nawaz Ranjha, M.M.A.; Sameen, A.; Zeng, X.-A.; et al. An Insight into Bisphenol A, Food Exposure and Its Adverse Effects on Health: A Review. Front. Nutr. 2022, 9, 1047827. [Google Scholar] [CrossRef] [PubMed]
- Vom Saal, F.S.; Myers, J.P. Bisphenol A and Risk of Metabolic Disorders. JAMA 2008, 300, 1353–1355. [Google Scholar] [CrossRef]
- Dallio, M.; Diano, N.; Masarone, M.; Gravina, A.G.; Patanè, V.; Romeo, M.; Di Sarno, R.; Errico, S.; Nicolucci, C.; Abenavoli, L.; et al. Chemical Effect of Bisphenol A on Non-Alcoholic Fatty Liver Disease. Int. J. Environ. Res. Public. Health 2019, 16, 3134. [Google Scholar] [CrossRef] [PubMed]
- An, S.J.; Yang, E.-J.; Oh, S.; Park, K.J.; Kim, T.; Hong, Y.-P.; Yang, Y.-J. The Association between Urinary Bisphenol A Levels and Nonalcoholic Fatty Liver Disease in Korean Adults: Korean National Environmental Health Survey (KoNEHS) 2015–2017. Environ. Health Prev. Med. 2021, 26, 91. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Yoo, E.R.; Li, A.A.; Cholankeril, G.; Tighe, S.P.; Kim, W.; Harrison, S.A.; Ahmed, A. Elevated Urinary Bisphenol A Levels Are Associated with Non-Alcoholic Fatty Liver Disease among Adults in the United States. Liver Int. Off. J. Int. Assoc. Study Liver 2019, 39, 1335–1342. [Google Scholar] [CrossRef]
- Ismael, L.Q.; Abdulhameed, A.R.; Keong, Y.Y.; Abdullah, M.N.H.; Bahari, H.; Jie, T.J.; Yin, K.B. Bisphenol A Is a Carcinogen That Induces Lipid Accumulation, Peroxisome Proliferator-activated Receptor-γ Expression and Liver Disease. Exp. Ther. Med. 2022, 24, 735. [Google Scholar] [CrossRef]
- Song, D.; Chen, Y.; Wang, B.; Li, D.; Xu, C.; Huang, H.; Huang, S.; Liu, R. Bisphenol A Inhibits Autophagosome-Lysosome Fusion and Lipid Droplet Degradation. Ecotoxicol. Environ. Saf. 2019, 183, 109492. [Google Scholar] [CrossRef]
- Grasselli, E.; Cortese, K.; Voci, A.; Vergani, L.; Fabbri, R.; Barmo, C.; Gallo, G.; Canesi, L. Direct Effects of Bisphenol A on Lipid Homeostasis in Rat Hepatoma Cells. Chemosphere 2013, 91, 1123–1129. [Google Scholar] [CrossRef]
- Dallio, M.; Masarone, M.; Errico, S.; Gravina, A.G.; Nicolucci, C.; Di Sarno, R.; Gionti, L.; Tuccillo, C.; Persico, M.; Stiuso, P.; et al. Role of Bisphenol A as Environmental Factor in the Promotion of Non-Alcoholic Fatty Liver Disease: In Vitro and Clinical Study. Aliment. Pharmacol. Ther. 2018, 47, 826–837. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; Gravina, A.G.; Diano, N.; Errico, S.; Masarone, M.; Romeo, M.; Tuccillo, C.; Stiuso, P.; Morisco, F.; et al. The Bisphenol A Induced Oxidative Stress in Non-Alcoholic Fatty Liver Disease Male Patients: A Clinical Strategy to Antagonize the Progression of the Disease. Int. J. Environ. Res. Public. Health 2020, 17, 3369. [Google Scholar] [CrossRef]
- Elswefy, S.E.-S.; Abdallah, F.R.; Atteia, H.H.; Wahba, A.S.; Hasan, R.A. Inflammation, Oxidative Stress and Apoptosis Cascade Implications in Bisphenol A-Induced Liver Fibrosis in Male Rats. Int. J. Exp. Pathol. 2016, 97, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-L.; Wang, Y.-C.; Hsu, Y.-A.; Chen, C.-S.; Weng, R.-C.; Lu, Y.-P.; Chuang, C.-Y.; Wan, L. Bisphenol A Coupled with a High-Fat Diet Promotes Hepatosteatosis through Reactive-Oxygen-Species-Induced CD36 Overexpression. Toxics 2022, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Zhang, H.; Jiang, X.; Zou, J.; Li, Q.; Mai, H.; Su, D.; Ling, W.; Feng, X. Bisphenol A Exposure Induces Gut Microbiota Dysbiosis and Consequent Activation of Gut-Liver Axis Leading to Hepatic Steatosis in CD-1 Mice. Environ. Pollut. 2020, 265, 114880. [Google Scholar] [CrossRef]
- Liu, R.; Liu, B.; Tian, L.; Jiang, X.; Li, X.; Cai, D.; Sun, J.; Bai, W.; Jin, Y. Exposure to Bisphenol A Caused Hepatoxicity and Intestinal Flora Disorder in Rats. Int. J. Mol. Sci. 2022, 23, 8042. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, L.S.; Oliveira, K.M.; Freitas, I.N.; Silva, J.A.; Silva, J.N.; Favero-Santos, B.C.; Bonfleur, M.L.; Carneiro, E.M.; Ribeiro, R.A. Bisphenol-A Exposure Worsens Hepatic Steatosis in Ovariectomized Mice Fed on a High-Fat Diet: Role of Endoplasmic Reticulum Stress and Fibrogenic Pathways. Life Sci. 2020, 256, 118012. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, E.; Poothong, S.; Koekkoek, J.; Lucattini, L.; Padilla-Sánchez, J.A.; Haugen, M.; Herzke, D.; Valdersnes, S.; Maage, A.; Cousins, I.T.; et al. Estimating Human Exposure to Perfluoroalkyl Acids via Solid Food and Drinks: Implementation and Comparison of Different Dietary Assessment Methods. Environ. Res. 2017, 158, 269–276. [Google Scholar] [CrossRef]
- Xu, Y.; Fletcher, T.; Pineda, D.; Lindh, C.H.; Nilsson, C.; Glynn, A.; Vogs, C.; Norström, K.; Lilja, K.; Jakobsson, K.; et al. Serum Half-Lives for Short- and Long-Chain Perfluoroalkyl Acids after Ceasing Exposure from Drinking Water Contaminated by Firefighting Foam. Environ. Health Perspect. 2020, 128, 077004. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, L.; Ducatman, A.; Deng, C.; von Stackelberg, K.E.; Danford, C.J.; Zhang, X. Association of Per- and Polyfluoroalkyl Substance Exposure with Fatty Liver Disease Risk in US Adults. JHEP Rep. Innov. Hepatol. 2023, 5, 100694. [Google Scholar] [CrossRef]
- Jin, R.; McConnell, R.; Catherine, C.; Xu, S.; Walker, D.I.; Stratakis, N.; Jones, D.P.; Miller, G.W.; Peng, C.; Conti, D.V.; et al. Perfluoroalkyl Substances and Severity of Nonalcoholic Fatty Liver in Children: An Untargeted Metabolomics Approach. Environ. Int. 2020, 134, 105220. [Google Scholar] [CrossRef]
- Costello, E.; Rock, S.; Stratakis, N.; Eckel, S.P.; Walker, D.I.; Valvi, D.; Cserbik, D.; Jenkins, T.; Xanthakos, S.A.; Kohli, R.; et al. Exposure to Per- and Polyfluoroalkyl Substances and Markers of Liver Injury: A Systematic Review and Meta-Analysis. Environ. Health Perspect. 2022, 130, 46001. [Google Scholar] [CrossRef]
- Wan, H.T.; Zhao, Y.G.; Wei, X.; Hui, K.Y.; Giesy, J.P.; Wong, C.K.C. PFOS-Induced Hepatic Steatosis, the Mechanistic Actions on β-Oxidation and Lipid Transport. Biochim. Biophys. Acta 2012, 1820, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Marques, E.; Pfohl, M.; Auclair, A.; Jamwal, R.; Barlock, B.J.; Sammoura, F.M.; Goedken, M.; Akhlaghi, F.; Slitt, A.L. Perfluorooctanesulfonic Acid (PFOS) Administration Shifts the Hepatic Proteome and Augments Dietary Outcomes Related to Hepatic Steatosis in Mice. Toxicol. Appl. Pharmacol. 2020, 408, 115250. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Gu, T.; Ling, J.; Luo, J.; Zhao, J.; Hu, B.; Hua, L.; Wan, C.; Jiang, S. PFOS Facilitates Liver Inflammation and Steatosis: An Involvement of NLRP3 Inflammasome-Mediated Hepatocyte Pyroptosis. J. Appl. Toxicol. JAT 2022, 42, 806–817. [Google Scholar] [CrossRef] [PubMed]
- Louisse, J.; Rijkers, D.; Stoopen, G.; Janssen, A.; Staats, M.; Hoogenboom, R.; Kersten, S.; Peijnenburg, A. Perfluorooctanoic Acid (PFOA), Perfluorooctane Sulfonic Acid (PFOS), and Perfluorononanoic Acid (PFNA) Increase Triglyceride Levels and Decrease Cholesterogenic Gene Expression in Human HepaRG Liver Cells. Arch. Toxicol. 2020, 94, 3137–3155. [Google Scholar] [CrossRef]
- Li, X.; Wang, Z.; Klaunig, J.E. The Effects of Perfluorooctanoate on High Fat Diet Induced Non-Alcoholic Fatty Liver Disease in Mice. Toxicology 2019, 416, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pfohl, M.; Marques, E.; Auclair, A.; Barlock, B.; Jamwal, R.; Goedken, M.; Akhlaghi, F.; Slitt, A.L. An ’Omics Approach to Unraveling the Paradoxical Effect of Diet on Perfluorooctanesulfonic Acid (PFOS) and Perfluorononanoic Acid (PFNA)-Induced Hepatic Steatosis. Toxicol. Sci. Off. J. Soc. Toxicol. 2021, 180, 277–294. [Google Scholar] [CrossRef]
- Das, K.P.; Wood, C.R.; Lin, M.T.; Starkov, A.A.; Lau, C.; Wallace, K.B.; Corton, J.C.; Abbott, B.D. Perfluoroalkyl Acids-Induced Liver Steatosis: Effects on Genes Controlling Lipid Homeostasis. Toxicology 2017, 378, 37–52. [Google Scholar] [CrossRef]
- Bjork, J.A.; Butenhoff, J.L.; Wallace, K.B. Multiplicity of Nuclear Receptor Activation by PFOA and PFOS in Primary Human and Rodent Hepatocytes. Toxicology 2011, 288, 8–17. [Google Scholar] [CrossRef]
- Rosen, M.B.; Das, K.P.; Rooney, J.; Abbott, B.; Lau, C.; Corton, J.C. PPARα-Independent Transcriptional Targets of Perfluoroalkyl Acids Revealed by Transcript Profiling. Toxicology 2017, 387, 95–107. [Google Scholar] [CrossRef]
- Kim, S.-J.; Heo, S.-H.; Lee, D.-S.; Hwang, I.G.; Lee, Y.-B.; Cho, H.-Y. Gender Differences in Pharmacokinetics and Tissue Distribution of 3 Perfluoroalkyl and Polyfluoroalkyl Substances in Rats. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2016, 97, 243–255. [Google Scholar] [CrossRef]
- Huang, M.C.; Dzierlenga, A.L.; Robinson, V.G.; Waidyanatha, S.; DeVito, M.J.; Eifrid, M.A.; Granville, C.A.; Gibbs, S.T.; Blystone, C.R. Toxicokinetics of Perfluorobutane Sulfonate (PFBS), Perfluorohexane-1-Sulphonic Acid (PFHxS), and Perfluorooctane Sulfonic Acid (PFOS) in Male and Female Hsd:Sprague Dawley SD Rats after Intravenous and Gavage Administration. Toxicol. Rep. 2019, 6, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, R. Sex Differences in the Association between Perfluoroalkyl Acids and Liver Function in US Adolescents: Analyses of NHANES 2013-2016. Environ. Pollut. 2019, 254, 113061. [Google Scholar] [CrossRef] [PubMed]
- Limei, E.; Zhang, S.; Jiang, X. Association between Perfluoroalkyl Substances Exposure and the Prevalence of Nonalcoholic Fatty Liver Disease in the Different Sexes: A Study from the National Health and Nutrition Examination Survey 2005–2018. Environ. Sci. Pollut. Res. Int. 2023, 30, 44292–44303. [Google Scholar] [CrossRef]
- Roth, K.; Yang, Z.; Agarwal, M.; Liu, W.; Peng, Z.; Long, Z.; Birbeck, J.; Westrick, J.; Liu, W.; Petriello, M.C. Exposure to a Mixture of Legacy, Alternative, and Replacement per- and Polyfluoroalkyl Substances (PFAS) Results in Sex-Dependent Modulation of Cholesterol Metabolism and Liver Injury. Environ. Int. 2021, 157, 106843. [Google Scholar] [CrossRef] [PubMed]
- Al-Eryani, L.; Wahlang, B.; Falkner, K.C.; Guardiola, J.J.; Clair, H.B.; Prough, R.A.; Cave, M. Identification of Environmental Chemicals Associated with the Development of Toxicant-Associated Fatty Liver Disease in Rodents. Toxicol. Pathol. 2015, 43, 482–497. [Google Scholar] [CrossRef]
- Li, W.; Xiao, H.; Wu, H.; Pan, C.; Deng, K.; Xu, X.; Zhang, Y. Analysis of Environmental Chemical Mixtures and Nonalcoholic Fatty Liver Disease: NHANES 1999–2014. Environ. Pollut. 2022, 311, 119915. [Google Scholar] [CrossRef]
- Rajak, S.; Raza, S.; Tewari, A.; Sinha, R.A. Environmental Toxicants and NAFLD: A Neglected yet Significant Relationship. Dig. Dis. Sci. 2022, 67, 3497–3507. [Google Scholar] [CrossRef]
- Armstrong, L.E.; Guo, G.L. Understanding Environmental Contaminants’ Direct Effects on Non-Alcoholic Fatty Liver Disease Progression. Curr. Environ. Health Rep. 2019, 6, 95–104. [Google Scholar] [CrossRef]
- Sang, H.; Lee, K.-N.; Jung, C.H.; Han, K.; Koh, E.H. Association between Organochlorine Pesticides and Nonalcoholic Fatty Liver Disease in the National Health and Nutrition Examination Survey 2003–2004. Sci. Rep. 2022, 12, 11590. [Google Scholar] [CrossRef]
- Jellali, R.; Jacques, S.; Essaouiba, A.; Gilard, F.; Letourneur, F.; Gakière, B.; Legallais, C.; Leclerc, E. Investigation of Steatosis Profiles Induced by Pesticides Using Liver Organ-on-Chip Model and Omics Analysis. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2021, 152, 112155. [Google Scholar] [CrossRef]
- Li, M.; Liu, T.; Yang, T.; Zhu, J.; Zhou, Y.; Wang, M.; Wang, Q. Gut Microbiota Dysbiosis Involves in Host Non-Alcoholic Fatty Liver Disease upon Pyrethroid Pesticide Exposure. Environ. Sci. Ecotechnol. 2022, 11, 100185. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tsakiridis, E.E.; Zhang, S.; Llanos, A.; Desjardins, E.M.; Yabut, J.M.; Green, A.E.; Day, E.A.; Smith, B.K.; Lally, J.S.V.; et al. The Pesticide Chlorpyrifos Promotes Obesity by Inhibiting Diet-Induced Thermogenesis in Brown Adipose Tissue. Nat. Commun. 2021, 12, 5163. [Google Scholar] [CrossRef] [PubMed]
- Wasef, L.; Nassar, A.M.K.; El-Sayed, Y.S.; Samak, D.; Noreldin, A.; Elshony, N.; Saleh, H.; Elewa, Y.H.A.; Hassan, S.M.A.; Saati, A.A.; et al. The Potential Ameliorative Impacts of Cerium Oxide Nanoparticles against Fipronil-Induced Hepatic Steatosis. Sci. Rep. 2021, 11, 1310. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, X.; Yue, L.; Hu, H.; Wei, X.; Guo, Q.; Zhang, B.; Fan, X.; Xin, Y.; Oh, Y.; et al. Thiamethoxam Induces Nonalcoholic Fatty Liver Disease in Mice via Methionine Metabolism Disturb via Nicotinamide N-Methyltransferase Overexpression. Chemosphere 2021, 273, 129727. [Google Scholar] [CrossRef]
- Taylor, R.; Armstrong, L.; Bhattacharya, A.; Henry, Z.; Brinker, A.; Buckley, B.; Kong, B.; Guo, G. Myclobutanil-Mediated Alteration of Liver-Gut FXR Signaling in Mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2023, 191, 387–399. [Google Scholar] [CrossRef]
- Stellavato, A.; Lamberti, M.; Pirozzi, A.V.A.; de Novellis, F.; Schiraldi, C. Myclobutanil Worsens Nonalcoholic Fatty Liver Disease: An in Vitro Study of Toxicity and Apoptosis on HepG2 Cells. Toxicol. Lett. 2016, 262, 100–104. [Google Scholar] [CrossRef]
- Pirozzi, A.V.A.; Stellavato, A.; La Gatta, A.; Lamberti, M.; Schiraldi, C. Mancozeb, a Fungicide Routinely Used in Agriculture, Worsens Nonalcoholic Fatty Liver Disease in the Human HepG2 Cell Model. Toxicol. Lett. 2016, 249, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Stossi, F.; Dandekar, R.D.; Johnson, H.; Lavere, P.; Foulds, C.E.; Mancini, M.G.; Mancini, M.A. Tributyltin Chloride (TBT) Induces RXRA down-Regulation and Lipid Accumulation in Human Liver Cells. PLoS ONE 2019, 14, e0224405. [Google Scholar] [CrossRef]
- González, N.; Domingo, J.L. Polychlorinated Dibenzo-p-Dioxins and Dibenzofurans (PCDD/Fs) in Food and Human Dietary Intake: An Update of the Scientific Literature. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2021, 157, 112585. [Google Scholar] [CrossRef]
- Shin, E.; Kim, J.; Choi, S.-D.; Kang, Y.-W.; Chang, Y.-S. Estimated Dietary Intake and Risk Assessment of Polychlorinated Dibenzo-p-Dioxins and Dibenzofurans and Dioxin-like Polychlorinated Biphenyls from Fish Consumption in the Korean General Population. Chemosphere 2016, 146, 419–425. [Google Scholar] [CrossRef]
- Houlahan, K.E.; Prokopec, S.D.; Sun, R.X.; Moffat, I.D.; Lindén, J.; Lensu, S.; Okey, A.B.; Pohjanvirta, R.; Boutros, P.C. Transcriptional Profiling of Rat White Adipose Tissue Response to 2,3,7,8-Tetrachlorodibenzo-ρ-Dioxin. Toxicol. Appl. Pharmacol. 2015, 288, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Nwanaji-Enwerem, J.C.; Jenkins, T.G.; Colicino, E.; Cardenas, A.; Baccarelli, A.A.; Boyer, E.W. Serum Dioxin Levels and Sperm DNA Methylation Age: Findings in Vietnam War Veterans Exposed to Agent Orange. Reprod. Toxicol. 2020, 96, 27–35. [Google Scholar] [CrossRef]
- Angrish, M.M.; Dominici, C.Y.; Zacharewski, T.R. TCDD-Elicited Effects on Liver, Serum, and Adipose Lipid Composition in C57BL/6 Mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2013, 131, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, N.; Wahlström, D.; Lundberg, R.; Nilsson, C.B.; Nilsson, K.C.; Stockling, K.; Hellmold, H.; Håkansson, H. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Alters the MRNA Expression of Critical Genes Associated with Cholesterol Metabolism, Bile Acid Biosynthesis, and Bile Transport in Rat Liver: A Microarray Study. Toxicol. Appl. Pharmacol. 2005, 207, 1–24. [Google Scholar] [CrossRef]
- Fader, K.A.; Nault, R.; Zhang, C.; Kumagai, K.; Harkema, J.R.; Zacharewski, T.R. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD)-Elicited Effects on Bile Acid Homeostasis: Alterations in Biosynthesis, Enterohepatic Circulation, and Microbial Metabolism. Sci. Rep. 2017, 7, 5921. [Google Scholar] [CrossRef]
- Fling, R.R.; Zacharewski, T.R. Aryl Hydrocarbon Receptor (AhR) Activation by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Dose-Dependently Shifts the Gut Microbiome Consistent with the Progression of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 12431. [Google Scholar] [CrossRef] [PubMed]
- Fling, R.R.; Doskey, C.M.; Fader, K.A.; Nault, R.; Zacharewski, T.R. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Dysregulates Hepatic One Carbon Metabolism during the Progression of Steatosis to Steatohepatitis with Fibrosis in Mice. Sci. Rep. 2020, 10, 14831. [Google Scholar] [CrossRef]
- Wahlang, B.; Hardesty, J.E.; Jin, J.; Falkner, K.C.; Cave, M.C. Polychlorinated Biphenyls and Nonalcoholic Fatty Liver Disease. Curr. Opin. Toxicol. 2019, 14, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Duval, C.; Teixeira-Clerc, F.; Leblanc, A.F.; Touch, S.; Emond, C.; Guerre-Millo, M.; Lotersztajn, S.; Barouki, R.; Aggerbeck, M.; Coumoul, X. Chronic Exposure to Low Doses of Dioxin Promotes Liver Fibrosis Development in the C57BL/6J Diet-Induced Obesity Mouse Model. Environ. Health Perspect. 2017, 125, 428–436. [Google Scholar] [CrossRef]
- Jin, J.; Wahlang, B.; Shi, H.; Hardesty, J.E.; Falkner, K.C.; Head, K.Z.; Srivastava, S.; Merchant, M.L.; Rai, S.N.; Cave, M.C.; et al. Dioxin-like and Non-Dioxin-like PCBs Differentially Regulate the Hepatic Proteome and Modify Diet-Induced Nonalcoholic Fatty Liver Disease Severity. Med. Chem. Res. Int. J. Rapid Commun. Des. Mech. Action Biol. Act. Agents 2020, 29, 1247–1263. [Google Scholar] [CrossRef]
- Wahlang, B.; Falkner, K.C.; Gregory, B.; Ansert, D.; Young, D.; Conklin, D.J.; Bhatnagar, A.; McClain, C.J.; Cave, M. Polychlorinated Biphenyl 153 Is a Diet-Dependent Obesogen That Worsens Nonalcoholic Fatty Liver Disease in Male C57BL6/J Mice. J. Nutr. Biochem. 2013, 24, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Prokopec, S.D.; Watson, J.D.; Sun, R.X.; Pohjanvirta, R.; Boutros, P.C. Male and Female Mice Show Significant Differences in Hepatic Transcriptomic Response to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. BMC Genomics 2015, 16, 625. [Google Scholar] [CrossRef] [PubMed]
- Nault, R.; Fader, K.A.; Harkema, J.R.; Zacharewski, T. Loss of Liver-Specific and Sexually Dimorphic Gene Expression by Aryl Hydrocarbon Receptor Activation in C57BL/6 Mice. PLoS ONE 2017, 12, e0184842. [Google Scholar] [CrossRef] [PubMed]
- Prokopec, S.D.; Watson, J.D.; Lee, J.; Pohjanvirta, R.; Boutros, P.C. Sex-Related Differences in Murine Hepatic Transcriptional and Proteomic Responses to TCDD. Toxicol. Appl. Pharmacol. 2015, 284, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Matteo, G.; Hoyeck, M.P.; Blair, H.L.; Zebarth, J.; Rick, K.R.C.; Williams, A.; Gagné, R.; Buick, J.K.; Yauk, C.L.; Bruin, J.E. Prolonged Low-Dose Dioxin Exposure Impairs Metabolic Adaptability to High-Fat Diet Feeding in Female but Not Male Mice. Endocrinology 2021, 162, bqab050. [Google Scholar] [CrossRef] [PubMed]
- Vega, N.; Pinteur, C.; Buffelan, G.; Loizon, E.; Vidal, H.; Naville, D.; Le Magueresse-Battistoni, B. Exposure to Pollutants Altered Glucocorticoid Signaling and Clock Gene Expression in Female Mice. Evidence of Tissue- and Sex-Specificity. Chemosphere 2021, 262, 127841. [Google Scholar] [CrossRef]
- Sampaio, G.R.; Guizellini, G.M.; da Silva, S.A.; de Almeida, A.P.; Pinaffi-Langley, A.C.C.; Rogero, M.M.; de Camargo, A.C.; Torres, E.A.F.S. Polycyclic Aromatic Hydrocarbons in Foods: Biological Effects, Legislation, Occurrence, Analytical Methods, and Strategies to Reduce Their Formation. Int. J. Mol. Sci. 2021, 22, 6010. [Google Scholar] [CrossRef]
- Goedtke, L.; Sprenger, H.; Hofmann, U.; Schmidt, F.F.; Hammer, H.S.; Zanger, U.M.; Poetz, O.; Seidel, A.; Braeuning, A.; Hessel-Pras, S. Polycyclic Aromatic Hydrocarbons Activate the Aryl Hydrocarbon Receptor and the Constitutive Androstane Receptor to Regulate Xenobiotic Metabolism in Human Liver Cells. Int. J. Mol. Sci. 2020, 22, 372. [Google Scholar] [CrossRef]
- Li, F.; Xiang, B.; Jin, Y.; Li, C.; Ren, S.; Wu, Y.; Li, J.; Luo, Q. Hepatotoxic Effects of Inhalation Exposure to Polycyclic Aromatic Hydrocarbons on Lipid Metabolism of C57BL/6 Mice. Environ. Int. 2020, 134, 105000. [Google Scholar] [CrossRef]
- Zhu, X.-Y.; Xia, H.-G.; Wang, Z.-H.; Li, B.; Jiang, H.-Y.; Li, D.-L.; Jin, R.; Jin, Y. In Vitro and in Vivo Approaches for Identifying the Role of Aryl Hydrocarbon Receptor in the Development of Nonalcoholic Fatty Liver Disease. Toxicol. Lett. 2020, 319, 85–94. [Google Scholar] [CrossRef]
- Kim, K.; Melough, M.M.; Vance, T.M.; Noh, H.; Koo, S.I.; Chun, O.K. Dietary Cadmium Intake and Sources in the US. Nutrients 2018, 11, 2. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Z.; Liu, L.; Zhan, N.; Qin, J.; Lu, X.; Cheng, M. Assessment of the Risks from Dietary Lead Exposure in China. J. Hazard. Mater. 2021, 418, 126134. [Google Scholar] [CrossRef] [PubMed]
- Cinnirella, S.; Hedgecock, I.M.; Sprovieri, F. Heavy Metals in the Environment: Sources, Interactions and Human Health. Environ. Sci. Pollut. Res. Int. 2014, 21, 3997–3998. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Lian, I.-B.; Kor, C.-T.; Chang, C.-C.; Su, P.-Y.; Chang, W.-T.; Liang, Y.-F.; Su, W.-W.; Soon, M.-S. Association between Soil Heavy Metals and Fatty Liver Disease in Men in Taiwan: A Cross Sectional Study. BMJ Open 2017, 7, e014215. [Google Scholar] [CrossRef] [PubMed]
- Sadighara, P.; Abedini, A.H.; Irshad, N.; Ghazi-Khansari, M.; Esrafili, A.; Yousefi, M. Association Between Non-Alcoholic Fatty Liver Disease and Heavy Metal Exposure: A Systematic Review. Biol. Trace Elem. Res. 2023. [Google Scholar] [CrossRef]
- Hong, D.; Min, J.-Y.; Min, K.-B. Association Between Cadmium Exposure and Liver Function in Adults in the United States: A Cross-Sectional Study. J. Prev. Med. Pub. Health 2021, 54, 471–480. [Google Scholar] [CrossRef]
- Park, E.; Kim, J.; Kim, B.; Park, E.Y. Association between Environmental Exposure to Cadmium and Risk of Suspected Non-Alcoholic Fatty Liver Disease. Chemosphere 2021, 266, 128947. [Google Scholar] [CrossRef]
- Li, Y.; Chen, C.; Lu, L.; Guo, W.; VanWagner, L.B.; Shikany, J.M.; Zhang, S.; Kahe, K. Cadmium Exposure in Young Adulthood Is Associated with Risk of Nonalcoholic Fatty Liver Disease in Midlife. Dig. Dis. Sci. 2022, 67, 689–696. [Google Scholar] [CrossRef]
- Xu, Z.; Weng, Z.; Liang, J.; Liu, Q.; Zhang, X.; Xu, J.; Xu, C.; Gu, A. Association between Urinary Cadmium Concentrations and Liver Function in Adolescents. Environ. Sci. Pollut. Res. Int. 2022, 29, 39768–39776. [Google Scholar] [CrossRef]
- Yang, C.; Li, Y.; Ding, R.; Xing, H.; Wang, R.; Zhang, M. Lead Exposure as a Causative Factor for Metabolic Associated Fatty Liver Disease (MAFLD) and a Lead Exposure Related Nomogram for MAFLD Prevalence. Front. Public Health 2022, 10, 1000403. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Kim, M.-S. Cadmium, Lead, and Mercury Mixtures Interact with Non-Alcoholic Fatty Liver Diseases. Environ. Pollut. 2022, 309, 119780. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Aimuzi, R.; Si, M.; Qu, Y.; Jiang, Y. Associations of Metal Mixtures with Metabolic-Associated Fatty Liver Disease and Non-Alcoholic Fatty Liver Disease: NHANES 2003–2018. Front. Public Health 2023, 11, 1133194. [Google Scholar] [CrossRef] [PubMed]
- Frediani, J.K.; Naioti, E.A.; Vos, M.B.; Figueroa, J.; Marsit, C.J.; Welsh, J.A. Arsenic Exposure and Risk of Nonalcoholic Fatty Liver Disease (NAFLD) among U.S. Adolescents and Adults: An Association Modified by Race/Ethnicity, NHANES 2005-2014. Environ. Health Glob. Access Sci. Source 2018, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The Effects of Cadmium Toxicity. Int. J. Environ. Res. Public. Health 2020, 17, 3782. [Google Scholar] [CrossRef]
- Zhao, M.; Ge, X.; Xu, J.; Li, A.; Mei, Y.; Yin, G.; Wu, J.; Liu, X.; Wei, L.; Xu, Q. Association between Urine Metals and Liver Function Biomarkers in Northeast China: A Cross-Sectional Study. Ecotoxicol. Environ. Saf. 2022, 231, 113163. [Google Scholar] [CrossRef]
- Gu, J.; Kong, A.; Guo, C.; Liu, J.; Li, K.; Ren, Z.; Zhou, Y.; Tang, M.; Shi, H. Cadmium Perturbed Lipid Profile and Induced Liver Dysfunction in Mice through Phosphatidylcholine Remodeling and Promoting Arachidonic Acid Synthesis and Metabolism. Ecotoxicol. Environ. Saf. 2022, 247, 114254. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, Y.; Chai, X.-X.; Zhou, J.; Shi, M.-J.; Zhao, Y.; Tian, Y.; Wang, X.-M.; Ying, T.-X.; Feng, Q.; et al. Chronic Exposure to Low-Dose Cadmium Facilitated Nonalcoholic Steatohepatitis in Mice by Suppressing Fatty Acid Desaturation. Ecotoxicol. Environ. Saf. 2022, 233, 113306. [Google Scholar] [CrossRef]
- Ren, C.; Ren, L.; Yan, J.; Bai, Z.; Zhang, L.; Zhang, H.; Xie, Y.; Li, X. Transcription Profiling of Cadmium-Exposed Livers Reveals Alteration of Lipid Metabolism and Predisposition to Hepatic Steatosis. Xenobiotica Fate Foreign Compd. Biol. Syst. 2021, 51, 1271–1281. [Google Scholar] [CrossRef]
- Young, J.L.; Cave, M.C.; Xu, Q.; Kong, M.; Xu, J.; Lin, Q.; Tan, Y.; Cai, L. Whole Life Exposure to Low Dose Cadmium Alters Diet-Induced NAFLD. Toxicol. Appl. Pharmacol. 2022, 436, 115855. [Google Scholar] [CrossRef]
- He, X.; Gao, J.; Hou, H.; Qi, Z.; Chen, H.; Zhang, X.-X. Inhibition of Mitochondrial Fatty Acid Oxidation Contributes to Development of Nonalcoholic Fatty Liver Disease Induced by Environmental Cadmium Exposure. Environ. Sci. Technol. 2019, 53, 13992–14000. [Google Scholar] [CrossRef]
- Obeng-Gyasi, E. Sources of Lead Exposure in Various Countries. Rev. Environ. Health 2019, 34, 25–34. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Cave, M.; Appana, S.; Patel, M.; Falkner, K.C.; McClain, C.J.; Brock, G. Polychlorinated Biphenyls, Lead, and Mercury Are Associated with Liver Disease in American Adults: NHANES 2003–2004. Environ. Health Perspect. 2010, 118, 1735–1742. [Google Scholar] [CrossRef]
- Betanzos-Robledo, L.; Cantoral, A.; Peterson, K.E.; Hu, H.; Hernández-Ávila, M.; Perng, W.; Jansen, E.; Ettinger, A.S.; Mercado-García, A.; Solano-González, M.; et al. Association between Cumulative Childhood Blood Lead Exposure and Hepatic Steatosis in Young Mexican Adults. Environ. Res. 2021, 196, 110980. [Google Scholar] [CrossRef] [PubMed]
- Vineeth Daniel, P.; Kamthan, M.; Gera, R.; Dogra, S.; Gautam, K.; Ghosh, D.; Mondal, P. Chronic Exposure to Pb2+ Perturbs ChREBP Transactivation and Coerces Hepatic Dyslipidemia. FEBS Lett. 2019, 593, 3084–3097. [Google Scholar] [CrossRef]
- Milosevic, N.; Maier, P. Lead Stimulates Intercellular Signalling between Hepatocytes and Kupffer Cells. Eur. J. Pharmacol. 2000, 401, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Wang, Y.; Zhang, H.; Zhang, K.; Chen, Y.; Chen, C.; Zhang, W.; Xia, F.; Wang, N.; Lu, Y. Chronic Lead Exposure Induces Fatty Liver Disease Associated with the Variations of Gut Microbiota. Ecotoxicol. Environ. Saf. 2022, 232, 113257. [Google Scholar] [CrossRef]
- Shi, X.; Wei, X.; Koo, I.; Schmidt, R.H.; Yin, X.; Kim, S.H.; Vaughn, A.; McClain, C.J.; Arteel, G.E.; Zhang, X.; et al. Metabolomic Analysis of the Effects of Chronic Arsenic Exposure in a Mouse Model of Diet-Induced Fatty Liver Disease. J. Proteome Res. 2014, 13, 547–554. [Google Scholar] [CrossRef]
- Jia, X.; Qiu, T.; Yao, X.; Jiang, L.; Wang, N.; Wei, S.; Tao, Y.; Pei, P.; Wang, Z.; Zhang, J.; et al. Arsenic Induces Hepatic Insulin Resistance via MtROS-NLRP3 Inflammasome Pathway. J. Hazard. Mater. 2020, 399, 123034. [Google Scholar] [CrossRef]
- Qiu, T.; Pei, P.; Yao, X.; Jiang, L.; Wei, S.; Wang, Z.; Bai, J.; Yang, G.; Gao, N.; Yang, L.; et al. Taurine Attenuates Arsenic-Induced Pyroptosis and Nonalcoholic Steatohepatitis by Inhibiting the Autophagic-Inflammasomal Pathway. Cell Death Dis. 2018, 9, 946. [Google Scholar] [CrossRef]
- Wei, S.; Qiu, T.; Wang, N.; Yao, X.; Jiang, L.; Jia, X.; Tao, Y.; Zhang, J.; Zhu, Y.; Yang, G.; et al. Ferroptosis Mediated by the Interaction between Mfn2 and IREα Promotes Arsenic-Induced Nonalcoholic Steatohepatitis. Environ. Res. 2020, 188, 109824. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhu, C.; Zhou, X. Effects of Lead and Cadmium Combined Heavy Metals on Liver Function and Lipid Metabolism in Mice. Biol. Trace Elem. Res. 2023, 201, 2864–2876. [Google Scholar] [CrossRef]
- Campanale, C.; Massarelli, C.; Savino, I.; Locaputo, V.; Uricchio, V.F. A Detailed Review Study on Potential Effects of Microplastics and Additives of Concern on Human Health. Int. J. Environ. Res. Public. Health 2020, 17, 1212. [Google Scholar] [CrossRef]
- Frias, J.P.G.L.; Nash, R. Microplastics: Finding a Consensus on the Definition. Mar. Pollut. Bull. 2019, 138, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Yee, M.S.-L.; Hii, L.-W.; Looi, C.K.; Lim, W.-M.; Wong, S.-F.; Kok, Y.-Y.; Tan, B.-K.; Wong, C.-Y.; Leong, C.-O. Impact of Microplastics and Nanoplastics on Human Health. Nanomaterials 2021, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Karbalaei, S.; Hanachi, P.; Walker, T.R.; Cole, M. Occurrence, Sources, Human Health Impacts and Mitigation of Microplastic Pollution. Environ. Sci. Pollut. Res. Int. 2018, 25, 36046–36063. [Google Scholar] [CrossRef]
- Yin, J.; Ju, Y.; Qian, H.; Wang, J.; Miao, X.; Zhu, Y.; Zhou, L.; Ye, L. Nanoplastics and Microplastics May Be Damaging Our Livers. Toxics 2022, 10, 586. [Google Scholar] [CrossRef]
- Prata, J.C.; da Costa, J.P.; Lopes, I.; Duarte, A.C.; Rocha-Santos, T. Environmental Exposure to Microplastics: An Overview on Possible Human Health Effects. Sci. Total Environ. 2020, 702, 134455. [Google Scholar] [CrossRef]
- Prata, J.C. Airborne Microplastics: Consequences to Human Health? Environ. Pollut. 2018, 234, 115–126. [Google Scholar] [CrossRef]
- De-la-Torre, G.E. Microplastics: An Emerging Threat to Food Security and Human Health. J. Food Sci. Technol. 2020, 57, 1601–1608. [Google Scholar] [CrossRef]
- Schwabl, P.; Köppel, S.; Königshofer, P.; Bucsics, T.; Trauner, M.; Reiberger, T.; Liebmann, B. Detection of Various Microplastics in Human Stool: A Prospective Case Series. Ann. Intern. Med. 2019, 171, 453–457. [Google Scholar] [CrossRef]
- Horvatits, T.; Tamminga, M.; Liu, B.; Sebode, M.; Carambia, A.; Fischer, L.; Püschel, K.; Huber, S.; Fischer, E.K. Microplastics Detected in Cirrhotic Liver Tissue. EBioMedicine 2022, 82, 104147. [Google Scholar] [CrossRef] [PubMed]
- Auguet, T.; Bertran, L.; Barrientos-Riosalido, A.; Fabregat, B.; Villar, B.; Aguilar, C.; Sabench, F. Are Ingested or Inhaled Microplastics Involved in Nonalcoholic Fatty Liver Disease? Int. J. Environ. Res. Public. Health 2022, 19, 13495. [Google Scholar] [CrossRef] [PubMed]
- Okamura, T.; Hamaguchi, M.; Hasegawa, Y.; Hashimoto, Y.; Majima, S.; Senmaru, T.; Ushigome, E.; Nakanishi, N.; Asano, M.; Yamazaki, M.; et al. Oral Exposure to Polystyrene Microplastics of Mice on a Normal or High-Fat Diet and Intestinal and Metabolic Outcomes. Environ. Health Perspect. 2023, 131, 027006. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, M.; He, C.; Wang, H.; Hu, Q. Polystyrene Nanoplastics Potentiate the Development of Hepatic Fibrosis in High Fat Diet Fed Mice. Environ. Toxicol. 2022, 37, 362–372. [Google Scholar] [CrossRef]
- Fan, X.; Wei, X.; Hu, H.; Zhang, B.; Yang, D.; Du, H.; Zhu, R.; Sun, X.; Oh, Y.; Gu, N. Effects of Oral Administration of Polystyrene Nanoplastics on Plasma Glucose Metabolism in Mice. Chemosphere 2022, 288, 132607. [Google Scholar] [CrossRef]
- Wang, H.; Shi, X.; Gao, Y.; Zhang, X.; Zhao, H.; Wang, L.; Zhang, X.; Chen, R. Polystyrene Nanoplastics Induce Profound Metabolic Shift in Human Cells as Revealed by Integrated Proteomic and Metabolomic Analysis. Environ. Int. 2022, 166, 107349. [Google Scholar] [CrossRef]
- He, Y.; Li, J.; Chen, J.; Miao, X.; Li, G.; He, Q.; Xu, H.; Li, H.; Wei, Y. Cytotoxic Effects of Polystyrene Nanoplastics with Different Surface Functionalization on Human HepG2 Cells. Sci. Total Environ. 2020, 723, 138180. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, H.; Wang, C.; Su, X.-L.; Song, Y.; Wu, P.; Yang, Z.; Wong, M.-H.; Cai, Z.; Zheng, C. Metabolomics Reveal Nanoplastic-Induced Mitochondrial Damage in Human Liver and Lung Cells. Environ. Sci. Technol. 2022, 56, 12483–12493. [Google Scholar] [CrossRef]
- Cheng, W.; Li, X.; Zhou, Y.; Yu, H.; Xie, Y.; Guo, H.; Wang, H.; Li, Y.; Feng, Y.; Wang, Y. Polystyrene Microplastics Induce Hepatotoxicity and Disrupt Lipid Metabolism in the Liver Organoids. Sci. Total Environ. 2022, 806, 150328. [Google Scholar] [CrossRef]
- Lai, W.; Xu, D.; Li, J.; Wang, Z.; Ding, Y.; Wang, X.; Li, X.; Xu, N.; Mai, K.; Ai, Q. Dietary Polystyrene Nanoplastics Exposure Alters Liver Lipid Metabolism and Muscle Nutritional Quality in Carnivorous Marine Fish Large Yellow Croaker (Larimichthys Crocea). J. Hazard. Mater. 2021, 419, 126454. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhang, Y.; Lemos, B.; Ren, H. Tissue Accumulation of Microplastics in Mice and Biomarker Responses Suggest Widespread Health Risks of Exposure. Sci. Rep. 2017, 7, 46687. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Sun, J.; Li, Z.; Zhang, W.; Liu, Z.; Li, C.; Peng, C.; Cui, G.; Shao, H.; Du, Z. Activation of Pyroptosis and Ferroptosis Is Involved in the Hepatotoxicity Induced by Polystyrene Microplastics in Mice. Chemosphere 2022, 291, 132944. [Google Scholar] [CrossRef]
- Chen, L.; Qi, M.; Zhang, L.; Yu, F.; Tao, D.; Xu, C.; Xu, S. Di(2-Ethylhexyl) Phthalate and Microplastics Cause Necroptosis and Apoptosis in Hepatocytes of Mice by Inducing Oxidative Stress. Environ. Toxicol. 2023. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, Q.; Cui, J.; Bao, B.; Deng, X.; Liu, L.; Guo, M.-Y. Polystyrene Microplastics Induce Endoplasmic Reticulum Stress, Apoptosis and Inflammation by Disrupting the Gut Microbiota in Carp Intestines. Environ. Pollut. 2023, 323, 121233. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.; Hanczko, R.; Telarico, T.; Oaks, Z.; Landas, S. Oxidative Stress, Inflammation and Carcinogenesis Are Controlled through the Pentose Phosphate Pathway by Transaldolase. Trends Mol. Med. 2011, 17, 395–403. [Google Scholar] [CrossRef]
- Lu, L.; Wan, Z.; Luo, T.; Fu, Z.; Jin, Y. Polystyrene Microplastics Induce Gut Microbiota Dysbiosis and Hepatic Lipid Metabolism Disorder in Mice. Sci. Total Environ. 2018, 631–632, 449–458. [Google Scholar] [CrossRef]
- Chen, X.; Zhuang, J.; Chen, Q.; Xu, L.; Yue, X.; Qiao, D. Chronic Exposure to Polyvinyl Chloride Microplastics Induces Liver Injury and Gut Microbiota Dysbiosis Based on the Integration of Liver Transcriptome Profiles and Full-Length 16S RRNA Sequencing Data. Sci. Total Environ. 2022, 839, 155984. [Google Scholar] [CrossRef]
- Huang, D.; Zhang, Y.; Long, J.; Yang, X.; Bao, L.; Yang, Z.; Wu, B.; Si, R.; Zhao, W.; Peng, C.; et al. Polystyrene Microplastic Exposure Induces Insulin Resistance in Mice via Dysbacteriosis and Pro-Inflammation. Sci. Total Environ. 2022, 838, 155937. [Google Scholar] [CrossRef]
- Yang, X.; Jiang, J.; Wang, Q.; Duan, J.; Chen, N.; Wu, D.; Xia, Y. Gender Difference in Hepatic AMPK Pathway Activated Lipid Metabolism Induced by Aged Polystyrene Microplastics Exposure. Ecotoxicol. Environ. Saf. 2022, 245, 114105. [Google Scholar] [CrossRef]
- Saeed, A.; Akhtar, M.F.; Saleem, A.; Akhtar, B.; Sharif, A. Reproductive and Metabolic Toxic Effects of Polystyrene Microplastics in Adult Female Wistar Rats: A Mechanistic Study. Environ. Sci. Pollut. Res. Int. 2023, 30, 63185–63199. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhuan, Q.; Zhang, L.; Meng, L.; Fu, X.; Hou, Y. Polystyrene Microplastics Induced Female Reproductive Toxicity in Mice. J. Hazard. Mater. 2022, 424, 127629. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Lu, L.; Zheng, M.; Zhang, X.; Tian, H.; Wang, W.; Ru, S. Polystyrene Microplastics Cause Tissue Damages, Sex-Specific Reproductive Disruption and Transgenerational Effects in Marine Medaka (Oryzias Melastigma). Environ. Pollut. 2019, 254, 113024. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Wang, Y.; Wang, S.; Xie, J.; Han, Q.; Chen, M. Comparing the Effects of Polystyrene Microplastics Exposure on Reproduction and Fertility in Male and Female Mice. Toxicology 2022, 465, 153059. [Google Scholar] [CrossRef]
- Thangavel, P.; Park, D.; Lee, Y.-C. Recent Insights into Particulate Matter (PM2.5)-Mediated Toxicity in Humans: An Overview. Int. J. Environ. Res. Public. Health 2022, 19, 7511. [Google Scholar] [CrossRef]
- Chen, H.; Oliver, B.G.; Pant, A.; Olivera, A.; Poronnik, P.; Pollock, C.A.; Saad, S. Effects of Air Pollution on Human Health—Mechanistic Evidence Suggested by in Vitro and in Vivo Modelling. Environ. Res. 2022, 212, 113378. [Google Scholar] [CrossRef] [PubMed]
- Manisalidis, I.; Stavropoulou, E.; Stavropoulos, A.; Bezirtzoglou, E. Environmental and Health Impacts of Air Pollution: A Review. Front. Public Health 2020, 8, 14. [Google Scholar] [CrossRef]
- Guo, B.; Guo, Y.; Nima, Q.; Feng, Y.; Wang, Z.; Lu, R.; Baimayangji; Ma, Y.; Zhou, J.; Xu, H.; et al. Exposure to Air Pollution Is Associated with an Increased Risk of Metabolic Dysfunction-Associated Fatty Liver Disease. J. Hepatol. 2022, 76, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yang, Q.; Zhou, Q.; Cao, W.; Yu, S.; Zhan, S.; Sun, F. Long-Term Exposure to Fine Particulate Matter and Non-Alcoholic Fatty Liver Disease: A Prospective Cohort Study. Gut 2022, 71, 443–445. [Google Scholar] [CrossRef]
- Li, F.-R.; Liao, J.; Zhu, B.; Li, X.; Cheng, Z.; Jin, C.; Mo, C.; Wu, X.; Li, Q.; Liang, F. Long-Term Exposure to Air Pollution and Incident Non-Alcoholic Fatty Liver Disease and Cirrhosis: A Cohort Study. Liver Int. Off. J. Int. Assoc. Study Liver 2023, 43, 299–307. [Google Scholar] [CrossRef]
- Chen, J.; Wu, L.; Yang, G.; Zhang, C.; Liu, X.; Sun, X.; Chen, X.; Wang, N. The Influence of PM2.5 Exposure on Non-Alcoholic Fatty Liver Disease. Life Sci. 2021, 270, 119135. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-X.; Ge, C.-X.; Qin, Y.-T.; Gu, T.-T.; Lou, D.-S.; Li, Q.; Hu, L.-F.; Feng, J.; Huang, P.; Tan, J. Prolonged PM2.5 Exposure Elevates Risk of Oxidative Stress-Driven Nonalcoholic Fatty Liver Disease by Triggering Increase of Dyslipidemia. Free Radic. Biol. Med. 2019, 130, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Yuan, C.; Si, B.; Wang, M.; Da, S.; Bai, L.; Wu, W. Combined Effects of Ambient Particulate Matter Exposure and a High-Fat Diet on Oxidative Stress and Steatohepatitis in Mice. PLoS ONE 2019, 14, e0214680. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Li, D.; Wu, Q.; Zhang, Y.; Gao, M.; Ji, A.; Jiang, Q.; Chen, R.; Zhang, R.; et al. Real Ambient Particulate Matter-Induced Lipid Metabolism Disorder: Roles of Peroxisome Proliferators-Activated Receptor Alpha. Ecotoxicol. Environ. Saf. 2022, 231, 113173. [Google Scholar] [CrossRef]
- Reyes-Caballero, H.; Rao, X.; Sun, Q.; Warmoes, M.O.; Lin, P.; Sussan, T.E.; Park, B.; Fan, T.W.-M.; Maiseyeu, A.; Rajagopalan, S.; et al. Air Pollution-Derived Particulate Matter Dysregulates Hepatic Krebs Cycle, Glucose and Lipid Metabolism in Mice. Sci. Rep. 2019, 9, 17423. [Google Scholar] [CrossRef] [PubMed]
- Ogino, N.; Miyagawa, K.; Nagaoka, K.; Sumida, K.; Kusanaga, M.; Oe, S.; Honma, Y.; Shibata, M.; Harada, M.; Suganuma, N.; et al. Airborne Fine Particulate Matter in Japan Induces Lipid Synthesis and Inhibits Autophagy in HepG2 Cells. Int. J. Biochem. Cell Biol. 2021, 141, 106099. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-H.; Fiel, M.I.; Sun, Q.; Guo, J.; Gordon, R.E.; Chen, L.-C.; Friedman, S.L.; Odin, J.A.; Allina, J. Kupffer Cell Activation by Ambient Air Particulate Matter Exposure May Exacerbate Non-Alcoholic Fatty Liver Disease. J. Immunotoxicol. 2009, 6, 266–275. [Google Scholar] [CrossRef]
- Xu, X.; Liu, C.; Xu, Z.; Tzan, K.; Zhong, M.; Wang, A.; Lippmann, M.; Chen, L.-C.; Rajagopalan, S.; Sun, Q. Long-Term Exposure to Ambient Fine Particulate Pollution Induces Insulin Resistance and Mitochondrial Alteration in Adipose Tissue. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 124, 88–98. [Google Scholar] [CrossRef]
- Long, M.-H.; Zhang, C.; Xu, D.-Q.; Fu, W.-L.; Gan, X.-D.; Li, F.; Wang, Q.; Xia, W.; Xu, D.-G. PM2.5 Aggravates Diabetes via the Systemically Activated IL-6-Mediated STAT3/SOCS3 Pathway in Rats’ Liver. Environ. Pollut. 2020, 256, 113342. [Google Scholar] [CrossRef]
- Pan, X.; Yu, Q.; Chen, S.; Li, Y.; Jiao, T.; Li, W.; Zhang, C.; Kureshi, A.; Cheng, L.; Xu, Q. Dissecting Contributions of Representative Heavy Metal Components in PM2.5 to Its Cytotoxicity. Ecotoxicol. Environ. Saf. 2023, 251, 114562. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, W.; Lu, Z.; Zhang, F.; Ding, W. Airborne PM2.5-Induced Hepatic Insulin Resistance by Nrf2/JNK-Mediated Signaling Pathway. Int. J. Environ. Res. Public. Health 2017, 14, 787. [Google Scholar] [CrossRef] [PubMed]
- Laing, S.; Wang, G.; Briazova, T.; Zhang, C.; Wang, A.; Zheng, Z.; Gow, A.; Chen, A.F.; Rajagopalan, S.; Chen, L.C.; et al. Airborne Particulate Matter Selectively Activates Endoplasmic Reticulum Stress Response in the Lung and Liver Tissues. Am. J. Physiol. Cell Physiol. 2010, 299, C736–C749. [Google Scholar] [CrossRef] [PubMed]
- Flessa, C.-M.; Kyrou, I.; Nasiri-Ansari, N.; Kaltsas, G.; Papavassiliou, A.G.; Kassi, E.; Randeva, H.S. Endoplasmic Reticulum Stress and Autophagy in the Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD): Current Evidence and Perspectives. Curr. Obes. Rep. 2021, 10, 134–161. [Google Scholar] [CrossRef] [PubMed]
- Kish, L.; Hotte, N.; Kaplan, G.G.; Vincent, R.; Tso, R.; Gänzle, M.; Rioux, K.P.; Thiesen, A.; Barkema, H.W.; Wine, E.; et al. Environmental Particulate Matter Induces Murine Intestinal Inflammatory Responses and Alters the Gut Microbiome. PLoS ONE 2013, 8, e62220. [Google Scholar] [CrossRef]
- Mutlu, E.A.; Engen, P.A.; Soberanes, S.; Urich, D.; Forsyth, C.B.; Nigdelioglu, R.; Chiarella, S.E.; Radigan, K.A.; Gonzalez, A.; Jakate, S.; et al. Particulate Matter Air Pollution Causes Oxidant-Mediated Increase in Gut Permeability in Mice. Part. Fibre Toxicol. 2011, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Ferro, D.; Baratta, F.; Pastori, D.; Cocomello, N.; Colantoni, A.; Angelico, F.; Del Ben, M. New Insights into the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Gut-Derived Lipopolysaccharides and Oxidative Stress. Nutrients 2020, 12, 2762. [Google Scholar] [CrossRef]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The Role of the Gut Microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Safari, Z.; Gérard, P. The Links between the Gut Microbiome and Non-Alcoholic Fatty Liver Disease (NAFLD). Cell. Mol. Life Sci. CMLS 2019, 76, 1541–1558. [Google Scholar] [CrossRef]
- Li, R.; Sun, Q.; Lam, S.M.; Chen, R.; Zhu, J.; Gu, W.; Zhang, L.; Tian, H.; Zhang, K.; Chen, L.-C.; et al. Sex-Dependent Effects of Ambient PM2.5 Pollution on Insulin Sensitivity and Hepatic Lipid Metabolism in Mice. Part. Fibre Toxicol. 2020, 17, 14. [Google Scholar] [CrossRef]
- Goettems-Fiorin, P.B.; Costa-Beber, L.C.; Dos Santos, J.B.; Friske, P.T.; Sulzbacher, L.M.; Frizzo, M.N.; Ludwig, M.S.; Rhoden, C.R.; Heck, T.G. Ovariectomy Predisposes Female Rats to Fine Particulate Matter Exposure’s Effects by Altering Metabolic, Oxidative, pro-Inflammatory, and Heat-Shock Protein Levels. Environ. Sci. Pollut. Res. Int. 2019, 26, 20581–20594. [Google Scholar] [CrossRef]
- Donnelly, M.C.; Stableforth, W.; Krag, A.; Reuben, A. The Negative Bidirectional Interaction between Climate Change and the Prevalence and Care of Liver Disease: A Joint BSG, BASL, EASL, and AASLD Commentary. J. Hepatol. 2022, 76, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Hadley, K.; Wheat, S.; Rogers, H.H.; Balakumar, A.; Gonzales-Pacheco, D.; Davis, S.S.; Linstadt, H.; Cushing, T.; Ziska, L.H.; Piper, C.; et al. Mechanisms Underlying Food Insecurity in the Aftermath of Climate-Related Shocks: A Systematic Review. Lancet Planet. Health 2023, 7, e242–e250. [Google Scholar] [CrossRef] [PubMed]
- Fanzo, J.C.; Downs, S.M. Climate Change and Nutrition-Associated Diseases. Nat. Rev. Dis. Primer 2021, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.A.; Sharda, P.; Patel, J.; Gubbi, S.; Bansal, R.; Bartel, M.J. Climate Change and Obesity. Horm. Metab. Res. 2021, 53, 575–587. [Google Scholar] [CrossRef]
- Krishnan, A.; Mou, X. A Brief Review of the Structure, Cytotoxicity, Synthesis, and Biodegradation of Microcystins. Water 2021, 13, 2147. [Google Scholar] [CrossRef]
- Chorus, I.; Fastner, J.; Welker, M. Cyanobacteria and Cyanotoxins in a Changing Environment: Concepts, Controversies, Challenges. Water 2021, 13, 2463. [Google Scholar] [CrossRef]
- Bui, T.; Dao, T.-S.; Vo, T.-G.; Lürling, M. Warming Affects Growth Rates and Microcystin Production in Tropical Bloom-Forming Microcystis Strains. Toxins 2018, 10, 123. [Google Scholar] [CrossRef]
- Lad, A.; Breidenbach, J.D.; Su, R.C.; Murray, J.; Kuang, R.; Mascarenhas, A.; Najjar, J.; Patel, S.; Hegde, P.; Youssef, M.; et al. As We Drink and Breathe: Adverse Health Effects of Microcystins and Other Harmful Algal Bloom Toxins in the Liver, Gut, Lungs and Beyond. Life 2022, 12, 418. [Google Scholar] [CrossRef]
- Arman, T.; Clarke, J. Microcystin Toxicokinetics, Molecular Toxicology, and Pathophysiology in Preclinical Rodent Models and Humans. Toxins 2021, 13, 537. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, H.; Du, X.; Shi, Z.; Liu, X.; Wang, R.; Zhang, S.; Tian, Z.; Shi, L.; Guo, H.; et al. Advances in the Toxicology Research of Microcystins Based on Omics Approaches. Environ. Int. 2021, 154, 106661. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, Y.; Xie, L.; Wang, L.; He, Y.; Wan, X.; Xue, Q. Long-Term Environmental Exposure to Microcystins Increases the Risk of Nonalcoholic Fatty Liver Disease in Humans: A Combined Fisher-Based Investigation and Murine Model Study. Environ. Int. 2020, 138, 105648. [Google Scholar] [CrossRef]
- Lad, A.; Su, R.; Breidenbach, J.; Stemmer, P.; Carruthers, N.; Sanchez, N.; Khalaf, F.; Zhang, S.; Kleinhenz, A.; Dube, P.; et al. Chronic Low Dose Oral Exposure to Microcystin-LR Exacerbates Hepatic Injury in a Murine Model of Non-Alcoholic Fatty Liver Disease. Toxins 2019, 11, 486. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Li, G.; Chen, J.; Lin, J.; Zeng, C.; Chen, J.; Deng, J.; Xie, P. Prolonged Exposure to Low-Dose Microcystin Induces Nonalcoholic Steatohepatitis in Mice: A Systems Toxicology Study. Arch. Toxicol. 2017, 91, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Arman, T.; Lynch, K.D.; Montonye, M.L.; Goedken, M.; Clarke, J.D. Sub-Chronic Microcystin-LR Liver Toxicity in Preexisting Diet-Induced Nonalcoholic Steatohepatitis in Rats. Toxins 2019, 11, 398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, X.-X.; Wu, B.; Yin, J.; Yu, Y.; Yang, L. Comprehensive Insights into Microcystin-LR Effects on Hepatic Lipid Metabolism Using Cross-Omics Technologies. J. Hazard. Mater. 2016, 315, 126–134. [Google Scholar] [CrossRef]
- Zheng, S.; Yang, Y.; Wen, C.; Liu, W.; Cao, L.; Feng, X.; Chen, J.; Wang, H.; Tang, Y.; Tian, L.; et al. Effects of Environmental Contaminants in Water Resources on Nonalcoholic Fatty Liver Disease. Environ. Int. 2021, 154, 106555. [Google Scholar] [CrossRef]
- Clarke, J.D.; Dzierlenga, A.; Arman, T.; Toth, E.; Li, H.; Lynch, K.D.; Tian, D.-D.; Goedken, M.; Paine, M.F.; Cherrington, N. Nonalcoholic Fatty Liver Disease Alters Microcystin-LR Toxicokinetics and Acute Toxicity. Toxicon 2019, 162, 1–8. [Google Scholar] [CrossRef]
- Mrdjen, I.; Morse, M.; Ruch, R.; Knobloch, T.; Choudhary, S.; Weghorst, C.; Lee, J. Impact of Microcystin-LR on Liver Function Varies by Dose and Sex in Mice. Toxins 2018, 10, 435. [Google Scholar] [CrossRef]
- Symonds, M.E.; Farhat, G.; Aldiss, P.; Pope, M.; Budge, H. Brown Adipose Tissue and Glucose Homeostasis—The Link between Climate Change and the Global Rise in Obesity and Diabetes. Adipocyte 2019, 8, 46–50. [Google Scholar] [CrossRef]
- Turner, J.B.; Kumar, A.; Koch, C.A. The Effects of Indoor and Outdoor Temperature on Metabolic Rate and Adipose Tissue—The Mississippi Perspective on the Obesity Epidemic. Rev. Endocr. Metab. Disord. 2016, 17, 61–71. [Google Scholar] [CrossRef]
- Hankir, M.K.; Klingenspor, M. Brown Adipocyte Glucose Metabolism: A Heated Subject. EMBO Rep. 2018, 19, e46404. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Q.A.; Liu, Y.; Jiang, L. Energy Metabolism in Brown Adipose Tissue. FEBS J. 2021, 288, 3647–3662. [Google Scholar] [CrossRef] [PubMed]
- Wibmer, A.G.; Becher, T.; Eljalby, M.; Crane, A.; Andrieu, P.C.; Jiang, C.S.; Vaughan, R.; Schöder, H.; Cohen, P. Brown Adipose Tissue Is Associated with Healthier Body Fat Distribution and Metabolic Benefits Independent of Regional Adiposity. Cell Rep. Med. 2021, 2, 100332. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, J.; Dai, H.; Duan, Y.; An, Y.; Shi, L.; Lv, Y.; Li, H.; Wang, C.; Ma, Q.; et al. Brown and Beige Adipose Tissue: A Novel Therapeutic Strategy for Obesity and Type 2 Diabetes Mellitus. Adipocyte 2021, 10, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Pace, N.P.; Vassallo, J.; Calleja-Agius, J. Gestational Diabetes, Environmental Temperature and Climate Factors—From Epidemiological Evidence to Physiological Mechanisms. Early Hum. Dev. 2021, 155, 105219. [Google Scholar] [CrossRef]
- Greenhill, C. Low Brown Adipose Tissue Activity Linked to NAFLD. Nat. Rev. Endocrinol. 2021, 17, 707. [Google Scholar] [CrossRef]
- Ahmed, B.A.; Ong, F.J.; Barra, N.G.; Blondin, D.P.; Gunn, E.; Oreskovich, S.M.; Szamosi, J.C.; Syed, S.A.; Hutchings, E.K.; Konyer, N.B.; et al. Lower Brown Adipose Tissue Activity Is Associated with Non-Alcoholic Fatty Liver Disease but Not Changes in the Gut Microbiota. Cell Rep. Med. 2021, 2, 100397. [Google Scholar] [CrossRef]
- Scheele, C.; Wolfrum, C. Brown Adipose Crosstalk in Tissue Plasticity and Human Metabolism. Endocr. Rev. 2020, 41, 53–65. [Google Scholar] [CrossRef]
- Gavaldà-Navarro, A.; Villarroya, J.; Cereijo, R.; Giralt, M.; Villarroya, F. The Endocrine Role of Brown Adipose Tissue: An Update on Actors and Actions. Rev. Endocr. Metab. Disord. 2022, 23, 31–41. [Google Scholar] [CrossRef]
- Keuper, M.; Jastroch, M. The Good and the BAT of Metabolic Sex Differences in Thermogenic Human Adipose Tissue. Mol. Cell. Endocrinol. 2021, 533, 111337. [Google Scholar] [CrossRef]
- Kaikaew, K.; Grefhorst, A.; Visser, J.A. Sex Differences in Brown Adipose Tissue Function: Sex Hormones, Glucocorticoids, and Their Crosstalk. Front. Endocrinol. 2021, 12, 652444. [Google Scholar] [CrossRef] [PubMed]
- Herz, C.T.; Kulterer, O.C.; Prager, M.; Marculescu, R.; Langer, F.B.; Prager, G.; Kautzky-Willer, A.; Haug, A.R.; Kiefer, F.W. Sex Differences in Brown Adipose Tissue Activity and Cold-Induced Thermogenesis. Mol. Cell. Endocrinol. 2021, 534, 111365. [Google Scholar] [CrossRef] [PubMed]
- Gómez-García, I.; Trepiana, J.; Fernández-Quintela, A.; Giralt, M.; Portillo, M.P. Sexual Dimorphism in Brown Adipose Tissue Activation and White Adipose Tissue Browning. Int. J. Mol. Sci. 2022, 23, 8250. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, L.A.; Kim, K.; Leitner, B.P.; Cassimatis, T.M.; O’Mara, A.E.; Johnson, J.W.; Halprin, M.S.; McGehee, S.M.; Brychta, R.J.; Cypess, A.M.; et al. Sexual Dimorphisms in Adult Human Brown Adipose Tissue. Obes. Silver Spring Md 2020, 28, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S.; Benedusi, V.; Fontana, R.; Maggi, A. Energy Metabolism and Fertility—A Balance Preserved for Female Health. Nat. Rev. Endocrinol. 2014, 10, 13–23. [Google Scholar] [CrossRef] [PubMed]
- McCartney, C.R.; Marshall, J.C. Polycystic Ovary Syndrome. N. Engl. J. Med. 2016, 375, 54–64. [Google Scholar] [CrossRef]
- Joham, A.E.; Norman, R.J.; Stener-Victorin, E.; Legro, R.S.; Franks, S.; Moran, L.J.; Boyle, J.; Teede, H.J. Polycystic Ovary Syndrome. Lancet Diabetes Endocrinol. 2022, 10, 668–680. [Google Scholar] [CrossRef]
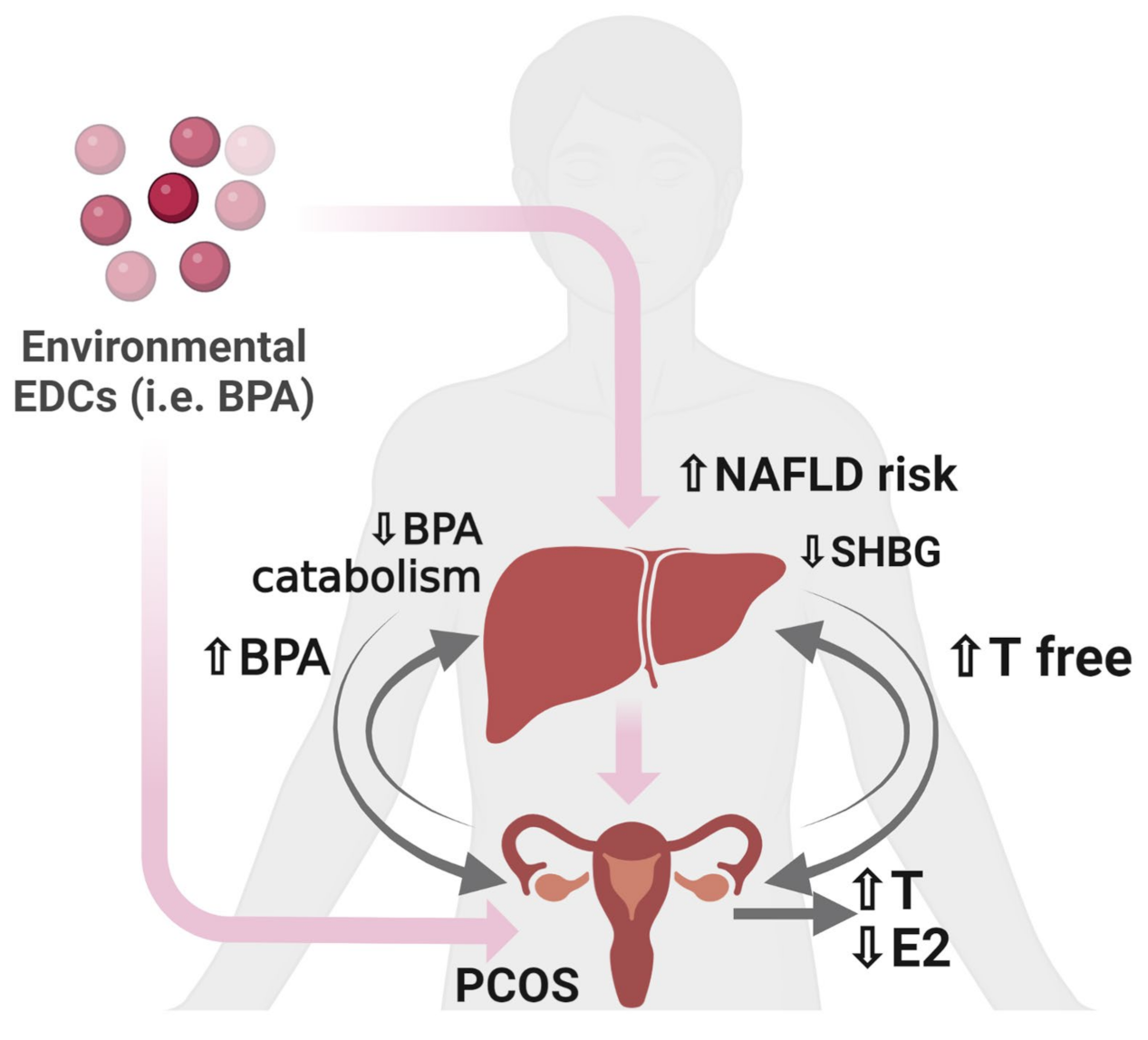
- Spremović Rađenović, S.; Pupovac, M.; Andjić, M.; Bila, J.; Srećković, S.; Gudović, A.; Dragaš, B.; Radunović, N. Prevalence, Risk Factors, and Pathophysiology of Nonalcoholic Fatty Liver Disease (NAFLD) in Women with Polycystic Ovary Syndrome (PCOS). Biomedicines 2022, 10, 131. [Google Scholar] [CrossRef]
- Falzarano, C.; Lofton, T.; Osei-Ntansah, A.; Oliver, T.; Southward, T.; Stewart, S.; Andrisse, S. Nonalcoholic Fatty Liver Disease in Women and Girls with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2022, 107, 258–272. [Google Scholar] [CrossRef]
- Sarkar, M.; Terrault, N.; Chan, W.; Cedars, M.I.; Huddleston, H.G.; Duwaerts, C.C.; Balitzer, D.; Gill, R.M. Polycystic Ovary Syndrome (PCOS) Is Associated with NASH Severity and Advanced Fibrosis. Liver Int. Off. J. Int. Assoc. Study Liver 2020, 40, 355–359. [Google Scholar] [CrossRef]
- Cui, P.; Hu, W.; Ma, T.; Hu, M.; Tong, X.; Zhang, F.; Shi, J.; Xu, X.; Li, X.; Shao, L.R.; et al. Long-Term Androgen Excess Induces Insulin Resistance and Non-Alcoholic Fatty Liver Disease in PCOS-like Rats. J. Steroid Biochem. Mol. Biol. 2021, 208, 105829. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, R.A.; Calogero, A.E.; Di Mauro, M.; Mongioi’, L.M.; Cannarella, R.; Rosta, G.; La Vignera, S. Androgen Excess and Metabolic Disorders in Women with PCOS: Beyond the Body Mass Index. J. Endocrinol. Investig. 2018, 41, 383–388. [Google Scholar] [CrossRef]
- Roy, S.; Abudu, A.; Salinas, I.; Sinha, N.; Cline-Fedewa, H.; Yaw, A.M.; Qi, W.; Lydic, T.A.; Takahashi, D.L.; Hennebold, J.D.; et al. Androgen-Mediated Perturbation of the Hepatic Circadian System Through Epigenetic Modulation Promotes NAFLD in PCOS Mice. Endocrinology 2022, 163, bqac127. [Google Scholar] [CrossRef] [PubMed]
- Palioura, E.; Diamanti-Kandarakis, E. Polycystic Ovary Syndrome (PCOS) and Endocrine Disrupting Chemicals (EDCs). Rev. Endocr. Metab. Disord. 2015, 16, 365–371. [Google Scholar] [CrossRef]
- Hammarstrand, S.; Jakobsson, K.; Andersson, E.; Xu, Y.; Li, Y.; Olovsson, M.; Andersson, E.M. Perfluoroalkyl Substances (PFAS) in Drinking Water and Risk for Polycystic Ovarian Syndrome, Uterine Leiomyoma, and Endometriosis: A Swedish Cohort Study. Environ. Int. 2021, 157, 106819. [Google Scholar] [CrossRef] [PubMed]
- Al-Saleh, I. The Relationship between Urinary Phthalate Metabolites and Polycystic Ovary Syndrome in Women Undergoing in Vitro Fertilization: Nested Case-Control Study. Chemosphere 2022, 286, 131495. [Google Scholar] [CrossRef]
- Kim, K.; Pollack, A.Z.; Nobles, C.J.; Sjaarda, L.A.; Zolton, J.R.; Radoc, J.G.; Schisterman, E.F.; Mumford, S.L. Associations between Blood Cadmium and Endocrine Features Related to PCOS-Phenotypes in Healthy Women of Reproductive Age: A Prospective Cohort Study. Environ. Health Glob. Access Sci. Source 2021, 20, 64. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, L.; Zhao, Y.; Wang, Y. Changes in Serum Heavy Metals in Polycystic Ovary Syndrome and Their Association with Endocrine, Lipid-Metabolism, Inflammatory Characteristics and Pregnancy Outcomes. Reprod. Toxicol. 2022, 111, 20–26. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Yang, Y.-C.; Chang, C.Y.-Y.; Lin, C.-C.; Hsu, W.-H.; Ju, S.-W.; Hsu, C.-Y.; Kao, C.-H. Risk of Polycystic Ovary Syndrome in Women Exposed to Fine Air Pollutants and Acidic Gases: A Nationwide Cohort Analysis. Int. J. Environ. Res. Public. Health 2019, 16, 4816. [Google Scholar] [CrossRef]
- Abudawood, M.; Tabassum, H.; Alanazi, A.H.; Almusallam, F.; Aljaser, F.; Ali, M.N.; Alenzi, N.D.; Alanazi, S.T.; Alghamdi, M.A.; Altoum, G.H.; et al. Antioxidant Status in Relation to Heavy Metals Induced Oxidative Stress in Patients with Polycystic Ovarian Syndrome (PCOS). Sci. Rep. 2021, 11, 22935. [Google Scholar] [CrossRef]
- Zhang, B.; Zhou, W.; Shi, Y.; Zhang, J.; Cui, L.; Chen, Z.-J. Lifestyle and Environmental Contributions to Ovulatory Dysfunction in Women of Polycystic Ovary Syndrome. BMC Endocr. Disord. 2020, 20, 19. [Google Scholar] [CrossRef] [PubMed]
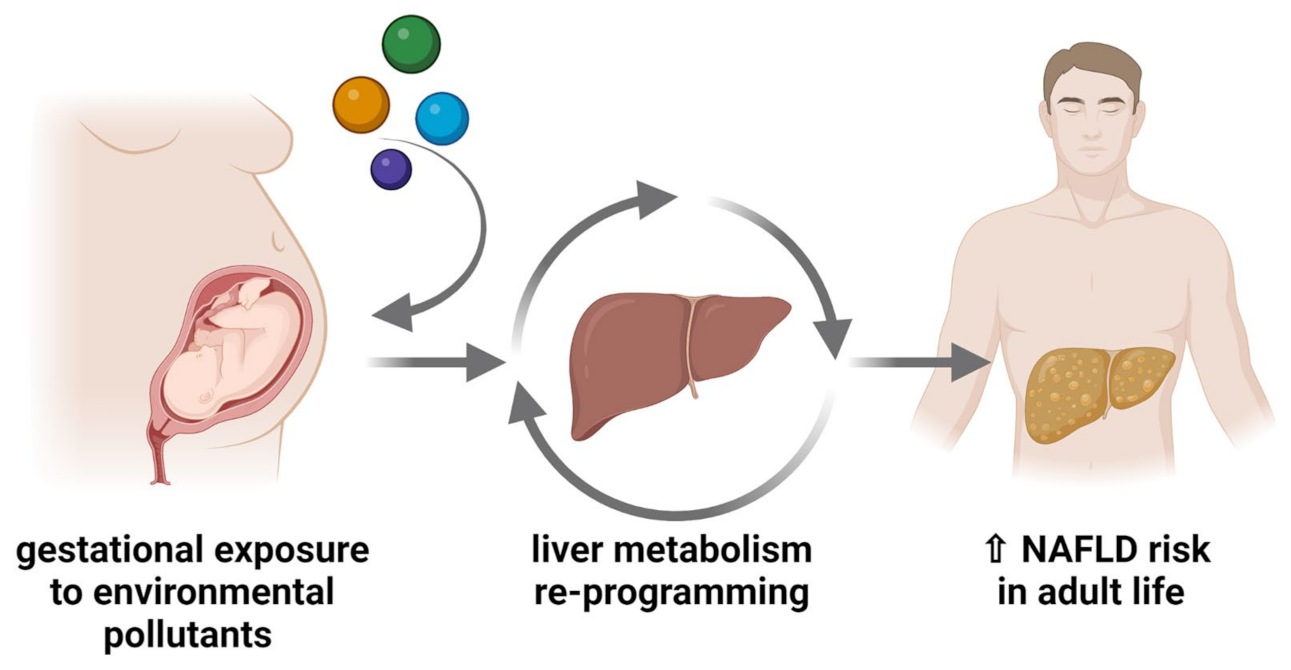
- Treviño, L.S.; Katz, T.A. Endocrine Disruptors and Developmental Origins of Nonalcoholic Fatty Liver Disease. Endocrinology 2018, 159, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.; Chan, C.S.; Drake, A.J. Early Life Programming and the Risk of Non-Alcoholic Fatty Liver Disease. J. Dev. Orig. Health Dis. 2017, 8, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Fan, J.; Wu, G.; Liu, X.; Wu, H.; Liu, J.; Chen, Y.; Su, S.; Cheng, X.; Xu, Z.; et al. Gestational Bisphenol A Exposure Induces Fatty Liver Development in Male Offspring Mice through the Inhibition of HNF1b and Upregulation of PPARγ. Cell Biol. Toxicol. 2021, 37, 65–84. [Google Scholar] [CrossRef]
- Lin, R.; Wu, D.; Wu, F.-J.; Meng, Y.; Zhang, J.-H.; Wang, X.-G.; Jia, L.-H. Non-Alcoholic Fatty Liver Disease Induced by Perinatal Exposure to Bisphenol a Is Associated With Activated MTOR and TLR4/NF-ΚB Signaling Pathways in Offspring Rats. Front. Endocrinol. 2019, 10, 620. [Google Scholar] [CrossRef]
- Shimpi, P.C.; More, V.R.; Paranjpe, M.; Donepudi, A.C.; Goodrich, J.M.; Dolinoy, D.C.; Rubin, B.; Slitt, A.L. Hepatic Lipid Accumulation and Nrf2 Expression Following Perinatal and Peripubertal Exposure to Bisphenol A in a Mouse Model of Nonalcoholic Liver Disease. Environ. Health Perspect. 2017, 125, 087005. [Google Scholar] [CrossRef] [PubMed]
- Dabeer, S.; Raisuddin, S. Perinatal Exposure to Environmental Endocrine Disruptor Bisphenol A Aggravates the Onset of Non-Alcoholic Fatty Liver Disease (NAFLD) in Weanling F1 Offspring of Obese Rats. Environ. Sci. Pollut. Res. 2023, 30, 3146–3165. [Google Scholar] [CrossRef]
- Wei, J.; Sun, X.; Chen, Y.; Li, Y.; Song, L.; Zhou, Z.; Xu, B.; Lin, Y.; Xu, S. Perinatal Exposure to Bisphenol A Exacerbates Nonalcoholic Steatohepatitis-like Phenotype in Male Rat Offspring Fed on a High-Fat Diet. J. Endocrinol. 2014, 222, 313–325. [Google Scholar] [CrossRef]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.-X.; Lezmi, S. Developmental Bisphenol A (BPA) Exposure Leads to Sex-Specific Modification of Hepatic Gene Expression and Epigenome at Birth That May Exacerbate High-Fat Diet-Induced Hepatic Steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef]
- Marchlewicz, E.; McCabe, C.; Djuric, Z.; Hoenerhoff, M.; Barks, J.; Tang, L.; Song, P.X.; Peterson, K.; Padmanabhan, V.; Dolinoy, D.C. Gestational Exposure to High Fat Diets and Bisphenol A Alters Metabolic Outcomes in Dams and Offspring, but Produces Hepatic Steatosis Only in Dams. Chemosphere 2022, 286, 131645. [Google Scholar] [CrossRef]
- Wang, D.; Yan, S.; Yan, J.; Teng, M.; Meng, Z.; Li, R.; Zhou, Z.; Zhu, W. Effects of Triphenyl Phosphate Exposure during Fetal Development on Obesity and Metabolic Dysfunctions in Adult Mice: Impaired Lipid Metabolism and Intestinal Dysbiosis. Environ. Pollut. 2019, 246, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Ditzel, E.J.; Nguyen, T.; Parker, P.; Camenisch, T.D. Effects of Arsenite Exposure during Fetal Development on Energy Metabolism and Susceptibility to Diet-Induced Fatty Liver Disease in Male Mice. Environ. Health Perspect. 2016, 124, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, N.; Conti, D.V.; Jin, R.; Margetaki, K.; Valvi, D.; Siskos, A.P.; Maitre, L.; Garcia, E.; Varo, N.; Zhao, Y.; et al. Prenatal Exposure to Perfluoroalkyl Substances Associated with Increased Susceptibility to Liver Injury in Children. Hepatology 2020, 72, 1758–1770. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, N.; Golden-Mason, L.; Margetaki, K.; Zhao, Y.; Valvi, D.; Garcia, E.; Maitre, L.; Andrusaityte, S.; Basagana, X.; Borràs, E.; et al. In Utero Exposure to Mercury Is Associated with Increased Susceptibility to Liver Injury and Inflammation in Childhood. Hepatology 2021, 74, 1546–1559. [Google Scholar] [CrossRef]
- Garcia, E.; Stratakis, N.; Valvi, D.; Maitre, L.; Varo, N.; Aasvang, G.M.; Andrusaityte, S.; Basagana, X.; Casas, M.; de Castro, M.; et al. Prenatal and Childhood Exposure to Air Pollution and Traffic and the Risk of Liver Injury in European Children. Environ. Epidemiol. 2021, 5, e153. [Google Scholar] [CrossRef]
- Lo, E.K.K.; Felicianna; Xu, J.-H.; Zhan, Q.; Zeng, Z.; El-Nezami, H. The Emerging Role of Branched-Chain Amino Acids in Liver Diseases. Biomedicines 2022, 10, 1444. [Google Scholar] [CrossRef]
- Grenier-Larouche, T.; Coulter Kwee, L.; Deleye, Y.; Leon-Mimila, P.; Walejko, J.M.; McGarrah, R.W.; Marceau, S.; Trahan, S.; Racine, C.; Carpentier, A.C.; et al. Altered Branched-Chain α-Keto Acid Metabolism Is a Feature of NAFLD in Individuals with Severe Obesity. JCI Insight 2022, 7, e159204. [Google Scholar] [CrossRef]
- van den Berg, E.H.; Flores-Guerrero, J.L.; Gruppen, E.G.; de Borst, M.H.; Wolak-Dinsmore, J.; Connelly, M.A.; Bakker, S.J.L.; Dullaart, R.P.F. Non-Alcoholic Fatty Liver Disease and Risk of Incident Type 2 Diabetes: Role of Circulating Branched-Chain Amino Acids. Nutrients 2019, 11, 705. [Google Scholar] [CrossRef]
- Lischka, J.; Schanzer, A.; Hojreh, A.; Ba Ssalamah, A.; Item, C.B.; de Gier, C.; Walleczek, N.-K.; Metz, T.F.; Jakober, I.; Greber-Platzer, S.; et al. A Branched-Chain Amino Acid-Based Metabolic Score Can Predict Liver Fat in Children and Adolescents with Severe Obesity. Pediatr. Obes. 2021, 16, e12739. [Google Scholar] [CrossRef]
- Yan, S.; Tian, S.; Meng, Z.; Teng, M.; Sun, W.; Jia, M.; Zhou, Z.; Bi, S.; Zhu, W. Exposure to Nitenpyram during Pregnancy Causes Colonic Mucosal Damage and Non-Alcoholic Steatohepatitis in Mouse Offspring: The Role of Gut Microbiota. Environ. Pollut. 2021, 271, 116306. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Z.; Wang, X.; Hu, W.; Chu, X.; Qian, M.; Wang, R.; Yu, S.; Wu, Q.; Tang, J.; et al. Effects of Polystyrene Nanoplastic Gestational Exposure on Mice. Chemosphere 2023, 324, 138255. [Google Scholar] [CrossRef]
- Sun, J.; Liu, H.; Zhang, C.; Liu, X.; Sun, X.; Chen, X.; Yang, G.; Wang, N. Predisposed Obesity and Long-Term Metabolic Diseases from Maternal Exposure to Fine Particulate Matter (PM2.5)—A Review of Its Effect and Potential Mechanisms. Life Sci. 2022, 310, 121054. [Google Scholar] [CrossRef]
- Wu, G.; Brown, J.; Zamora, M.L.; Miller, A.; Satterfield, M.C.; Meininger, C.J.; Steinhauser, C.B.; Johnson, G.A.; Burghardt, R.C.; Bazer, F.W.; et al. Adverse Organogenesis and Predisposed Long-Term Metabolic Syndrome from Prenatal Exposure to Fine Particulate Matter. Proc. Natl. Acad. Sci. USA 2019, 116, 11590–11595. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chen, L.; Bennett, E.; Wheeler, A.J.; Southam, K.; Yen, S.; Johnston, F.; Zosky, G.R. Can Maternal Exposure to Air Pollution Affect Post-Natal Liver Development? Toxics 2023, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Pejhan, A.; Agah, J.; Adli, A.; Mehrabadi, S.; Raoufinia, R.; Mokamel, A.; Abroudi, M.; Ghalenovi, M.; Sadeghi, Z.; Bolghanabadi, Z.; et al. Exposure to Air Pollution during Pregnancy and Newborn Liver Function. Chemosphere 2019, 226, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Zhu, P.; Wu, Y.; Jin, Y.; Yu, S.; Wei, H.; Qian, M.; Cao, W.; Xu, S.; et al. Prenatal Exposure to Diesel Exhaust PM2.5 Programmed Non-Alcoholic Fatty Liver Disease Differently in Adult Male Offspring of Mice Fed Normal Chow and a High-Fat Diet. Environ. Pollut. 2019, 255, 113366. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited After a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef]
- Chen, H.; Van Reyk, D.; Oliveira, A.; Chan, Y.L.; Town, S.E.; Rayner, B.; Pollock, C.A.; Saad, S.; George, J.; Padula, M.P.; et al. Sex-Dependent Responses to Maternal Exposure to PM2.5 in the Offspring. Antioxidants 2022, 11, 2255. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolce, A.; Della Torre, S. Sex, Nutrition, and NAFLD: Relevance of Environmental Pollution. Nutrients 2023, 15, 2335. https://doi.org/10.3390/nu15102335
Dolce A, Della Torre S. Sex, Nutrition, and NAFLD: Relevance of Environmental Pollution. Nutrients. 2023; 15(10):2335. https://doi.org/10.3390/nu15102335
Chicago/Turabian StyleDolce, Arianna, and Sara Della Torre. 2023. "Sex, Nutrition, and NAFLD: Relevance of Environmental Pollution" Nutrients 15, no. 10: 2335. https://doi.org/10.3390/nu15102335
APA StyleDolce, A., & Della Torre, S. (2023). Sex, Nutrition, and NAFLD: Relevance of Environmental Pollution. Nutrients, 15(10), 2335. https://doi.org/10.3390/nu15102335