
Characteristics of the Gut Microbiome and Serum Metabolome in Patients with Functional Constipation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants and Sample Collection
2.2. DNA Extraction and Sequencing from Fecal Samples
2.3. 16S rRNA Data Processing
2.4. LC/MS Non-Targeted Metabolomics Analysis
2.5. Statistical Analysis
3. Results
3.1. Basic Characteristics of the Study Cohort
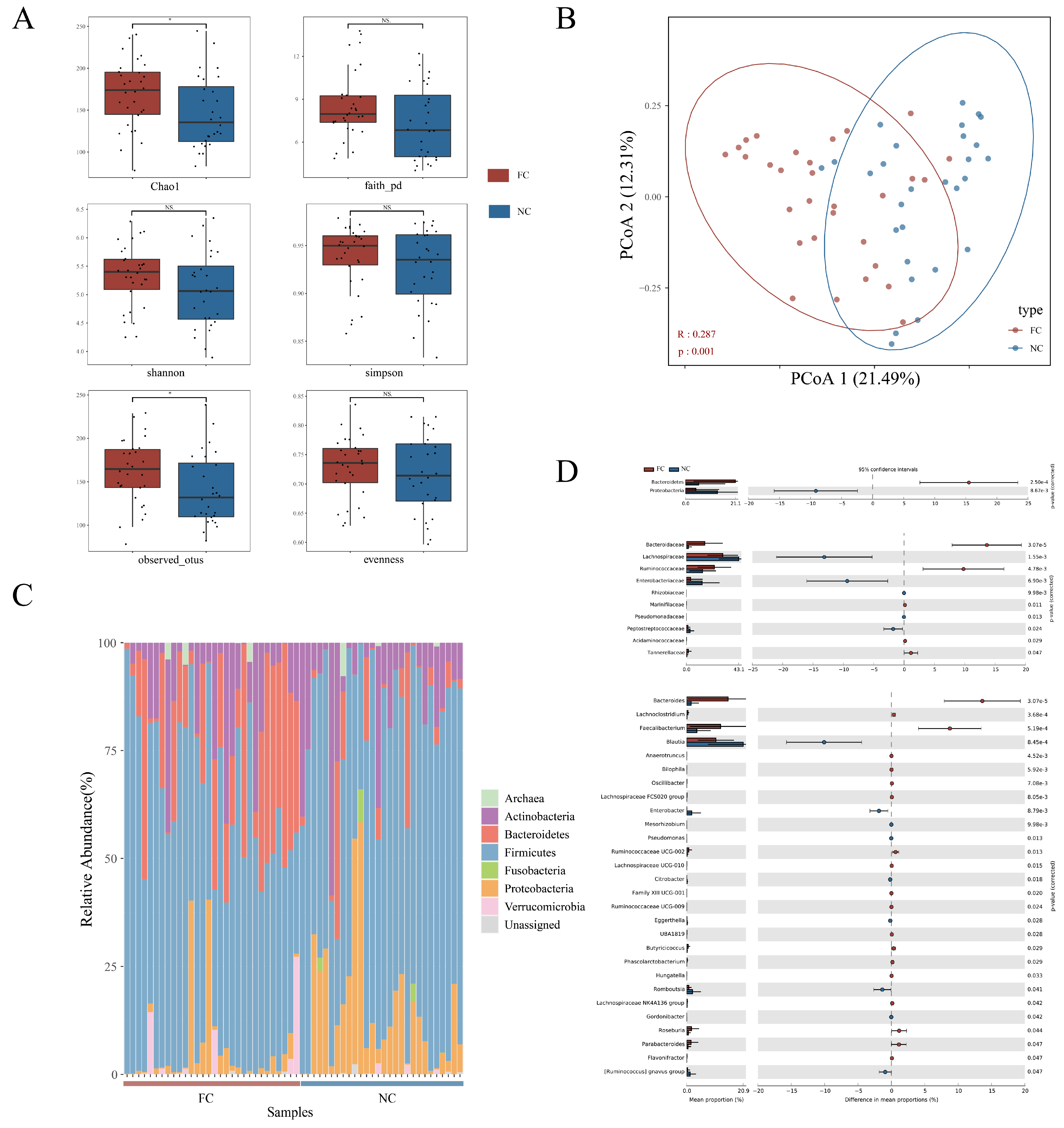
3.2. Changes in Intestinal Microbial Composition of Functional Constipation Patients
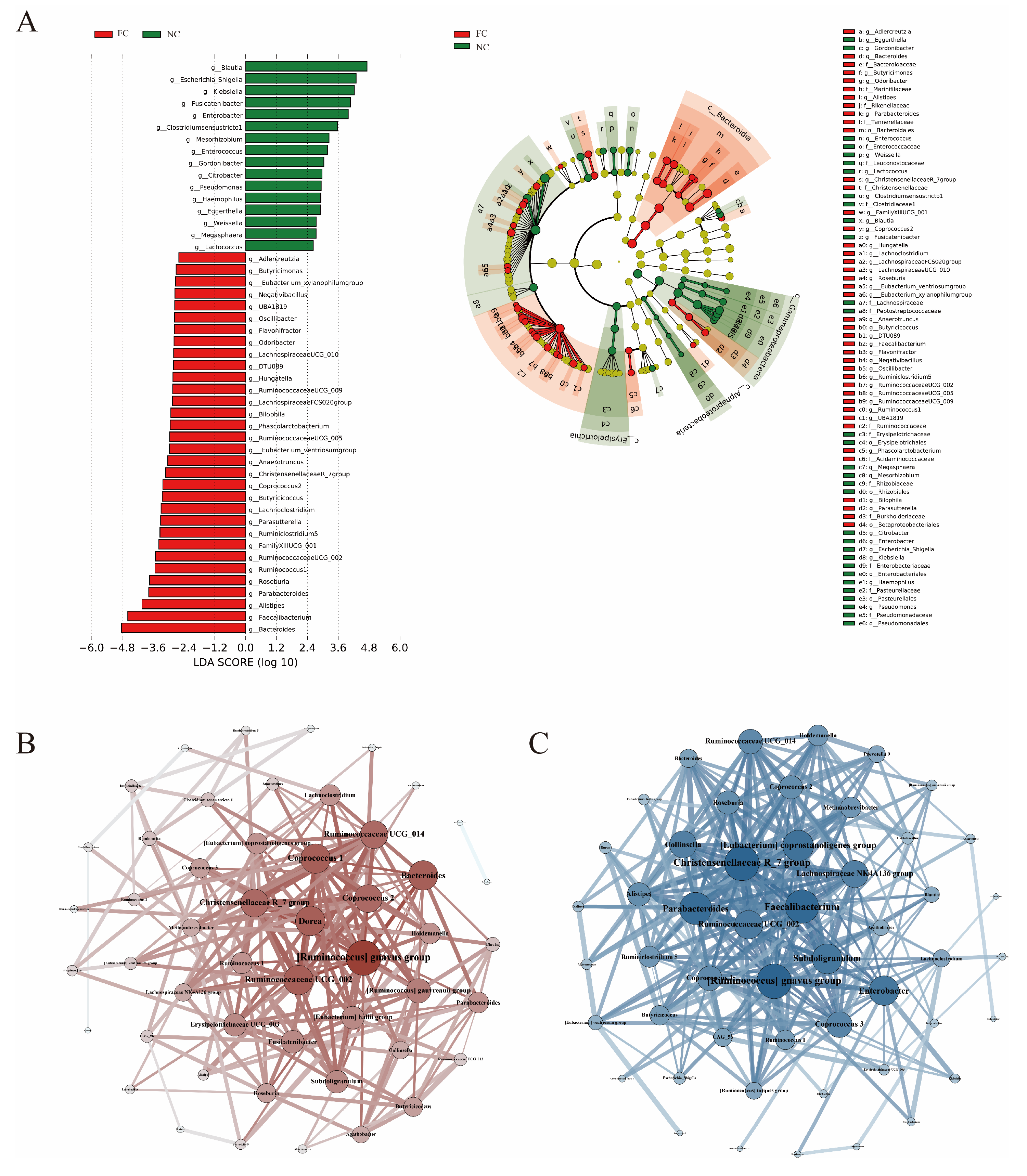
3.3. Identification of Differences in Intestinal Microbiota
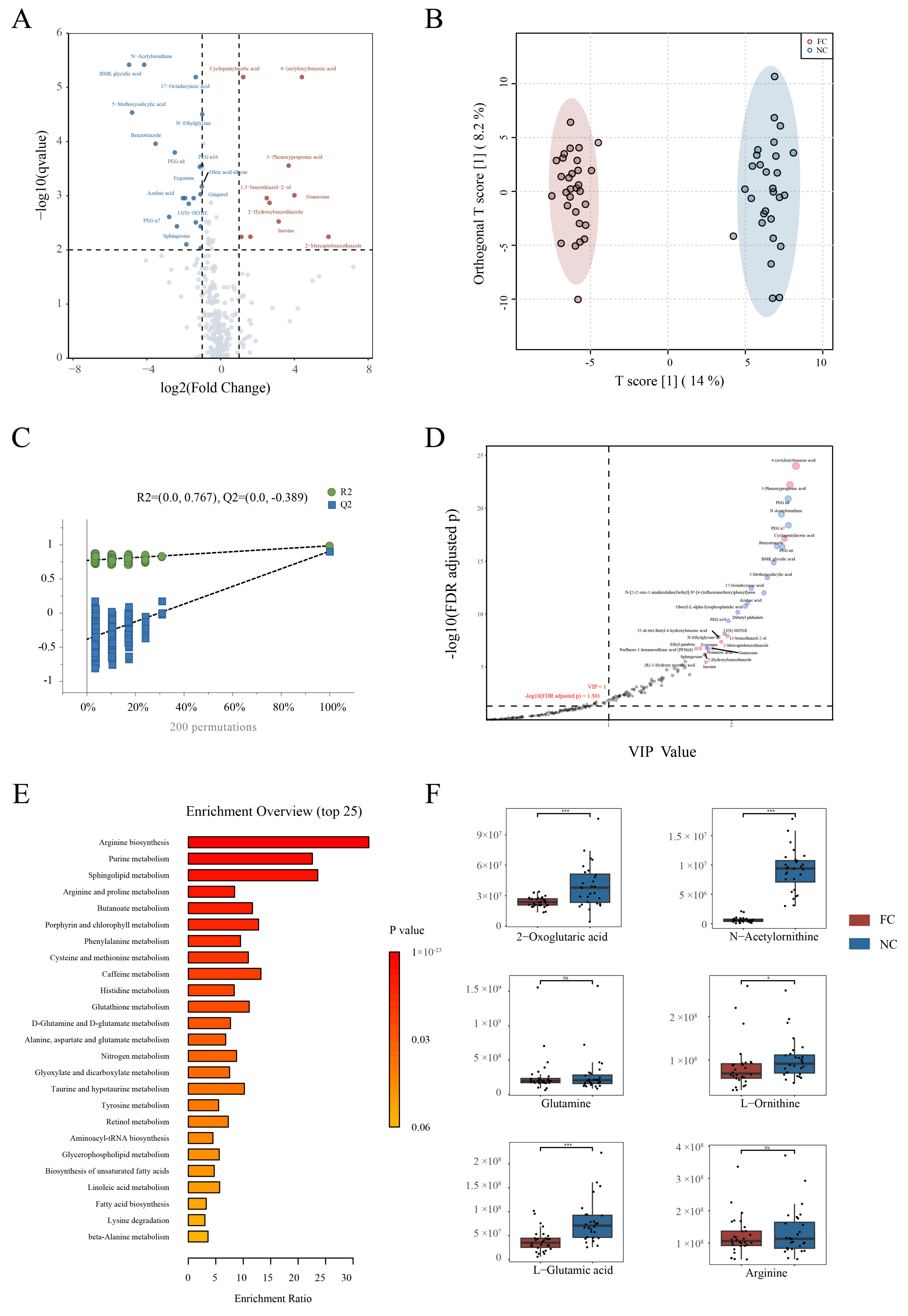
3.4. Changes in Serum Metabolite Profile and Crucial Metabolites in Constipation Patients
3.5. Conjoint Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vriesman, M.H.; Koppen, I.J.N.; Camilleri, M.; Di Lorenzo, C.; Benninga, M.A. Management of functional constipation in children and adults. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Barberio, B.; Judge, C.; Savarino, E.V.; Ford, A.C. Global prevalence of functional constipation according to the Rome criteria: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Rattanakovit, K.; Patcharatrakul, T. Diagnosis and management of chronic constipation in adults. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Bharucha, A.E.; Lacy, B.E. Mechanisms, Evaluation, and Management of Chronic Constipation. Gastroenterology 2020, 158, 1232–1249 e1233. [Google Scholar] [CrossRef]
- Pan, R.; Wang, L.; Xu, X.; Chen, Y.; Wang, H.; Wang, G.; Zhao, J.; Chen, W. Crosstalk between the Gut Microbiome and Colonic Motility in Chronic Constipation: Potential Mechanisms and Microbiota Modulation. Nutrients 2022, 14, 3704. [Google Scholar] [CrossRef]
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Turroni, F.; Mangifesta, M.; Viappiani, A.; Ticinesi, A.; Nouvenne, A.; Meschi, T.; van Sinderen, D.; et al. Unveiling the gut microbiota composition and functionality associated with constipation through metagenomic analyses. Sci. Rep. 2017, 7, 9879. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Chen, J.; Ren, X.; Yang, C.; Liu, S.; Bai, X.; Shan, S.; Dong, X. Gut Microbiota Composition Changes in Constipated Women of Reproductive Age. Front. Cell Infect. Microbiol. 2020, 10, 557515. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, R.; Li, D.; Zhao, L.; Zhu, L. Role of gut microbiota in functional constipation. Gastroenterol. Rep. 2021, 9, 392–401. [Google Scholar] [CrossRef]
- Wong, R.K.; Palsson, O.S.; Turner, M.J.; Levy, R.L.; Feld, A.D.; von Korff, M.; Whitehead, W.E. Inability of the Rome III criteria to distinguish functional constipation from constipation-subtype irritable bowel syndrome. Am. J. Gastroenterol. 2010, 105, 2228–2234. [Google Scholar] [CrossRef] [Green Version]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016, 65, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Wichmann, A.; Allahyar, A.; Greiner, T.U.; Plovier, H.; Lunden, G.O.; Larsson, T.; Drucker, D.J.; Delzenne, N.M.; Cani, P.D.; Backhed, F. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe 2013, 14, 582–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Gallausiaux, C.; Marinelli, L.; Blottiere, H.M.; Larraufie, P.; Lapaque, N. SCFA: Mechanisms and functional importance in the gut. Proc. Nutr. Soc. 2021, 80, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Xu, X.; Miwa, H. Role of Gut Microbiota-Gut Hormone Axis in the Pathophysiology of Functional Gastrointestinal Disorders. J. Neurogastroenterol. Motil. 2018, 24, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimidi, E.; Christodoulides, S.; Scott, S.M.; Whelan, K. Mechanisms of Action of Probiotics and the Gastrointestinal Microbiota on Gut Motility and Constipation. Adv. Nutr. 2017, 8, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Wong, B.S.; Camilleri, M.; McKinzie, S.; Burton, D.; Graffner, H.; Zinsmeister, A.R. Effects of A3309, an ileal bile acid transporter inhibitor, on colonic transit and symptoms in females with functional constipation. Am. J. Gastroenterol. 2011, 106, 2154–2164. [Google Scholar] [CrossRef]
- Zhu, G.; Guo, M.; Zhao, J.; Zhang, H.; Wang, G.; Chen, W. Integrative Metabolomic Characterization Reveals the Mediating Effect of Bifidobacterium breve on Amino Acid Metabolism in a Mouse Model of Alzheimer’s Disease. Nutrients 2022, 14, 735. [Google Scholar] [CrossRef]
- Guo, M.; Yao, J.; Yang, F.; Liu, W.; Bai, H.; Ma, J.; Ma, X.; Zhang, J.; Fang, Y.; Miao, Y.; et al. The composition of intestinal microbiota and its association with functional constipation of the elderly patients. Future Microbiol. 2020, 15, 163–175. [Google Scholar] [CrossRef]
- Zafar, H.; Saier, M.H., Jr. Gut Bacteroides species in health and disease. Gut Microbes 2021, 13, 1–20. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Strandwitz, P.; Kim, K.H.; Terekhova, D.; Liu, J.K.; Sharma, A.; Levering, J.; McDonald, D.; Dietrich, D.; Ramadhar, T.R.; Lekbua, A.; et al. GABA-modulating bacteria of the human gut microbiota. Nat. Microbiol. 2019, 4, 396–403. [Google Scholar] [CrossRef]
- Auteri, M.; Zizzo, M.G.; Serio, R. GABA and GABA receptors in the gastrointestinal tract: From motility to inflammation. Pharmacol. Res. 2015, 93, 11–21. [Google Scholar] [CrossRef]
- Mizutani, T.; Aboagye, S.Y.; Ishizaka, A.; Afum, T.; Mensah, G.I.; Asante-Poku, A.; Asandem, D.A.; Parbie, P.K.; Abana, C.Z.; Kushitor, D.; et al. Gut microbiota signature of pathogen-dependent dysbiosis in viral gastroenteritis. Sci. Rep. 2021, 11, 13945. [Google Scholar] [CrossRef]
- Tian, H.; Ye, C.; Yang, B.; Cui, J.; Zheng, Z.; Wu, C.; Zhou, S.; Lv, X.; Qin, N.; Qin, H.; et al. Gut Metagenome as a Potential Diagnostic and Predictive Biomarker in Slow Transit Constipation. Front. Med. (Lausanne) 2021, 8, 777961. [Google Scholar] [CrossRef]
- Fu, X.; Liu, Z.; Zhu, C.; Mou, H.; Kong, Q. Nondigestible carbohydrates, butyrate, and butyrate-producing bacteria. Crit. Rev. Food Sci. Nutr. 2018, 59, S130–S152. [Google Scholar] [CrossRef]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef]
- Canani, R.B. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519. [Google Scholar] [CrossRef]
- Bach Knudsen, K.; Lærke, H.; Hedemann, M.; Nielsen, T.; Ingerslev, A.; Gundelund Nielsen, D.; Theil, P.; Purup, S.; Hald, S.; Schioldan, A.; et al. Impact of Diet-Modulated Butyrate Production on Intestinal Barrier Function and Inflammation. Nutrients 2018, 10, 1499. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Xu, C.; Fan, Y.; Li, Y.; Wang, Y.; Zhang, X.; Yu, S.; Wang, J.; Chai, R.; Zhao, Z.; et al. Effect of fecal microbiota transplantation in patients with slow transit constipation and the relative mechanisms based on the protein digestion and absorption pathway. J. Transl. Med. 2021, 19, 490. [Google Scholar] [CrossRef]
- Xu, Y.; Labedan, B.; Glansdorff, N. Surprising arginine biosynthesis: A reappraisal of the enzymology and evolution of the pathway in microorganisms. Microbiol. Mol. Biol. Rev. 2007, 71, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Nakato, J.; Ho, Y.Y.; Omae, R.; Mizushige, T.; Uchida, K.; Tominaga, M.; Kim, M.; Goto, T.; Takahashi, N.; Kawada, T.; et al. l-Ornithine and l-lysine stimulate gastrointestinal motility via transient receptor potential vanilloid 1. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, C.; Li, H.; Bai, X.; Hu, T.; Xue, X.; An, J.; Zhang, Y.; Dong, X. Alteration of Serum Metabolites in Women of Reproductive Age with Chronic Constipation. Med. Sci. Monit. 2022, 28, e934117. [Google Scholar] [CrossRef] [PubMed]
- Sugitani, Y.; Inoue, R.; Inatomi, O.; Nishida, A.; Morishima, S.; Imai, T.; Kawahara, M.; Naito, Y.; Andoh, A. Mucosa-associated gut microbiome in Japanese patients with functional constipation. J. Clin. Biochem. Nutr. 2021, 68, 187–192. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Constipation (n = 30) | Health (n = 28) | p-Value | |
|---|---|---|---|---|
| Age, Mean ± SD | 42 ± 10.12 | 46.43 ± 10.06 | 0.101 | |
| Sex, n (%) | Female | 28(93.3) | 23(82.1) | 0.246 |
| Male | 2(6.7) | 5(17.9) | ||
| BMI, Mean ± SD | 22.56 ± 2.81 | 23.41 ± 2.84 | 0.127 | |
| Defecation frequency, Mean ± SD (times/day) | 0.31 ± 0.13 | 0.70 ± 0.38 | 8.988 × 10−6 | |
| Bristol stool typing, Mean ± SD | 1.93 ± 0.91 | 3.86 ± 1.41 | 1.826 × 10−7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Wang, L.; Yu, Q.; Tang, N.; Mei, C.; Zhang, H.; Wang, G.; Lu, J.; Chen, W. Characteristics of the Gut Microbiome and Serum Metabolome in Patients with Functional Constipation. Nutrients 2023, 15, 1779. https://doi.org/10.3390/nu15071779
Wang J, Wang L, Yu Q, Tang N, Mei C, Zhang H, Wang G, Lu J, Chen W. Characteristics of the Gut Microbiome and Serum Metabolome in Patients with Functional Constipation. Nutrients. 2023; 15(7):1779. https://doi.org/10.3390/nu15071779
Chicago/Turabian StyleWang, Jialiang, Linlin Wang, Qiangqing Yu, Nan Tang, Chunxia Mei, Hao Zhang, Gang Wang, Jian Lu, and Wei Chen. 2023. "Characteristics of the Gut Microbiome and Serum Metabolome in Patients with Functional Constipation" Nutrients 15, no. 7: 1779. https://doi.org/10.3390/nu15071779
APA StyleWang, J., Wang, L., Yu, Q., Tang, N., Mei, C., Zhang, H., Wang, G., Lu, J., & Chen, W. (2023). Characteristics of the Gut Microbiome and Serum Metabolome in Patients with Functional Constipation. Nutrients, 15(7), 1779. https://doi.org/10.3390/nu15071779