The Role of Intestinal Bacteria Overgrowth in Obesity-Related Nonalcoholic Fatty Liver Disease

Abstract
:1. Introduction
2. Relationship between the Gut and the Liver
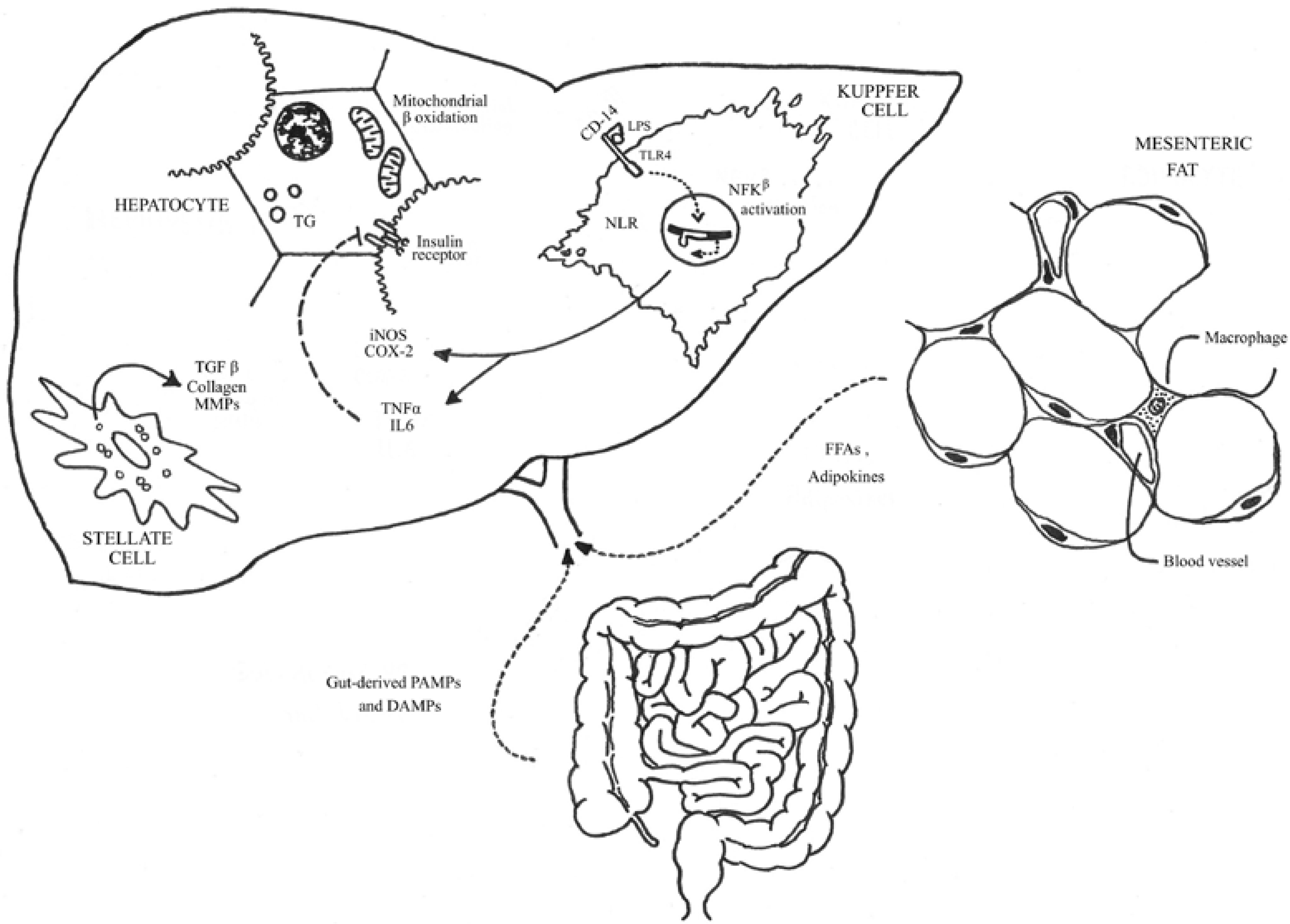
3. Biological and Molecular Basis of SIBO in NAFLD

4. Treatment of SIBO in NAFLD Patients

{kind=link}
| Study | Hepatic Disorder and Sample Size | Variables | Results |
|---|---|---|---|
| Wigg et al., 2001 [10] | 22 NASH (23% DM) vs. 23 controls (4% DM) | SIBO, gut permeability, endotoxin, TNF-α | NASH group: higher prevalence of SIBO (50% vs. 22%; p = 0.048); higher mean TNF-α levels (p = 0.01). |
| Sajjad et al., 2005 [11] | 12 NASH (41.6% DM) vs. 11 healthy controls | SIBO, ghrelin, insulin, ethanol | NASH group: higher prevalence of SIBO (50% vs. 9.1%; p = 0.025; lower plasma levels of acylated ghrelin (p = 0.015); higher fasting insulin concentrations (p < 0.006). |
| Soza et al., 2005 [57] | 10 nondiabetic NAFLD vs. 10 healthy controls | OCTT, EndoCAb IgG, IgM | NAFLD group: higher basal breathed H2 (p = 0.0084); prolonged OCTT (p = 0.0037). |
| Fu et al., 2006 [58] | 10 nondiabetic NASH vs. 10 healthy controls | OCTT, EndoCAb IgG | NASH group: prolonged OCTT (p = 0.00032); higher EndoCAb IgG titers (p = 0.011). |
| Sabaté et al., 2008 [12] | 146 morbidly obese referred for bariatric surgery vs. 40 healthy controls | SIBO, liver biopsy | Obese group: higher prevalence of SIBO (17.1% vs. 2.5%; p = 0.031). SIBO (p = 0.005) and MS (p = 0.006) were independently associated with severe hepatic steatosis. |
| Thuy et al., 2008 [59] | 12 nondiabetic NAFLD and 6 healthy controls | Diet, endotoxin, TLR4, PAI-1 plasma and liver | NAFLD group: consumed more fructose (p < 0.05); higher plasma levels of endotoxin (p < 0.05), PAI-1(p < 0.05), hepatic TLR4 (p < 0.05) and PAI-1 mRNA expression (p < 0.05). PAI-1 concentrations correlated with endotoxin levels (r = 0.83; p < 0.005) and with hepatic TLR4 mRNA expression (r = 0.54; p < 0.05). Hepatic mRNA expression of PAI-1 correlated with dietary intakes of carbohydrates (r = 0.67; p < 0.01), fructose (r = 0.58; p < 0.01), glucose (r = 0.58; p < 0.01) and sucrose (r = 0.70; p < 0.01). |
| Miele et al., 2009 [13] | 35 NAFLD (34% MS) vs. 27 untreated celiac disease (14.5% MS) vs. 24 healthy controls | SIBO, gut permeability, tight junctions, liver biopsy | NAFLD group: higher prevalence of SIBO (60% vs. 20.8%; p < 0.001), higher gut permeability (p < 0.001). SIBO and gut permeability correlated with the severity of steatosis (p < 0.001 and p < 0.05, respectively). |
| Shanab et al., 2011 [14] | 18 NASH (33%DM) vs. 16 healthy controls | SIBO, LBP, TLR2 and 4 on CD14+ cells, IL-1β, IL-6, IL-8, and TNF-α | NASH group: higher prevalence of SIBO (77.78% vs. 31.25%; p < 0.0001), higher TLR4 on CD14+ cells expression (p < 0.05); higher levels of IL-8 (p = 0.04), which correlated positively with TLR4 expression (r = 0.5123, p = 0.036). |
| Volynets et al., 2012 [15] | 20 NAFLD (25% pre-diabetic) vs. 10 healthy controls | Diet, SIBO, OCTT, gut permeability, blood alcohol, endotoxin, PAI-1 | NAFLD group: higher gut permeability, blood alcohol and endotoxin levels (for all, p < 0.05). Consumed more energy, carbohydrate, fructose, sucrose (for all, p < 0.05) and more glucose, protein and animal-derived protein (for all, p < 0.01). |
| Study | Hepatic Disorder and Sample Size | Intervention | Outcome |
|---|---|---|---|
| Loguercio et al., 2002 [68] | 10 NASH; 12 chronic HCV infection; 10 alcoholic cirrhosis | Mixture of Lactobacillus and Bifidobacterium + FOS + vitamins and minerals for 2 months | NASH patients: decrease in ALT, GGT, MDA, 4-HNE, TNF-α levels. |
| Loguercio et al., 2005 [69] | 22 NAFLD; 20 alcoholic cirrhosis; 20 HCV-related chronic hepatitis; 16 HCV-related cirrhosis | VSL#3 formula for 3 months | NAFLD and alcoholic cirrhosis groups: decrease in MDA, 4-HNE levels. All groups: decrease in S-NO levels. |
| Vajro et al., 2011 [70] | 20 NAFLD children (10 probiotic; 10 placebo) | L. rhamnosus for 8 weeks | Experimental group: decrease in ALT, antipeptidoglycan-polysaccharide antibodies levels. |
| Aller et al., 2011 [71] | 28 NAFLD (14 probiotic; 14 placebo) | L. bulgaricus and Streptococcus thermophiles for 3 months | Experimental group: decrease in AST, ALT, GGT levels. |
| Malaguarnera et al., 2012 [37] | 66 NAFLD (34 probiotic; 32 placebo) | B. longum + FOS for 24 weeks | Experimental group: decrease in TNF-α, CRP, AST, HOMA-IR, endotoxin levels; steatosis; NASH activity index. |
| Wong VW et al., 2013 [72] | 20 NAFLD (10 probiotic; 10 placebo) | Lepicol probiotic formula | Experimental group: decrease in AST levels; IHTG. |
| Eslamparast et al., 2014 [73] | 52 NAFLD (26 synbiotic; 26 placebo) | Synbiotic formula for 28 weeks | Experimental group: decrease in ALT, AST, GGT, CRP, TNF-α, nuclear factor κ-B levels; improvement fibrosis score. |
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Day, C. Pathogenesis of steatohepatitis. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, S.; Abeygunasekera, S.; Farrell, G.C.; Holmes-Walker, J.; Hui, J.M.; Fung, C.; Karim, R.; Lin, R.; Samarasinghe, D.; Liddle, C.; et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 2002, 35, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Iacono, A.; Raso, G.M.; Canani, R.B.; Calignano, A.; Meli, R. Probiotics as an emerging therapeutic strategy to treat NAFLD: Focus on molecular and biochemical mechanisms. J. Nutr. Biochem. 2011, 22, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Roh, Y.S.; Seki, E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroenterol. Hepatol. 2013, 28, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, I.D.; McGilvray, S.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Sajjad, A.; Mottershead, M.; Syn, W.K.; Jones, R.; Smith, S.; Nwokolo, C.U. Ciprofloxacin suppresses bacterial overgrowth, increases fasting insulin but does not correct low acylated ghrelin concentration in non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2005, 22, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Sabaté, J.M.; Jouët, P.; Harnois, F.; Mechler, C.; Msika, S.; Grossin, M.; Coffin, B. High prevalence of small intestinal bacterial overgrowth in patients with morbid obesity: A contributor to severe hepatic steatosis. Obes. Surg. 2008, 18, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; la Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Shanab, A.A.; Scully, P.; Crosbie, O.; Buckley, M.; O’Mahony, L.; Shanahan, F.; Gazareen, S.; Murphy, E.; Quigley, E.M. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: Association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig. Dis. Sci. 2011, 56, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Volynets, V.; Küper, M.A.; Strahl, S.; Maier, I.B.; Spruss, A.; Wagnerberger, S.; Königsrainer, A.; Bischoff, S.C.; Bergheim, I. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 2012, 57, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.G.; Casafont, F.; Crespo, J.; Cayón, A.; Mayorga, M.; Estebanez, A.; Fernadez-Escalante, J.C.; Pons-Romero, F. Lipopolysaccharide-binding protein plasma levels and liver TNF-α gene expression in obese patients: Evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes. Surg. 2007, 17, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Neish, A.S. Microbes in gastrointestinal health and disease. Gastroenterology 2009, 136, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Duseja, A.; Chawla, Y.K. Obesity and NAFLD: The Role of Bacteria and Microbiota. Clin. Liver Dis. 2014, 18, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Zoetendal, E.G.; Vaughan, E.E.; de Vos, W.M. A microbial world within us. Mol. Microbiol. 2006, 59, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Vajro, P.; Paolella, G.; Fasano, A. Microbiota and gut-liver axis: Their influences on obesity and obesity-related liver disease. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Bellot, P.; Francés, R.; Such, J. Pathological bacterial translocation in cirrhosis: Pathophysiology, diagnosis and clinical implications. Liver Int. 2013, 33, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, D.; Ahmed, A.; Li, X.; Ma, Y.; Tang, L.; Mo, D.; Ma, Y.; Xin, Y. Comparison of the gut microbe profiles and numbers between patients with liver cirrhosis and healthy individuals. Curr. Microbiol. 2012, 65, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.S.; Shah, V.H. The role of gut-liver axis in the pathogenesis of liver cirrhosis and portal hypertension. Clin. Mol. Hepatol. 2012, 18, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dhiman, R.K.; Kumari, S.; Rana, S.; Agarwal, R.; Duseja, A.; Chawla, Y. Role of small intestinal bacterial overgrowth and delayed gastrointestinal transit time in cirrhotic patients with minimal hepatic encephalopathy. J. Hepatol. 2010, 53, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M. Non-alcoholic fatty liver disease: The bile acid-activated farnesoid X receptor as an emerging treatment target. J. Lipids 2012. [Google Scholar] [CrossRef]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Zúñiga, V.; Bartolí, R.; Planas, R.; Hofmann, A.F.; Viñado, B.; Hagey, L.R.; Hernández, J.M.; Mañé, J.; Alvarez, M.A.; Ausina, V.; et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology 2003, 37, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhong, W.; Li, H.; Li, Q.; Qiu, Y.; Zheng, X.; Chen, H.; Zhao, X.; Zhang, S.; Zhou, Z.; et al. Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. FASEB J. 2013, 27, 3583–3593. [Google Scholar] [CrossRef] [PubMed]
- Caricilli, A.M.; Saad, M.J. The role of gut microbiota on insulin resistance. Nutrients 2013, 5, 829–851. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, J.L.; May, T. The effect of a methanogen, Methanobrevibacter smithii, on the growth rate, organic acid production, and specific ATP activity of three predominant ruminal cellulolytic bacteria. Curr. Microbiol. 2000, 40, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E.; et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Vacante, M.; Antic, T.; Giordano, M.; Chisari, G.; Acquaviva, R.; Mastrojeni, S.; Malaguarnera, G.; Mistretta, A.; Li Volti, G.; et al. Bifidobacterium longum with fructo-oligosaccharides in patients with nonalcoholic steatohepatitis. Dig. Dis. Sci. 2012, 57, 545–553. [Google Scholar]
- Madrid, A.M.; Hurtado, C.; Gatica, S.; Chacon, I.; Toyos, A.; Defilippi, C. Endogenous ethanol production in patients with liver cirrhosis, motor alteration and bacterial overgrowth. Rev. Med. Chile 2002, 130, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Mandrekar, P.; Dolganiuc, A. Innate immune response and hepatic inflammation. Semin. Liver Dis. 2007, 27, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Paik, Y.H.; Seki, E. Toll-like receptor signaling and liver fibrosis. Gastroenterol. Res. Pract. 2010. [Google Scholar] [CrossRef]
- Meli, R.; Raso, G.M.; Calignano, A. Role of innate immune response in non-alcoholic fatty liver disease: metabolic complications and therapeutic tools. Front. Immunol. 2014, 5, 177. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A. Toll-like receptors and adaptor molecules in liver disease: Update. Hepatology 2008, 48, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Kramer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Bashmakov, Y.; Horton, J.D. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem. 1999, 274, 30028–30032. [Google Scholar] [CrossRef] [PubMed]
- Matusoka, M.; Tsukamoto, H. Stimulation of hepatic lipocyte collagen production by Kupffer cell-derived transforming growth factor β: Implication for a pathogenic role. Hepatology 1990, 11, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Fukunishi, S.; Sujishi, T.; Takeshita, A.; Ohama, H.; Tsuchimoto, Y.; Asai, A.; Tsuda, Y.; Higuchi, K. Lipopolysaccharides accelerate hepatic steatosis in the development of nonalcoholic fatty liver disease in Zucker rats. J. Clin. Biochem. Nutr. 2014, 54, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Bondar, G.F.; Pisesky, W. Complications of small intestinal shortcircuiting for obesity. Arch. Surg. 1967, 94, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.G.; Richards, R.C.; Albo, D., Jr. Fatty degeneration of the liver after intestinal bypass for obesity. Am. J. Surg. 1968, 116, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Nazim, M.; Stamp, G.; Hodgson, H.J.F. Non-alcoholic steatohepatitis associated with small intestinal diverticulosis and bacterial overgrowth. Hepatogastroenterology 1989, 36, 349–351. [Google Scholar] [PubMed]
- Riordan, S.M.; Duncombe, V.M.; Thomas, M.C.; Nagree, A.; Bolin, T.D.; McIver, C.J.; Williams, R. Small intestinal bacterial overgrowth, intestinal permeability, and non-alcoholic steatohepatitis. Gut 2002, 50, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Khoshini, R.; Dai, S.C.; Lezcano, S.; Pimentel, M. A systematic review of diagnostic tests for small intestinal bacterial overgrowth. Dig. Dis. Sci. 2008, 53, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Horváth, V.J.; Izbéki, F.; Lengyel, C.; Kempler, P.; Várkonyi, T. Diabetic gastroparesis: Functional/morphologic background, diagnosis, and treatment options. Curr. Diabetes Rep. 2014, 14, 527. [Google Scholar] [CrossRef]
- Soza, A.; Riquelme, A.; González, R.; Alvarez, M.; Pérez-Ayuso, R.M.; Glasinovic, J.C.; Arrese, M. Increased orocecal transit time in patients with nonalcoholic fatty liver disease. Dig. Dis. Sci. 2005, 50, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.S.; Jiang, F. Cisapride decreasing orocecal transit time in patients with nonalcoholic steatohepatitis. Hepatobiliary Pancreat. Dis. Int. 2006, 5, 534–537. [Google Scholar] [PubMed]
- Thuy, S.; Ladurner, R.; Volynets, V.; Wagner, S.; Strahl, S.; Königsrainer, A.; Maier, K.P.; Bischoff, S.C.; Bergheim, I. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J. Nutr. 2008, 138, 1452–1455. [Google Scholar] [PubMed]
- Alessi, M.C.; Bastelica, D.; Mavri, A.; Morange, P.; Berthet, B.; Grino, M.; Juhan-Vague, I. Plasma PAI-1 levels are more strongly related to liver steatosis than to adipose tissue accumulation. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Guo, L.; Davis, M.A.; Lambert, J.C.; Beier, J.I.; Duveau, I.; Luyendyk, J.P.; Roth, R.A.; Arteel, G.E. Metformin prevents alcohol-induced liver injury in the mouse: Critical role of plasminogen activator inhibitor-1. Gastroenterology 2006, 130, 2099–2112. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Guo, L.; Davis, M.A.; Duveau, I.; Arteel, G.E. Critical role of plasminogen activator inhibitor-1 in cholestatic liver injury and fibrosis. J. Pharmacol. Exp. Ther. 2006, 316, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; de Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Fagà, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Volynets, V.; Machann, J.; Küper, M.A.; Maier, I.B.; Spruss, A.; Königsrainer, A.; Bischoff, S.C.; Bergheim, I. A moderate weight reduction through dietary intervention decreases hepatic fat content in patients with non-alcoholic fatty liver disease (NAFLD): A pilot study. Eur. J. Nutr. 2013, 52, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.W.; Lê, K.A.; Davis, J.; Alderete, T.L.; Cherry, R.; Lebel, S.; Goran, M.I. High rates of fructose malabsorption are associated with reduced liver fat in obese African Americans. J. Am. Coll. Nutr. 2012, 31, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.C.; Zhao, W.; Li, S. Small intestinal bacteria overgrowth decreases small intestinal motility in the NASH rats. World J. Gastroenterol. 2008, 14, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; de Simone, T.; Federico, A.; Terracciano, F.; Tuccillo, C.; di Chicco, M.; Cartenì, M. Gut-liver axis: A new point of attack to treat chronic liver damage? Am. J. Gastroenterol. 2002, 97, 2144–2146. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; Federico, A.; Tuccillo, C.; Terracciano, F.; D’Auria, M.V.; de Simone, C.; del Vecchio Blanco, C. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J. Clin. Gastroenterol. 2005, 39, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Vajro, P.; Mandato, C.; Licenziati, M.R.; Franzese, A.; Vitale, D.F.; Lenta, S.; Caropreso, M.; Vallone, G.; Meli, R. Effects of Lactobacillus rhamnosus strain GG in pediatric obesity-related liver disease. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; de Luis, D.A.; Izaola, O.; Conde, R.; Gonzalez Sagrado, M.; Primo, D.; de la Fuente, B.; Gonzalez, J. Effect of a probiotic on liver aminotransferases in nonalcoholic fatty liver disease patients: A double blind randomized clinical trial. Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 1090–1095. [Google Scholar] [PubMed]
- Wong, V.W.; Won, G.L.; Chim, A.M.; Chu, W.C.; Yeung, D.K.; Li, K.C.; Chan, H.L. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann. Hepatol. 2013, 12, 256–262. [Google Scholar] [PubMed]
- Eslamparast, T.; Poustchi, H.; Zamani, F.; Sharafkhah, M.; Malekzadeh, R.; Hekmatdoost, A. Synbiotic supplementation in nonalcoholic fatty liver disease: A randomized, double-blind, placebo-controlled pilot study. Am. J. Clin. Nutr. 2014, 99, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; Li, L.; Yu, CH.; Shen, Z.; Chen, L.H.; Li, Y.M. Effects of probiotics on nonalcoholic fatty liver disease: A meta-analysis. World J. Gastroenterol. 2013, 19, 6911–6918. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferolla, S.M.; Armiliato, G.N.A.; Couto, C.A.; Ferrari, T.C.A. The Role of Intestinal Bacteria Overgrowth in Obesity-Related Nonalcoholic Fatty Liver Disease. Nutrients 2014, 6, 5583-5599. https://doi.org/10.3390/nu6125583
Ferolla SM, Armiliato GNA, Couto CA, Ferrari TCA. The Role of Intestinal Bacteria Overgrowth in Obesity-Related Nonalcoholic Fatty Liver Disease. Nutrients. 2014; 6(12):5583-5599. https://doi.org/10.3390/nu6125583
Chicago/Turabian StyleFerolla, Silvia M., Geyza N. A. Armiliato, Cláudia A. Couto, and Teresa C. A. Ferrari. 2014. "The Role of Intestinal Bacteria Overgrowth in Obesity-Related Nonalcoholic Fatty Liver Disease" Nutrients 6, no. 12: 5583-5599. https://doi.org/10.3390/nu6125583
APA StyleFerolla, S. M., Armiliato, G. N. A., Couto, C. A., & Ferrari, T. C. A. (2014). The Role of Intestinal Bacteria Overgrowth in Obesity-Related Nonalcoholic Fatty Liver Disease. Nutrients, 6(12), 5583-5599. https://doi.org/10.3390/nu6125583