
Insufficient Evidence of a Breastmilk Microbiota at Six-Weeks Postpartum: A Pilot Study

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. DNA Extraction from Breastmilk
2.3. Shotgun Metagenomic Sequencing and Analysis
2.4. Metagenomic Sequencing Quality Control and Composition
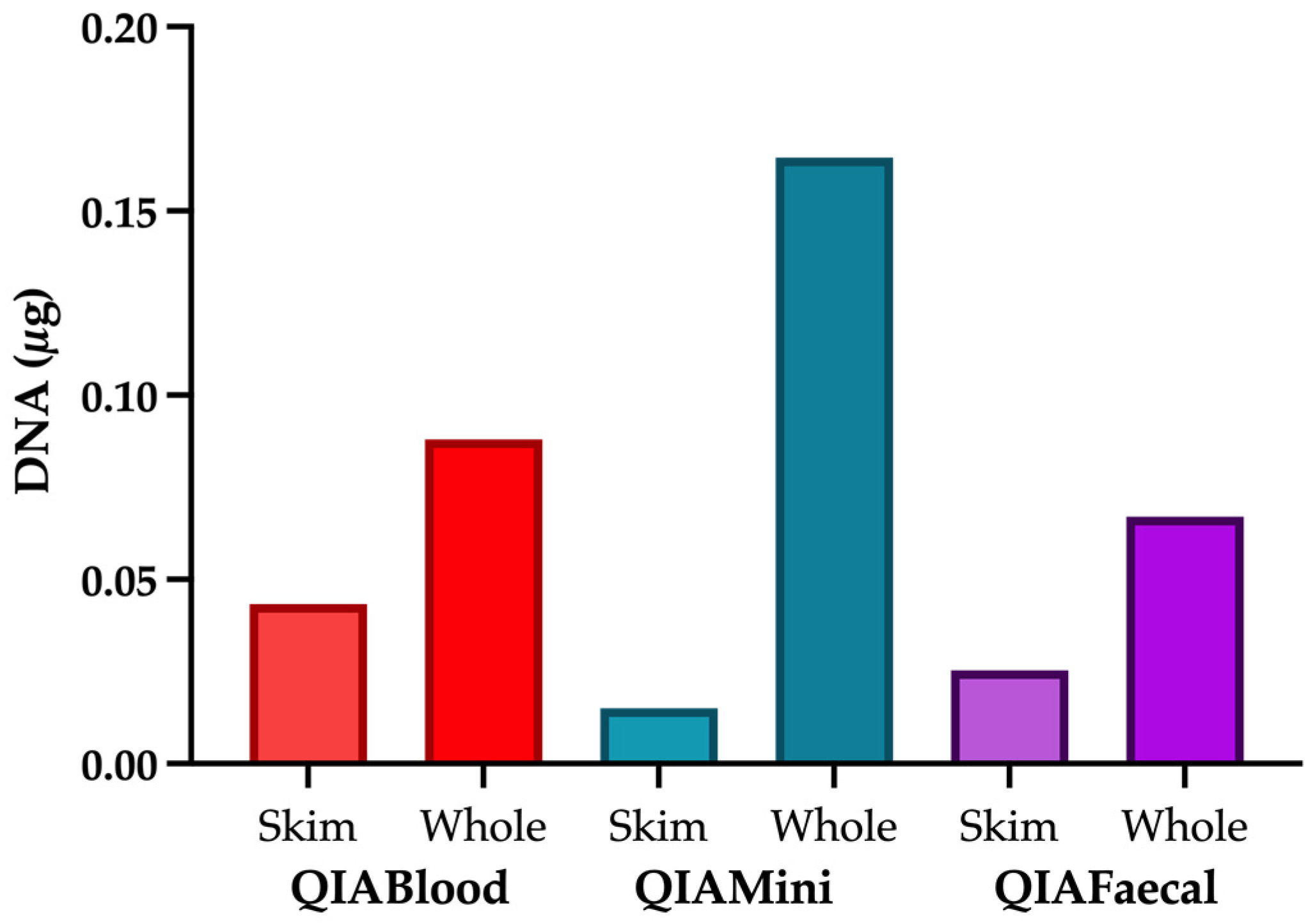
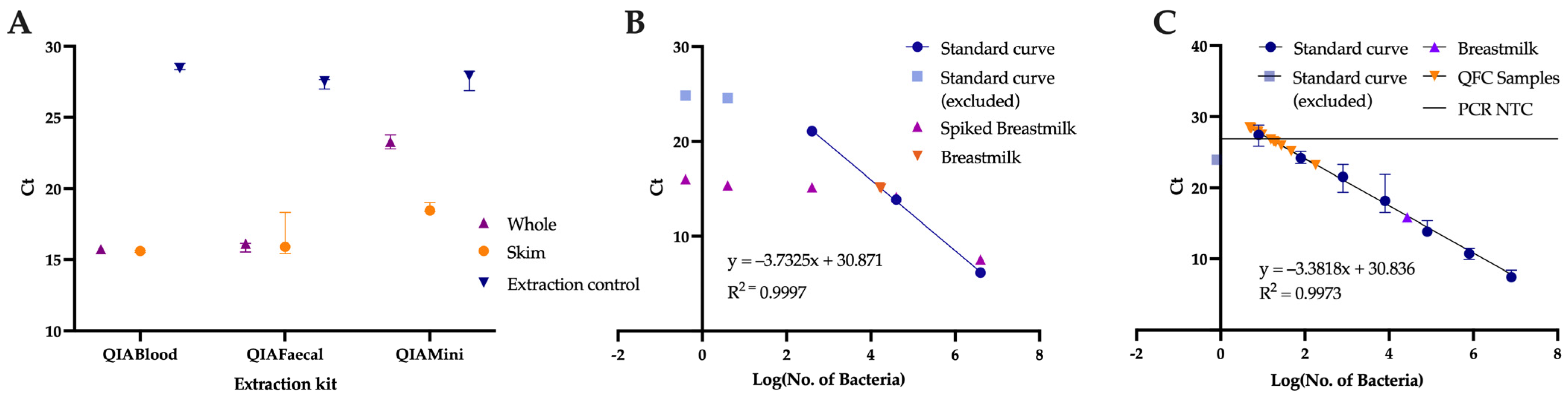
2.5. Comparison of Extraction Kits
2.6. Quantification of Breastmilk Microbial Load
2.7. Statistical Analysis
3. Results
3.1. Role of the Extraction Method
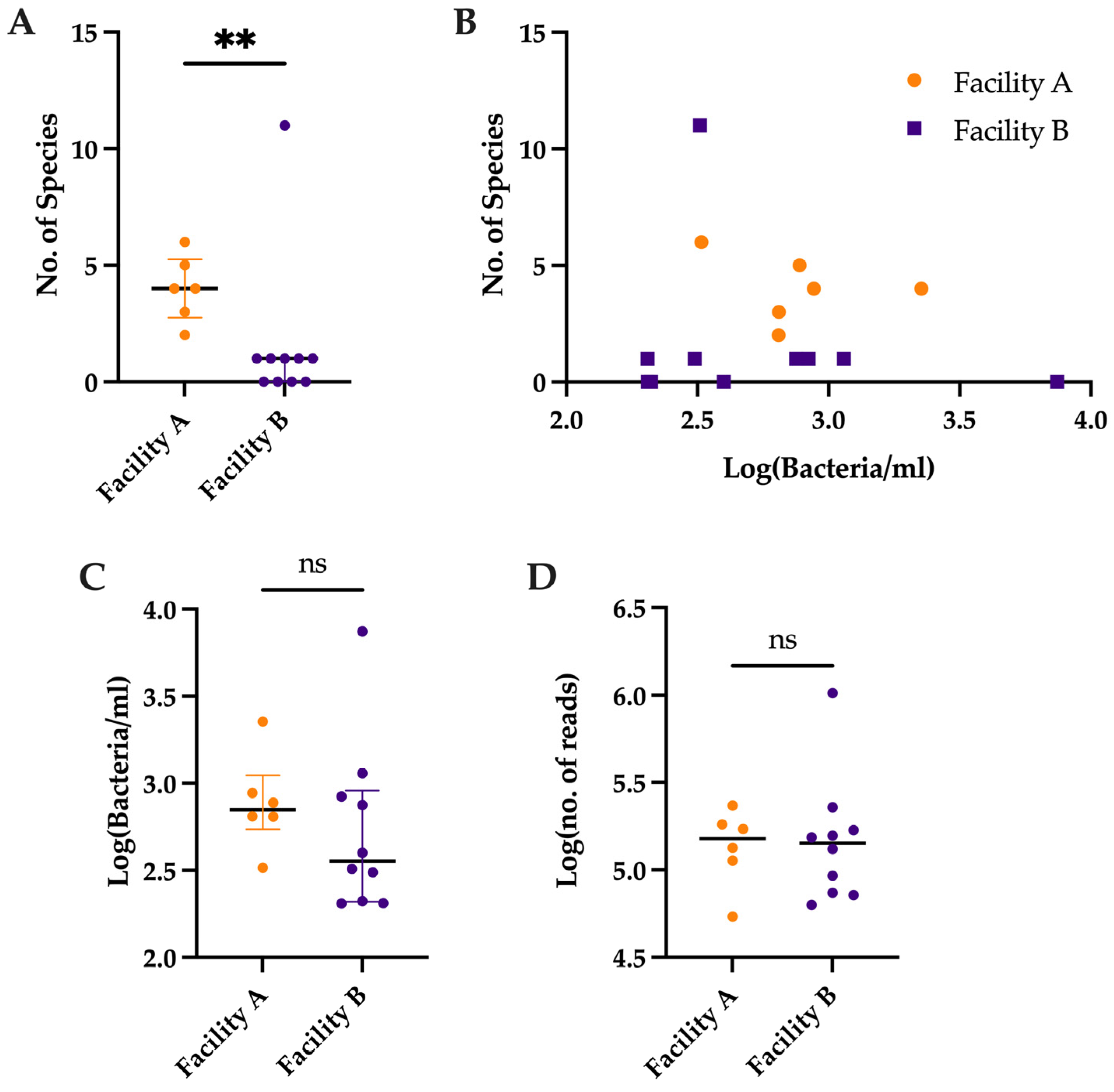
3.2. Comparison of Number of Species Detected by Metagenomic Sequencing vs. Bacterial Load
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The World Health Organisation. Breastfeeding. 2021. Available online: https://www.who.int/health-topics/breastfeeding (accessed on 16 February 2021).
- Azad, M.B.; Konya, T.; Maughan, H.; Guttman, D.S.; Field, C.J.; Chari, R.S.; Sears, M.R.; Becker, A.B.; Scott, J.A.; Kozyrskyj, A.L. Gut microbiota of healthy Canadian infants: Profiles by mode of delivery and infant diet at 4 months. Can. Med. Assoc. J. 2013, 185, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzan-Yulzari, A.; Turta, O.; Belogolovski, A.; Ziv, O.; Kunz, C.; Perschbacher, S.; Neuman, H.; Pasolli, E.; Oz, A.; Ben-Amram, H.; et al. Neonatal antibiotic exposure impairs child growth during the first six years of life by perturbing intestinal microbial colonization. Nat. Commun. 2021, 12, 443. [Google Scholar] [CrossRef]
- Aversa, Z.; Atkinson, E.J.; Schafer, M.J.; Theiler, R.N.; Rocca, W.A.; Blaser, M.J.; LeBrasseur, N.K. Association of Infant Antibiotic Exposure With Childhood Health Outcomes. Mayo Clin. Proc. 2021, 96, 66–77. [Google Scholar] [CrossRef]
- Stanislawski, M.A.; Dabelea, D.; Wagner, B.D.; Iszatt, N.; Dahl, C.; Sontag, M.K.; Knight, R.; Lozupone, C.A.; Eggesbø, M. Gut Microbiota in the First 2 Years of Life and the Association with Body Mass Index at Age 12 in a Norwegian Birth Cohort. mBio 2018, 9, e01751-18. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.-M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef] [Green Version]
- Borewicz, K.; Gu, F.; Saccenti, E.; Hechler, C.; Beijers, R.; de Weerth, C.; van Leeuwen, S.S.; Schols, H.A.; Smidt, H. The association between breastmilk oligosaccharides and faecal microbiota in healthy breastfed infants at two, six, and twelve weeks of age. Sci. Rep. 2020, 10, 4270. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.C.C.; Lewis, Z.T.; Krishnan, S.; Bernstein, R.M.; Moore, S.E.; Prentice, A.M.; Mills, D.A.; Lebrilla, C.B.; Zivkovic, A.M. Growth and Morbidity of Gambian Infants are Influenced by Maternal Milk Oligosaccharides and Infant Gut Microbiota. Sci. Rep. 2017, 7, 40466. [Google Scholar] [CrossRef] [Green Version]
- Lewis, Z.T.; Totten, S.M.; Smilowitz, J.T.; Popovic, M.; Parker, E.; Lemay, D.G.; Van Tassell, M.L.; Miller, M.J.; Jin, Y.-S.; German, J.B.; et al. Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome 2015, 3, 13. [Google Scholar] [CrossRef]
- Sela, D.A.; Chapman, J.; Adeuya, A.; Kim, J.H.; Chen, F.; Whitehead, T.R.; Lapidus, A.; Rokhsar, D.S.; Lebrilla, C.B.; German, J.B.; et al. The genome sequence of Bifidobacterium longum subsp. reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 18964. [Google Scholar] [CrossRef] [Green Version]
- Marcobal, A.; Barboza, M.; Sonnenburg, E.D.; Pudlo, N.; Martens, E.C.; Desai, P.; Lebrilla, C.B.; Weimer, B.C.; Mills, D.A.; German, J.B.; et al. Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe 2011, 10, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Cheema, A.S.; Gridneva, Z.; Furst, A.J.; Roman, A.S.; Trevenen, M.L.; Turlach, B.A.; Lai, C.T.; Stinson, L.F.; Bode, L.; Payne, M.S.; et al. Human Milk Oligosaccharides and Bacterial Profile Modulate Infant Body Composition during Exclusive Breastfeeding. Int. J. Mol. Sci. 2022, 23, 2865. [Google Scholar] [CrossRef]
- Kordy, K.; Gaufin, T.; Mwangi, M.; Li, F.; Cerini, C.; Lee, D.J.; Adisetiyo, H.; Woodward, C.; Pannaraj, P.S.; Tobin, N.H.; et al. Contributions to human breast milk microbiome and enteromammary transfer of Bifidobacterium breve. PLoS ONE 2020, 15, e0219633. [Google Scholar] [CrossRef] [Green Version]
- Martin, V.; Maldonado-Barragan, A.; Moles, L.; Rodriguez-Banos, M.; Campo, R.D.; Fernandez, L.; Rodriguez, J.M.; Jimenez, E. Sharing of Bacterial Strains Between Breast Milk and Infant Feces. J. Hum. Lact. 2012, 28, 36–44. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Schloter, M.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Zimmermann, P.; Curtis, N. Breast milk microbiota: A review of the factors that influence composition. J. Infect. 2020, 81, 17–47. [Google Scholar] [CrossRef]
- Edwards, C.; Van Loo-Bouwman, C.; Van Diepen, J.; Schoemaker, M.; Ozanne, S.; Venema, K.; Stanton, C.; Marinello, V.; Rueda, R.; Flourakis, M.; et al. A systematic review of breast milk microbiota composition and the evidence for transfer to and colonisation of the infant gut. Benef. Microbes 2022, 13, 365–381. [Google Scholar] [CrossRef]
- Moossavi, S.; Sepehri, S.; Robertson, B.; Bode, L.; Goruk, S.; Field, C.J.; Lix, L.M.; de Souza, R.J.; Becker, A.B.; Mandhane, P.J.; et al. Composition and Variation of the Human Milk Microbiota Are Influenced by Maternal and Early-Life Factors. Cell Host Microbe 2019, 25, 324–335.e4. [Google Scholar] [CrossRef] [Green Version]
- Stinson, L.F.; Trevenen, M.L.; Geddes, D.T. The Viable Microbiome of Human Milk Differs from the Metataxonomic Profile. Nutrients 2021, 13, 4445. [Google Scholar] [CrossRef]
- Damaceno, Q.S.; Souza, J.P.; Nicoli, J.R.; Paula, R.L.; Assis, G.B.; Figueiredo, H.C.; Azevedo, V.; Martins, F.S. Evaluation of Potential Probiotics Isolated from Human Milk and Colostrum. Probiotics Antimicrob Proteins 2017, 9, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, C.; Gloor, G.B.; Brackstone, M.; Scott, L.; Tangney, M.; Reid, G. The Microbiota of Breast Tissue and Its Association with Breast Cancer. Appl. Environ. Microbiol. 2016, 82, 5039–5048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinson, L.F.; Sindi, A.S.M.; Cheema, A.S.; Lai, C.T.; Mühlhäusler, B.S.; Wlodek, M.E.; Payne, M.S.; Geddes, D.T. The human milk microbiome: Who, what, when, where, why, and how? Nutr. Rev. 2020, 79, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M. The origin of human milk bacteria: Is there a bacterial entero-mammary pathway during late pregnancy and lactation? Adv. Nutr. 2014, 5, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Zeng, Z.; Liang, X.; Tang, X.; Luo, H.; Wang, D.; Zhou, J.; Xiao, X. Effect of breast milk with or without bacteria on infant gut microbiota. BMC Pregnancy Childbirth 2022, 22, 595. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, P.; Pasolli, E.; Tett, A.; Asnicar, F.; Gorfer, V.; Fedi, S.; Armanini, F.; Truong, D.T.; Manara, S.; Zolfo, M.; et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 2018, 24, 133–145.e5. [Google Scholar] [CrossRef]
- Jiménez, E.; de Andrés, J.; Manrique, M.; Pareja-Tobes, P.; Tobes, R.; Martínez-Blanch, J.F.; Codoñer, F.M.; Ramón, D.; Fernández, L.; Rodríguez, J.M. Metagenomic Analysis of Milk of Healthy and Mastitis-Suffering Women. J. Hum. Lact. 2015, 31, 406–415. [Google Scholar] [CrossRef]
- Ward, T.L.; Hosid, S.; Ioshikhes, I.; Altosaar, I. Human milk metagenome: A functional capacity analysis. BMC Microbiol. 2013, 13, 116. [Google Scholar] [CrossRef] [Green Version]
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, G.; Lau, H.C.-H.; Yu, J. Metagenomic Sequencing for Microbial DNA in Human Samples: Emerging Technological Advances. Int. J. Mol. Sci. 2022, 23, 2181. [Google Scholar] [CrossRef]
- Borg, D.; Rae, K.; Fiveash, C.; Schagen, J.; James-McAlpine, J.; Friedlander, F.; Thurston, C.; Oliveri, M.; Harmey, T.; Cavanagh, E.; et al. Queensland Family Cohort: A study protocol. BMJ Open 2021, 11, e044463. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Lahti, L.S.S. Microbiome R Package (2012–2019). Available online: http://microbiome.github.io (accessed on 13 October 2021).
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes 2014. Available online: https://CRAN.R-project.org/package=RColorBrewer (accessed on 13 October 2021).
- Kassambara, A. Ggpubr: ‘Ggplot2’ Based Publication Ready Plots 2020. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 13 October 2021).
- Wickham, H.F.; Henry, L.; Muller, K. Dplyr: A Grammar of Data Manipulation 2021. Available online: https://CRAN.R-project.org/package=dplyr (accessed on 13 October 2021).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis 2016. Available online: https://ggplot2.tidyverse.org (accessed on 13 October 2021).
- Firke, S. Janitor: SImple Tools for Examining and Cleaning Dirty Data 2021. Available online: https://CRAN.R-project.org/package=janitor (accessed on 13 October 2021).
- Ojo-Okunola, A.; Claassen-Weitz, S.; Mwaikono, K.S.; Gardner-Lubbe, S.; Zar, H.J.; Nicol, M.P.; Du Toit, E. The Influence of DNA Extraction and Lipid Removal on Human Milk Bacterial Profiles. Methods Protoc. 2020, 3, 39. [Google Scholar] [CrossRef]
- Zhang, B.; Brock, M.; Arana, C.; Dende, C.; van Oers, N.S.; Hooper, L.V.; Raj, P. Impact of Bead-Beating Intensity on the Genus- and Species-Level Characterization of the Gut Microbiome Using Amplicon and Complete 16S rRNA Gene Sequencing. Front. Cell. Infect. Microbiol. 2021, 11, 678522. [Google Scholar] [CrossRef]
- Perez-Muñoz, M.E.; Arrieta, M.C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauder, A.P.; Roche, A.M.; Sherrill-Mix, S.; Bailey, A.; Laughlin, A.L.; Bittinger, K.; Leite, R.; Elovitz, M.A.; Parry, S.; Bushman, F.D. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiby, J.S.; McCormick, K.; Sherrill-Mix, S.; Clarke, E.L.; Kessler, L.R.; Taylor, L.J.; Hofstaedter, C.E.; Roche, A.M.; Mattei, L.M.; Bittinger, K.; et al. Lack of detection of a human placenta microbiome in samples from preterm and term deliveries. Microbiome 2018, 6, 196. [Google Scholar] [CrossRef]
- Theis, K.R.; Romero, R.; Winters, A.D.; Greenberg, J.M.; Gomez-Lopez, N.; Alhousseini, A.; Bieda, J.; Maymon, E.; Pacora, P.; Fettweis, J.M.; et al. Does the human placenta delivered at term have a microbiota? Results of cultivation, quantitative real-time PCR, 16S rRNA gene sequencing, and metagenomics. Am. J. Obstet. Gynecol. 2019, 220, 267.e1–267.e39. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention. How Much and How Often to Breastfeed. 2022. Available online: https://www.cdc.gov/nutrition/infantandtoddlernutrition/breastfeeding/how-much-and-how-often.html (accessed on 11 April 2022).
- World Health Organisation. Infant and Young Child Feeding: Model Chapter for Textbooks for Medical Students and Allied Health Professionals. 2009. Available online: https://apps.who.int/iris/handle/10665/44117 (accessed on 1 December 2022).
- Ballard, O.; Morrow, A.L. Human milk composition: Nutrients and bioactive factors. Pediatr. Clin. N. Am. 2013, 60, 49–74. [Google Scholar] [CrossRef] [Green Version]
- Fehr, K.; Moossavi, S.; Sbihi, H.; Boutin, R.C.; Bode, L.; Robertson, B.; Yonemitsu, C.; Field, C.J.; Becker, A.B.; Mandhane, P.J.; et al. Breastmilk Feeding Practices Are Associated with the Co-Occurrence of Bacteria in Mothers’ Milk and the Infant Gut: The CHILD Cohort Study. Cell Host Microbe 2020, 28, 285–297.e4. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Weyrich, L.S.; Farrer, A.G.; Eisenhofer, R.; Arriola, L.A.; Young, J.; Selway, C.A.; Handsley-Davis, M.; Adler, C.J.; Breen, J.; Cooper, A. Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 2019, 19, 982–996. [Google Scholar] [CrossRef] [Green Version]
- Hornung, B.V.H.; Zwittink, R.D.; Kuijper, E.J. Issues and current standards of controls in microbiome research. FEMS Microbiol. Ecol. 2019, 95, fiz045. [Google Scholar] [CrossRef]
- Brito, I.L.; Gurry, T.; Zhao, S.; Huang, K.; Young, S.K.; Shea, T.P.; Naisilisili, W.; Jenkins, A.P.; Jupiter, S.D.; Gevers, D.; et al. Transmission of human-associated microbiota along family and social networks. Nat. Microbiol. 2019, 4, 964–971. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Facility A (n = 6) | Facility B (n = 10) | p-Value (Facility A vs. Facility B) | |
|---|---|---|---|
| Maternal age (years) | 30.67 ± 5.24 | 32.10 ± 3.81 | 0.54 |
| Maternal ethnicity | |||
| Caucasian | 4 | 9 | 0.52 |
| North-East Asian | 2 | 0 | 0.13 |
| Southern and Central Asian | 0 | 1 | >0.99 |
| Male infants N (%) | 3 (50) | 5 (50) | >0.99 |
| Gestational age at birth (days) | 277 (272–283) | 273 (265–274) | 0.10 |
| Exclusively breastfed N (%) | 3 (50) | 5 (50) | >0.99 |
| Pre-pregnancy BMI (kg/m2) | 23.0 ± 1.3 | 22.8 ± 3.2 | 0.91 |
| Parity (previous pregnancies > 20 weeks) * | 0.5 (0–1) | 1 (1–3) | 0.16 |
| Antibiotic use 2 weeks pre-conception to 6 weeks post-partum N (%) | 2 (33) | 8 (80) | 0.12 |
| Probiotic use 2 weeks pre-concenption to 6 weeks post-partum N (%) | 1 (17) | 1 (10) | >0.99 |
| Mastitis in the previous 6 weeks N (%) | 1 (17) | 0 (0) | 0.38 |
| Caesarean section N (%) | 0 (0) | 8 (80) | 0.007 |
| Chronic Disease | |||
| Allergies (no anaphylaxis) N | 1 | 4 | 0.59 |
| Mental health disorder N | 2 | 2 | 0.60 |
| Endocrine disorder (inc. thyroid) N | 1 | 2 | >0.99 |
| Asthma diagnosed ever N | 0 | 4 | 0.23 |
| Endometriosis N | 0 | 2 | 0.50 |
| Polycystic Ovarian Syndrome (PCOS) N | 0 | 1 | >0.99 |
| Osteoarthritis N | 0 | 1 | >0.99 |
| Dermatitis/eczema N | 0 | 1 | >0.99 |
| Gilbert’s Syndrome N | 0 | 1 | >0.99 |
| Gestational diabetes mellitus N | 0 | 0 | >0.99 |
| Affected by >1 of the above N | 1 | 5 | 0.31 |
| Facility A | Facility B | |
|---|---|---|
| Sequencing Platform | NovaSeq6000 (Illumina) | NovaSeq6000 (Illumina) |
| Sequencing length | 2 × 150 bp | 2 × 150 bp |
| Library preparation kit | Nextera DNA Flex Library Kit (Illumina #20018705) | Illumina DNA Prep (Illumina #20018705) |
| Library preparation platform | Mantis Liquid Handler (Formulatrix) and Epmotion (Eppendorf #507500301) | Zephyr NGS (Perkin Elmer) |
| Library quantification and quality control | Quanti-iT dsDNA HS Assay Kit (Invitrogen) and Agilent D1000 HS tapes (#5067-5582) on the TapeStation 4200 (Agilent # G2991AA) | Quant-iT ds DNA HS Assay kit (ThermoFisher Scientific) and QIAxcel Advanced System (#9002123) using QIAxcel DNA High Resolution Kit (#929002) |
| Library pooling | 2nM per library | 2nM per library |
| Library pool quantification | Qubit dsDNA HS assay kit (Invitrogen) | Qubit dsDNA HS assay kit (Invitrogen) on Qubit Flex Fluorometer |
| Library pool quality control | Agilent D1000 HS tapes (#5067-5582) on the TapeStation 4200 (Agilent #G2991AA) | QIAxcel Advanced System (#9002123) using QIAxcel DNA High Resolution Kit (#929002) |
| Species | CFU/Capsule |
|---|---|
| Lactobacillus rhamnosus Lr-32 | 6 × 109 |
| Lactobacillus rhamnosus HN001 | 1 × 109 |
| Lactobacillus plantarum Lp-115 | 4.2 × 109 |
| Lactobacillus rhamnosus GG | 4 × 109 |
| Lactobacillus gasseri Lg-36 | 5 × 108 |
| Lactobacillus casei Lc-11 | 3.2 × 109 |
| Lactobacillus delbrueckii subsp. bulgaricus Lb-87 | 2 × 108 |
| Lactobacillus paracasei Lpc-37 | 1.7 × 109 |
| Lactobacillus reuteri 1E1 | 2 × 108 |
| Bifidobacterium animalis subsp. lactis Bl-04 | 5 × 109 |
| Bifidobacterium breve Bb-18 | 8 × 108 |
| Bifidobacterium longum Bl-05 | 5 × 108 |
| Bifidobacterium longum subsp. infantis Bi-26 | 3 × 108 |
| Bifidobacterium animalis subsp. lactis HN019 | 1 × 109 |
| Streptococcus thermophilus St-21 | 3.4 × 109 |
| Species | NegCon | BM A | BM B | BM C | BM D | BM E | BM F |
|---|---|---|---|---|---|---|---|
| Cutibacterium acnes | 0 | 6.28 | 0 | 0 | 0 | 0 | 0 |
| Porphyromonas gingivalis | 2.06 | 0.70 | 0 | 2.04 | 2.09 | 0.74 | 1.08 |
| Gemella haemolysans | 0 | 0 | 0 | 0 | 0 | 3.89 | 0 |
| Staphylococcus aureus | 0 | 0 | 0 | 0 | 0 | 42.88 | 3.27 |
| Staphylococcus epidermidis | 0 | 2.81 | 4.85 | 1.02 | 1.87 | 2.50 | 3.18 |
| Enterococcus faecalis | 27.92 | 22.66 | 17.68 | 21.76 | 21.68 | 6.44 | 18.64 |
| Streptococcus agalactiae | 0 | 0 | 0 | 0 | 0 | 0.94 | 0 |
| Streptococcus mitis | 0 | 1.15 | 0 | 0 | 0 | 16.17 | 0 |
| Streptococcus mutans | 2.92 | 0.70 | 0 | 2.35 | 1.10 | 0.77 | 1.25 |
| Streptococcus oralis | 0 | 0 | 0 | 0 | 0 | 1.67 | 0 |
| Flavonifractor plautii | 0 | 0 | 0 | 0 | 0 | 0 | 10.10 |
| Veillonella atypica | 0 | 5.78 | 23.08 | 9.95 | 0 | 0 | 0 |
| Veillonella seminalis | 0 | 0 | 0 | 0 | 7.20 | 0 | 0 |
| Rhodobacter sphaeroides | 0 | 0.60 | 0 | 2.23 | 2.40 | 0 | 0.85 |
| Escherichia coli | 0.70 | 0 | 0 | 3.72 | 2.35 | 0 | 0.70 |
| Acinetobacter baumannii | 0 | 0 | 0 | 0 | 0.27 | 0 | 0 |
| Acinetobacter baylyi | 66.40 | 59.32 | 54.40 | 56.94 | 61.04 | 23.99 | 60.92 |
| Total contaminants | 100 | 83.38 | 72.08 | 86.81 | 88.26 | 31.95 | 82.60 |
| BM G | BM H | BM I | BM J | BM K | BM L | BM M | BM N | BM O | BM P | |
|---|---|---|---|---|---|---|---|---|---|---|
| Bifidobacterium breve | 0 | 3.03 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Bifidobacterium longum | 0 | 2.03 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Corynebacterium kroppenstedtii | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rothia mucilaginosa | 0 | 3.46 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cutibacterium acnes | 100 | 20.39 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 0 |
| Gemella haemolysans | 0 | 2.44 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Staphylococcus epidermidis | 0 | 5.34 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 |
| Staphylococcus hominis | 0 | 1.98 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Streptococcus mitis | 0 | 52.38 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 0 |
| Streptococcus oralis | 0 | 4.24 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Streptococcus parasanguinis | 0 | 2.37 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Streptococcus salivarius | 0 | 2.35 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leech, S.M.; Gilbert, M.C.; Clifton, V.L.; Kumar, S.; Rae, K.M.; Borg, D.; Dekker Nitert, M. Insufficient Evidence of a Breastmilk Microbiota at Six-Weeks Postpartum: A Pilot Study. Nutrients 2023, 15, 696. https://doi.org/10.3390/nu15030696
Leech SM, Gilbert MC, Clifton VL, Kumar S, Rae KM, Borg D, Dekker Nitert M. Insufficient Evidence of a Breastmilk Microbiota at Six-Weeks Postpartum: A Pilot Study. Nutrients. 2023; 15(3):696. https://doi.org/10.3390/nu15030696
Chicago/Turabian StyleLeech, Sophie M., Morgan C. Gilbert, Vicki L. Clifton, Sailesh Kumar, Kym M. Rae, Danielle Borg, and Marloes Dekker Nitert. 2023. "Insufficient Evidence of a Breastmilk Microbiota at Six-Weeks Postpartum: A Pilot Study" Nutrients 15, no. 3: 696. https://doi.org/10.3390/nu15030696
APA StyleLeech, S. M., Gilbert, M. C., Clifton, V. L., Kumar, S., Rae, K. M., Borg, D., & Dekker Nitert, M. (2023). Insufficient Evidence of a Breastmilk Microbiota at Six-Weeks Postpartum: A Pilot Study. Nutrients, 15(3), 696. https://doi.org/10.3390/nu15030696