
Different Effects of High-Fat/High-Sucrose and High-Fructose Diets on Advanced Glycation End-Product Accumulation and on Mitochondrial Involvement in Heart and Skeletal Muscle in Mice
 , , , ,
, , , ,  ,
,  , , and
, , and
Abstract
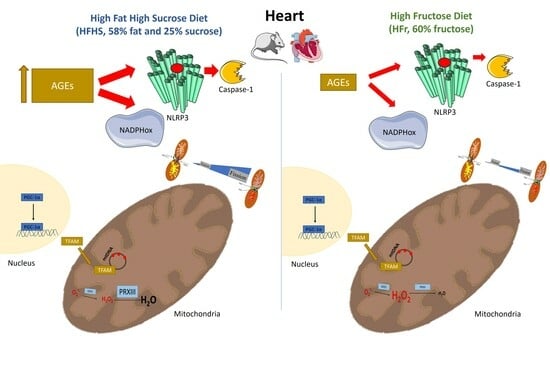
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Oral Glucose Tolerance Test (OGTT)
2.3. Insulin Tolerance Test (ITT)
2.4. Evaluation of Biochemical Markers
2.5. Nε-(Carboxymethyl)-Lysine (CML) Levels in Plasma and Tissues
2.6. Western Blot Analysis
2.7. Determination of Relative mtDNA Copy Number
2.8. Modified Purines Analysis
2.9. Reagents
2.10. Statistical Analysis
3. Results
3.1. Characteristics of Dietary Interventions
3.2. Effects of HFHS and HFr Diets on Metabolic Indicators
3.3. Inflammation and Oxidative Stress in Heart
3.4. AGEs in Plasma, Heart, and TA Muscle Tissues
3.5. Evaluation of Mitochondrial Oxidative Stress and Analysis of Relative mtDNA Content
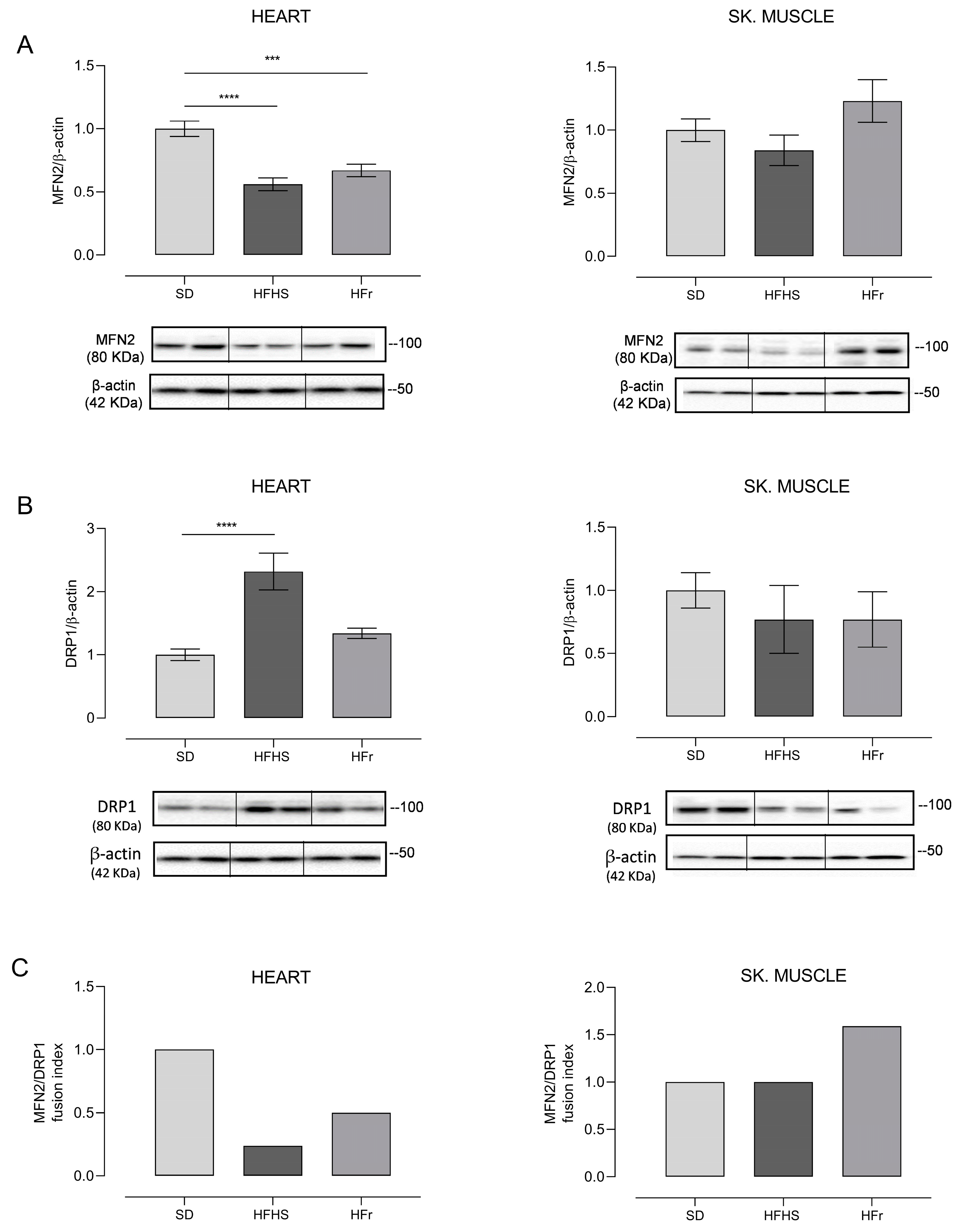
3.6. Evaluation of Mitochondrial Biogenesis and Dynamics
4. Discussion
4.1. Systemic Profile in HFHS Diet
4.1.1. Hearts of HFHS Mice
4.1.2. TA of HFHS Mice
4.2. HFr Diet
4.2.1. Hearts of HFr Mice
4.2.2. TA of HFr Mice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ofei, F. Obesity—A preventable disease. Ghana Med. J. 2005, 39, 98–101. [Google Scholar]
- Rodríguez-Correa, E.; González-Pérez, I.; Clavel-Pérez, P.I.; Contreras-Vargas, Y.; Carvajal, K. Biochemical and nutritional overview of diet-induced metabolic syndrome models in rats: What is the best choice? Nutr. Diabetes 2020, 10, 24. [Google Scholar] [CrossRef]
- West, I.C. Radicals and oxidative stress in diabetes. Diabet. Med. 2000, 17, 171–180. [Google Scholar] [CrossRef]
- Castro, A.M.; Macedo-de la Concha, L.E.; Pantoja-Meléndez, C.A. Low-grade inflammation and its relation to obesity and chronic degenerative diseases. Rev. Med. Hosp. Gen. Méx. 2017, 80, 101–105. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Stenvinkel, P. Obesity—A disease with many aetiologies disguised in the same oversized phenotype: Has the overeating theory failed? Nephrol. Dial. Transplant. 2015, 30, 1656–1664. [Google Scholar] [CrossRef]
- Tappy, L.; Lê, K.-A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef]
- Tappy, L. Fructose-containing caloric sweeteners as a cause of obesity and metabolic disorders. J. Exp. Biol. 2018, 221, jeb164202. [Google Scholar] [CrossRef]
- Mellor, K.M.; Ritchie, R.H.; Delbridge, L.M.D. Reactive oxygen species and insulin-resistant cardiomyopathy. Clin. Exp. Pharmacol. Physiol. 2010, 37, 222–228. [Google Scholar] [CrossRef]
- Taskinen, M.-R.; Packard, C.J.; Borén, J. Dietary Fructose and the Metabolic Syndrome. Nutrients 2019, 11, 1987. [Google Scholar] [CrossRef]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef]
- Sloan, C.; Tuinei, J.; Nemetz, K.; Frandsen, J.; Soto, J.; Wride, N.; Sempokuya, T.; Alegria, L.; Bugger, H.; Abel, E.D. Central Leptin Signaling Is Required to Normalize Myocardial Fatty Acid Oxidation Rates in Caloric-Restricted ob/ob Mice. Diabetes 2011, 60, 1424–1434. [Google Scholar] [CrossRef]
- Palomer, X.; Álvarez-Guardia, D.; Rodríguez-Calvo, R.; Coll, T.; Laguna, J.C.; Davidson, M.M.; Chan, T.O.; Feldman, A.M.; Vázquez-Carrera, M. TNF-α reduces PGC-1α expression through NF-κB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovasc. Res. 2009, 81, 703–712. [Google Scholar] [CrossRef]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef]
- Twarda-Clapa, A.; Olczak, A.; Białkowska, A.M.; Koziołkiewicz, M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Varela, A.T.; Rolo, A.P.; Palmeira, C.M. High-fat and obesogenic diets: Current and future strategies to fight obesity and diabetes. Genes Nutr. 2014, 9, 406. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Moffatt, T.L.; O’Hara, K.A.; Xiang, B.; Dolinsky, V.W.; Duhamel, T.A. Muscle-specific sirtuin 3 overexpression does not attenuate the pathological effects of high-fat/high-sucrose feeding but does enhance cardiac SERCA2a activity. Physiol. Rep. 2021, 9, e14961. [Google Scholar] [CrossRef]
- D’Souza, K.; Nzirorera, C.; Cowie, A.M.; Varghese, G.P.; Trivedi, P.; Eichmann, T.O.; Biswas, D.; Touaibia, M.; Morris, A.J.; Aidinis, V.; et al. Autotaxin-LPA signaling contributes to obesity-induced insulin resistance in muscle and impairs mitochondrial metabolism. J. Lipid Res. 2018, 59, 1805–1817. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Mattox, T.A.; Thayne, K.; Katunga, L.A.; La Favor, J.D.; Neufer, P.D.; Hickner, R.C.; Wingard, C.J.; Anderson, E.J. Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J. Physiol. 2013, 591, 3471–3486. [Google Scholar] [CrossRef]
- Feillet-Coudray, C.; Sutra, T.; Fouret, G.; Ramos, J.; Wrutniak-Cabello, C.; Cabello, G.; Cristol, J.; Coudray, C. Oxidative stress in rats fed a high-fat high-sucrose diet and preventive effect of polyphenols: Involvement of mitochondrial and NAD(P)H oxidase systems. Free. Radic. Biol. Med. 2009, 46, 624–632. [Google Scholar] [CrossRef]
- Sarikaya, B.; Runa, M.; Dayanir, D.; Gündüztepe, Y.; Pinar, L. Paraoxonase and oxidative stress changes in left and right ventricles of exhaustively exercised rats. Can. J. Physiol. Pharmacol. 2021, 99, 752–759. [Google Scholar] [CrossRef]
- Felicetti, F.; Aimaretti, E.; Dal Bello, F.; Gatti, F.; Godono, A.; Saba, F.; Einaudi, G.; Collino, M.; Fagioli, F.; Aragno, M.; et al. Advanced glycation end products and their related signaling cascades in adult survivors of childhood Hodgkin lymphoma: A possible role in the onset of late complications. Free Radic. Biol. Med. 2022, 178, 76–82. [Google Scholar] [CrossRef]
- Chimienti, G.; Picca, A.; Fracasso, F.; Russo, F.; Orlando, A.; Riezzo, G.; Leeuwenburgh, C.; Pesce, V.; Lezza, A.M.S. The Age-Sensitive Efficacy of Calorie Restriction on Mitochondrial Biogenesis and mtDNA Damage in Rat Liver. Int. J. Mol. Sci. 2021, 22, 1665. [Google Scholar] [CrossRef]
- Chimienti, G.; Picca, A.; Fracasso, F.; Marzetti, E.; Calvani, R.; Leeuwenburgh, C.; Russo, F.; Lezza, A.M.S.; Pesce, V. Differences in Liver TFAM Binding to mtDNA and mtDNA Damage between Aged and Extremely Aged Rats. Int. J. Mol. Sci. 2019, 20, 2601. [Google Scholar] [CrossRef]
- Picca, A.; Pesce, V.; Fracasso, F.; Joseph, A.-M.; Leeuwenburgh, C.; Lezza, A.M.S. Aging and Calorie Restriction Oppositely Affect Mitochondrial Biogenesis through TFAM Binding at Both Origins of Mitochondrial DNA Replication in Rat Liver. PLoS ONE 2013, 8, e74644. [Google Scholar] [CrossRef]
- Matias, A.M.; Müller Estevam, W.; Murucci Coelho, P.; Haese, D.; Kobi, J.B.B.S.; Lima-Leopoldo, A.P.; Soares Leopoldo, A. Differential Effects of High Sugar, High Lard or a Combination of Both on Nutritional, Hormonal and Cardiovascular Metabolic Profiles of Rodents. Nutrients 2018, 10, 1071. [Google Scholar] [CrossRef]
- Marín-Royo, G.; Rodríguez, C.; Le Pape, A.; Jurado-Lopez, R.; Luaces, M.; Antequera, A.; Martínez-Gonzalez, J.; Souza-Neto, F.V.; Nieto, M.L.; Martínez-Martínez, E.; et al. The role of mitochondrial oxidative stress in the metabolic alterations in diet-induced obesity in rats. FASEB J. 2019, 33, 12060–12072. [Google Scholar] [CrossRef]
- Pranprawit, A.; Wolber, F.M.; Heyes, J.A.; Molan, A.L.; Kruger, M.C. Short-term and long-term effects of excessive consumption of saturated fats and/or sucrose on metabolic variables in Sprague Dawley rats: A pilot study. J. Sci. Food Agric. 2013, 93, 3191–3197. [Google Scholar] [CrossRef]
- Moorkens, K.; Leroy, J.L.M.R.; Verheyen, S.; Marei, W.F.A. Effects of an obesogenic diet on the oviduct depend on the duration of feeding. PLoS ONE 2022, 17, e0275379. [Google Scholar] [CrossRef]
- Abu Bakar, M.H.; Shariff, K.A.; Tan, J.S.; Lee, L.K. Celastrol attenuates inflammatory responses in adipose tissues and improves skeletal muscle mitochondrial functions in high fat diet-induced obese rats via upregulation of AMPK/SIRT1 signaling pathways. Eur. J. Pharmacol. 2020, 883, 173371. [Google Scholar] [CrossRef]
- Williams, L.M.; Campbell, F.M.; Drew, J.E.; Koch, C.; Hoggard, N.; Rees, W.D.; Kamolrat, T.; Ngo, H.T.; Steffensen, I.-L.; Gray, S.R.; et al. The development of diet-induced obesity and glucose intolerance in C57BL/6 mice on a high-fat diet consists of distinct phases. PLoS ONE 2014, 9, e106159. [Google Scholar] [CrossRef]
- Kotzé-Hörstmann, L.; Cois, A.; Johnson, R.; Mabasa, L.; Shabalala, S.; Van Jaarsveld, P.J.; Gijsen, H.S.-V. Characterization and Comparison of the Divergent Metabolic Consequences of High-Sugar and High-Fat Diets in Male Wistar Rats. Front. Physiol. 2022, 13, 904366. [Google Scholar] [CrossRef]
- Benetti, E.; Mastrocola, R.; Vitarelli, G.; Cutrin, J.C.; Nigro, D.; Chiazza, F.; Mayoux, E.; Collino, M.; Fantozzi, R. Empagliflozin Protects against Diet-Induced NLRP-3 Inflammasome Activation and Lipid Accumulation. J. Pharmacol. Exp. Ther. 2016, 359, 45–53. [Google Scholar] [CrossRef]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef]
- Li, S.; Liu, Y.; Sigmon, V.K.; McCort, A.; Ren, J. High-fat diet enhances visceral advanced glycation end products, nuclear O-Glc-Nac modification, p38 mitogen-activated protein kinase activation and apoptosis. Diabetes Obes. Metab. 2005, 7, 448–454. [Google Scholar] [CrossRef]
- Chimienti, G.; Picca, A.; Sirago, G.; Fracasso, F.; Calvani, R.; Bernabei, R.; Russo, F.; Carter, C.S.; Leeuwenburgh, C.; Pesce, V.; et al. Increased TFAM binding to mtDNA damage hot spots is associated with mtDNA loss in aged rat heart. Free Radic. Biol. Med. 2018, 124, 447–453. [Google Scholar] [CrossRef]
- Yuzefovych, L.V.; Musiyenko, S.I.; Wilson, G.L.; Rachek, L.I. Mitochondrial DNA Damage and Dysfunction, and Oxidative Stress Are Associated with Endoplasmic Reticulum Stress, Protein Degradation and Apoptosis in High Fat Diet-Induced Insulin Resistance Mice. PLoS ONE 2013, 8, e54059. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Scorrano, L. Determinants and outcomes of mitochondrial dynamics. Mol. Cell 2023, 83, 857–876. [Google Scholar] [CrossRef]
- Sonn, S.K.; Song, E.J.; Seo, S.; Kim, Y.Y.; Um, J.-H.; Yeo, F.J.; Lee, D.S.; Jeon, S.; Lee, M.-N.; Jin, J.; et al. Peroxiredoxin 3 deficiency induces cardiac hypertrophy and dysfunction by impaired mitochondrial quality control. Redox Biol. 2022, 51, 102275. [Google Scholar] [CrossRef]
- Tong, M.; Zablocki, D.; Sadoshima, J. The role of Drp1 in mitophagy and cell death in the heart. J. Mol. Cell. Cardiol. 2020, 142, 138–145. [Google Scholar] [CrossRef]
- Zheng, Y.; Gibb, A.A.; Xu, H.; Liu, S.; Hill, B.G. The metabolic state of the heart regulates mitochondrial supercomplex abundance in mice. Redox Biol. 2023, 63, 102740. [Google Scholar] [CrossRef]
- Luptak, I.; Qin, F.; Sverdlov, A.L.; Pimentel, D.R.; Panagia, M.; Croteau, D.; Siwik, D.A.; Bachschmid, M.M.; He, H.; Balschi, J.A.; et al. Energetic Dysfunction Is Mediated by Mitochondrial Reactive Oxygen Species and Precedes Structural Remodeling in Metabolic Heart Disease. Antioxid. Redox Signal. 2019, 31, 539–549. [Google Scholar] [CrossRef]
- Rasool, S.; Geetha, T.; Broderick, T.L.; Babu, J.R. High Fat With High Sucrose Diet Leads to Obesity and Induces Myodegeneration. Front. Physiol. 2018, 9, 1054. [Google Scholar] [CrossRef]
- Chanseaume, E.; Malpuech-Brugère, C.; Patrac, V.; Bielicki, G.; Rousset, P.; Couturier, K.; Salles, J.; Renou, J.-P.; Boirie, Y.; Morio, B. Diets High in Sugar, Fat, and Energy Induce Muscle Type–Specific Adaptations in Mitochondrial Functions in Rats. J. Nutr. 2006, 136, 2194–2200. [Google Scholar] [CrossRef]
- Jung, S.; Bae, H.; Song, W.-S.; Jang, C. Dietary Fructose and Fructose-Induced Pathologies. Annu. Rev. Nutr. 2022, 42, 45–66. [Google Scholar] [CrossRef]
- Herman, M.A.; Birnbaum, M.J. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab. 2021, 33, 2329–2354. [Google Scholar] [CrossRef]
- Collotta, D.; Lucarini, L.; Chiazza, F.; Cento, A.S.; Durante, M.; Sgambellone, S.; Chini, J.; Baratta, F.; Aragno, M.; Mastrocola, R.; et al. Reduced Susceptibility to Sugar-Induced Metabolic Derangements and Impairments of Myocardial Redox Signaling in Mice Chronically Fed with D-Tagatose when Compared to Fructose. Oxidative Med. Cell. Longev. 2018, 2018, 5042428. [Google Scholar] [CrossRef]
- Pesce, V.; Fracasso, F.; Cassano, P.; Lezza, A.M.S.; Cantatore, P.; Gadaleta, M.N.; Yu-Wai-Man, P.; Votruba, M.; Moore, A.T.; Chinnery, P.F.; et al. Acetyl-L-carnitine supplementation to old rats partially reverts the age-related mitochondrial decay of soleus muscle by activating peroxisome proliferator-activated receptor gamma coactivator-1alpha-dependent mitochondrial biogenesis. Rejuvenation Res. 2010, 13, 148–151. [Google Scholar] [CrossRef]
- Federico, M.; Zavala, M.; Vico, T.; López, S.; Portiansky, E.; Alvarez, S.; Abrille, M.C.V.; Palomeque, J. CaMKII activation in early diabetic hearts induces altered sarcoplasmic reticulum-mitochondria signaling. Sci. Rep. 2021, 11, 20025. [Google Scholar] [CrossRef]
- Meneses, M.J.; Sousa-Lima, I.; Jarak, I.; Raposo, J.F.; Alves, M.G.; Macedo, M.P. Distinct impacts of fat and fructose on the liver, muscle, and adipose tissue metabolome: An integrated view. Front. Endocrinol. 2022, 13, 898471. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SD (n = 15) | HFHS (n = 15) | HFr (n = 15) | |
|---|---|---|---|
| Diet energy supply (kcal/g) | 3.850 | 5.560 | 3.850 |
| Food intake (g/day/mouse) | 2.889 ± 0.308 | 3.104 ± 0.553 | 3.109 ± 0.411 |
| Total calorie intake (kcal/day/mouse) | 11.124 ± 1.186 | 17.259 ± 3.072 a | 11.968 ± 1.583 b |
| Fat calorie intake (kcal/day/mouse) | 0.478 ± 0.051 | 10.010 ± 1.782 a | 0.515 ± 0.068 b |
| Sucrose or fructose calorie intake (kcal/day/mouse) | 0.000 ± 0.000 | 1.291 ± 0.230 a | 7.181 ± 0.950 a,b |
| SD (n = 15) | HFHS (n = 15) | HFr (n = 15) | |
|---|---|---|---|
| Triglycerides (mg/dL) | 23.61 ± 1.78 | 41.60 ± 4.22 a | 37.30 ± 4.63 a |
| Cholesterol (mg/dL) | 85.45 ± 11.94 | 173.6 ± 10.9 a | 252.3 ± 27.91 a,b |
| IL-6 (pg/mL) | 4.36 ± 0.16 | 11.87 ± 0.34 a | 9.61 ± 0.86 a |
| LDH (U/L) | 230.70 ± 19.73 | 303.60 ± 25.91 a | 297.20 ± 25.01 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aimaretti, E.; Chimienti, G.; Rubeo, C.; Di Lorenzo, R.; Trisolini, L.; Dal Bello, F.; Moradi, A.; Collino, M.; Lezza, A.M.S.; Aragno, M.; et al. Different Effects of High-Fat/High-Sucrose and High-Fructose Diets on Advanced Glycation End-Product Accumulation and on Mitochondrial Involvement in Heart and Skeletal Muscle in Mice. Nutrients 2023, 15, 4874. https://doi.org/10.3390/nu15234874
Aimaretti E, Chimienti G, Rubeo C, Di Lorenzo R, Trisolini L, Dal Bello F, Moradi A, Collino M, Lezza AMS, Aragno M, et al. Different Effects of High-Fat/High-Sucrose and High-Fructose Diets on Advanced Glycation End-Product Accumulation and on Mitochondrial Involvement in Heart and Skeletal Muscle in Mice. Nutrients. 2023; 15(23):4874. https://doi.org/10.3390/nu15234874
Chicago/Turabian StyleAimaretti, Eleonora, Guglielmina Chimienti, Chiara Rubeo, Rosa Di Lorenzo, Lucia Trisolini, Federica Dal Bello, Atefeh Moradi, Massimo Collino, Angela Maria Serena Lezza, Manuela Aragno, and et al. 2023. "Different Effects of High-Fat/High-Sucrose and High-Fructose Diets on Advanced Glycation End-Product Accumulation and on Mitochondrial Involvement in Heart and Skeletal Muscle in Mice" Nutrients 15, no. 23: 4874. https://doi.org/10.3390/nu15234874
APA StyleAimaretti, E., Chimienti, G., Rubeo, C., Di Lorenzo, R., Trisolini, L., Dal Bello, F., Moradi, A., Collino, M., Lezza, A. M. S., Aragno, M., & Pesce, V. (2023). Different Effects of High-Fat/High-Sucrose and High-Fructose Diets on Advanced Glycation End-Product Accumulation and on Mitochondrial Involvement in Heart and Skeletal Muscle in Mice. Nutrients, 15(23), 4874. https://doi.org/10.3390/nu15234874