Understanding the Role of Maternal Diet on Kidney Development; an Opportunity to Improve Cardiovascular and Renal Health for Future Generations

{kind=link}
{kind=link}
Abstract
:1. Maternal Nutrition and the Kidney
1.1. Maternal Protein Restriction Programs Reduced Nephron Number but the Physiological Consequences are Unclear
1.2. Maternal Overnutrition Has Subtle Impacts upon Kidney Development and Function in the Offspring
1.3. Maternal Micronutrient Status and Its Impact on Offspring Kidney Health
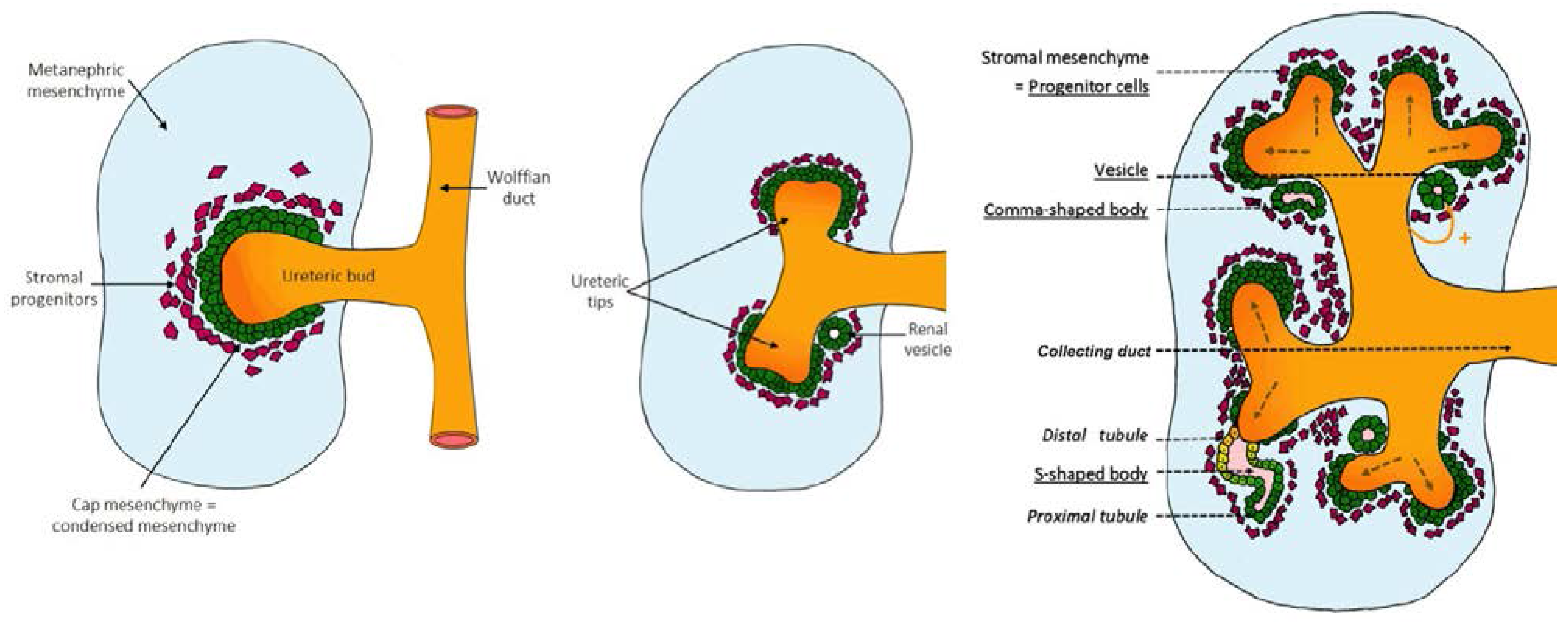
2. Kidney Development
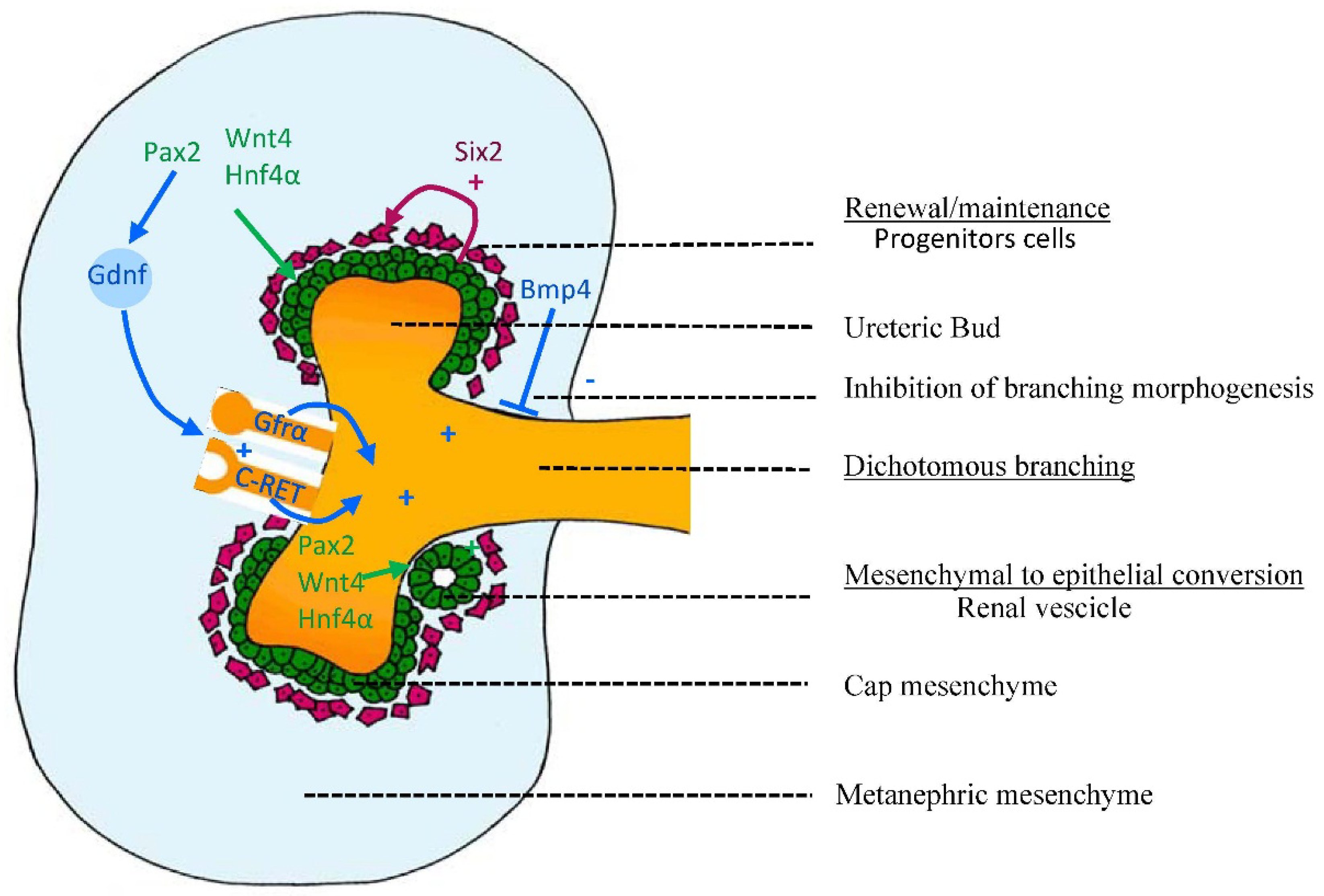
2.1. Early Signaling: Specification of the Ureteric Bud and Metanephric Mesenchyme

2.2. Genetic Control of Kidney Development
2.2.1. Control of Branching: Molecules Involved in Branching and Inhibiting Lateral Branching

2.2.2. Control of Nephrogenesis: Epithelial-Mesenchymal Transition (EMT)
3. Specification of Nephron Structures
4. Arcade Formation
5. Final Nephron Endowment
Early Investigations Supporting Changes during Kidney Development (Genes and Pathways, Cells and Timing)
6. Maternal Diet and the Offspring Epigenome
7. The Impact of Nephron Endowment on Adult Cardiovascular and Renal Health
8. Conclusions
Author Contributions
Conflicts of Interest
References
- Lucas, A. Programming by early nutrition in man. Ciba Found. Symp. 1991, 156, 38–50. [Google Scholar]
- Yajnik, C.S.; Deshpande, S.S.; Jackson, A.A.; Refsum, H.; Rao, S.; Fisher, D.J.; Bhat, D.S.; Naik, S.S.; Coyaji, K.J.; Joglekar, C.V.; et al. Vitamin B12 and folate concentrations during pregnancy and insulin resistance in the offspring: The pune maternal nutrition study. Diabetologia 2008, 51, 29–38. [Google Scholar]
- Cleal, J.K.; Poore, K.R.; Boullin, J.P.; Khan, O.; Chau, R.; Hambidge, O.; Torrens, C.; Newman, J.P.; Poston, L.; Noakes, D.E.; et al. Mismatched pre- and postnatal nutrition leads to cardiovascular dysfunction and altered renal function in adulthood. PNAS 2007, 104, 9529–9533. [Google Scholar]
- Poston, L. Influences of maternal nutritional status on vascular function in the offspring. Curr. Drug Targets 2007, 8, 914–922. [Google Scholar]
- Pettitt, D.J.; Jovanovic, L. Birth weight as a predictor of type 2 diabetes mellitus: The U-shaped curve. Curr. Diab. Rep. 2001, 1, 78–81. [Google Scholar]
- Van der Spuy, Z.M. Nutrition and reproduction. Clin. Obstet. Gynaecol. 1985, 12, 579–604. [Google Scholar]
- Van der Spuy, Z.M.; Jones, D.L.; Wright, C.S.; Piura, B.; Paintin, D.B.; James, V.H.; Jacobs, H.S. Inhibition of 3-beta-hydroxy steroid dehydrogenase activity in first trimester human pregnancy with trilostane and win 32729. Clin. Endocrinol. (Oxf.) 1983, 19, 521–531. [Google Scholar]
- Imdad, A.; Bhutta, Z.A. Maternal nutrition and birth outcomes: Effect of balanced protein-energy supplementation. Paediatr. Perinat. Epidemiol. 2012, 26 (Suppl. 1), 178–190. [Google Scholar]
- Widdowson, E.M. Prenatal nutrition. Ann. New York Acad. Sci. 1977, 300, 188–196. [Google Scholar]
- McCance, R.A.; Widdowson, E.M.; Verdon-Roe, C.M. A study of english diets by the individual method: III. Pregnant women at different economic levels. J. Hyg. (Lond.) 1938, 38, 596–622. [Google Scholar]
- Widdowson, E.M.; McCance, R.A. The effect of finite periods of undernutrition at different ages on the composition and subsequent development of the rat. Proc. R. Soc. Lond. Ser. B Containing Papers Biol. Character. R. Soc. 1963, 158, 329–342. [Google Scholar]
- Baird, A.; Widdowson, E.M.; Cowley, J.J. Effects of calorie and protein deficiencies early in life on the subsequent learning ability of rats. Br. J. Nutr. 1971, 25, 391–403. [Google Scholar]
- Dickerson, J.W.; Merat, A.; Widdowson, E.M. Intra-uterine growth retardation in the pig. 3. The chemical structure of the brain. Biol. Neonate 1971, 19, 354–362. [Google Scholar]
- Widdowson, E.M. Changes in pigs due to undernutrition before birth, and for one, two, and three years afterwards, and the effects of rehabilitation. Adv. Exp. Med. Biol. 1974, 49, 165–181. [Google Scholar]
- Armitage, J.A.; Khan, I.Y.; Taylor, P.D.; Nathanielsz, P.W.; Poston, L. Developmental programming of the metabolic syndrome by maternal nutritional imbalance: How strong is the evidence from experimental models in mammals? J. Physiol. 2004, 561, 355–377. [Google Scholar]
- Armitage, J.A.; Lakasing, L.; Taylor, P.D.; Balachandran, A.A.; Jensen, R.I.; Dekou, V.; Ashton, N.; Nyengaard, J.R.; Poston, L. Developmental programming of aortic and renal structure in offspring of rats fed fat-rich diets in pregnancy. J. Physiol. 2005, 565, 171–184. [Google Scholar]
- Tomat, A.L.; Costa Mde, L.; Arranz, C.T. Zinc restriction during different periods of life: Influence in renal and cardiovascular diseases. Nutrition 2011, 27, 392–398. [Google Scholar]
- El-Khashab, E.K.; Hamdy, A.M.; Maher, K.M.; Fouad, M.A.; Abbas, G.Z. Effect of maternal vitamin A deficiency during pregnancy on neonatal kidney size. J. Perinat. Med. 2013, 41, 199–203. [Google Scholar]
- Cooke, C.L.; Zhao, L.; Gysler, S.; Arany, E.; Regnault, T.R. Sex-specific effects of low protein diet on in utero programming of renal g-protein coupled receptors. J. Dev. Orig. Health Dis. 2014, 5, 36–44. [Google Scholar]
- Brenner, B.M.; Garcia, D.L.; Anderson, S. Glomeruli and blood pressure: Less of one, more the other? Am. J. Hypertens. 1988, 1, 335–347. [Google Scholar]
- Hoppe, C.C.; Evans, R.G.; Bertram, J.F.; Moritz, K.M. Effects of dietary protein restriction on nephron number in the mouse. Ame. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1768–R1774. [Google Scholar]
- Hoppe, C.C.; Evans, R.G.; Moritz, K.M.; Cullen-McEwen, L.A.; Fitzgerald, S.M.; Dowling, J.; Bertram, J.F. Combined prenatal and postnatal protein restriction influences adult kidney structure, function, and arterial pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R462–R469. [Google Scholar]
- Nwagwu, M.O.; Cook, A.; Langley-Evans, S.C. Evidence of progressive deterioration of renal function in rats exposed to a maternal low-protein diet in utero. Br. J. Nutr. 2000, 83, 79–85. [Google Scholar]
- Vehaskari, V.M.; Aviles, D.H.; Manning, J. Prenatal programming of adult hypertension in the rat. Kidney Int. 2001, 59, 238–245. [Google Scholar]
- Vehaskari, V.M.; Woods, L.L. Prenatal programming of hypertension: Lessons from experimental models. J. Am. Soc. Nephrol. 2005, 16, 2545–2556. [Google Scholar]
- Woods, L.L.; Ingelfinger, J.R.; Nyengaard, J.R.; Rasch, R. Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr. Res. 2001, 49, 460–467. [Google Scholar]
- Woods, L.L.; Ingelfinger, J.R.; Rasch, R. Modest maternal protein restriction fails to program adult hypertension in female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1131–R1136. [Google Scholar]
- Woods, L.L.; Weeks, D.A.; Rasch, R. Programming of adult blood pressure by maternal protein restriction: Role of nephrogenesis. Kidney Int. 2004, 65, 1339–1348. [Google Scholar]
- Chong, E.; Yosypiv, I.V. Developmental programming of hypertension and kidney disease. Int. J. Nephrol. 2012, 2012, 760580. [Google Scholar]
- Zimanyi, M.A.; Denton, K.M.; Forbes, J.M.; Thallas-Bonke, V.; Thomas, M.C.; Poon, F.; Black, M.J. A developmental nephron deficit in rats is associated with increased susceptibility to a secondary renal injury due to advanced glycation end-products. Diabetologia 2006, 49, 801–810. [Google Scholar]
- Do Carmo Franco, M.; Ponzio, B.F.; Gomes, G.N.; Gil, F.Z.; Tostes, R.; Carvalho, M.H.; Fortes, Z.B. Micronutrient prenatal supplementation prevents the development of hypertension and vascular endothelial damage induced by intrauterine malnutrition. Life Sci. 2009, 85, 327–333. [Google Scholar]
- Reyes-Castro, L.A.; Rodriguez, J.S.; Charco, R.; Bautista, C.J.; Larrea, F.; Nathanielsz, P.W.; Zambrano, E. Maternal protein restriction in the rat during pregnancy and/or lactation alters cognitive and anxiety behaviors of female offspring. Int. J. Dev. Neurosci. 2011, 30, 39–45. [Google Scholar]
- Bertram, C.; Trowern, A.R.; Copin, N.; Jackson, A.A.; Whorwood, C.B. The maternal diet during pregnancy programs altered expression of the glucocorticoid receptor and type 2 11beta-hydroxysteroid dehydrogenase: Potential molecular mechanisms underlying the programming of hypertension in utero. Endocrinology 2001, 142, 2841–2853. [Google Scholar]
- Dagan, A.; Habib, S.; Gattineni, J.; Dwarakanath, V.; Baum, M. Prenatal programming of rat thick ascending limb chloride transport by low-protein diet and dexamethasone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R93–R99. [Google Scholar]
- Ozanne, S.E.; Hales, C.N. Lifespan: Catch-up growth and obesity in male mice. Nature 2004, 427, 411–412. [Google Scholar]
- Ozanne, S.E.; Hales, C.N. Poor fetal growth followed by rapid postnatal catch-up growth leads to premature death. Mech. Ageing Dev. 2005, 126, 852–854. [Google Scholar]
- Roseboom, T.J.; van der Meulen, J.H.; Osmond, C.; Barker, D.J.; Ravelli, A.C.; Schroeder-Tanka, J.M.; van Montfrans, G.A.; Michels, R.P.; Bleker, O.P. Coronary heart disease after prenatal exposure to the Dutch famine, 1944–1945. Heart 2000, 84, 595–598. [Google Scholar]
- Roseboom, T.J.; van der Meulen, J.H.; Ravelli, A.C.; Osmond, C.; Barker, D.J.; Bleker, O.P. Effects of prenatal exposure to the Dutch famine on adult disease in later life: An overview. Twin Res. 2001, 4, 293–298. [Google Scholar]
- Roseboom, T.J.; van der Meulen, J.H.; Ravelli, A.C.; van Montfrans, G.A.; Osmond, C.; Barker, D.J.; Bleker, O.P. Blood pressure in adults after prenatal exposure to famine. J. Hypertens. 1999, 17, 325–330. [Google Scholar]
- Jarvie, E.; Hauguel-de-Mouzon, S.; Nelson, S.M.; Sattar, N.; Catalano, P.M.; Freeman, D.J. Lipotoxicity in obese pregnancy and its potential role in adverse pregnancy outcome and obesity in the offspring. Clin. Sci. (Lond.) 2010, 119, 123–129. [Google Scholar]
- Zhang, S.; Rattanatray, L.; McMillen, I.C.; Suter, C.M.; Morrison, J.L. Periconceptional nutrition and the early programming of a life of obesity or adversity. Prog. Biophys. Mol. Biol. 2011, 106, 307–314. [Google Scholar]
- Taylor, P.D.; Samuelsson, A.M.; Poston, L. Maternal obesity and the developmental programming of hypertension: A role for leptin. Acta Physiol. (Oxf.) 2014, 210, 508–523. [Google Scholar]
- Henry, S.L.; Barzel, B.; Wood-Bradley, R.J.; Burke, S.L.; Head, G.A.; Armitage, J.A. Developmental origins of obesity-related hypertension. Clin. Exp. Pharmacol. Physiol. 2012, 39, 799–806. [Google Scholar]
- Henry, S.L.; Bensley, J.G.; Wood-Bradley, R.J.; Cullen-McEwen, L.A.; Bertram, J.F.; Armitage, J.A. White adipocytes: More than just fat depots. Int. J. Biochem. Cell Biol. 2012, 44, 435–440. [Google Scholar]
- Tarantal, A.F.; Berglund, L. Obesity and lifespan health—Importance of the fetal environment. Nutrients 2014, 6, 1725–1736. [Google Scholar]
- Shasa, D.R.; Odhiambo, J.F.; Long, N.M.; Tuersunjiang, N.; Nathanielsz, P.W.; Ford, S.P. Multigenerational impact of maternal overnutrition/obesity in the sheep on the neonatal leptin surge in granddaughters. Int. J. Obes. (Lond.) 2014. [Google Scholar] [CrossRef]
- Reynolds, R.M.; Allan, K.M.; Raja, E.A.; Bhattacharya, S.; McNeill, G.; Hannaford, P.C.; Sarwar, N.; Lee, A.J.; Bhattacharya, S.; Norman, J.E. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: Follow-up of 1,323,275 person years. BMJ 2013, 347, f4539. [Google Scholar]
- Catalano, P.M.; Presley, L.; Minium, J.; Hauguel-de Mouzon, S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 2009, 32, 1076–1080. [Google Scholar]
- Boney, C.M.; Verma, A.; Tucker, R.; Vohr, B.R. Metabolic syndrome in childhood: Association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 2005, 115, e290–e296. [Google Scholar]
- Prior, L.J.; Davern, P.J.; Burke, S.L.; Lim, K.; Armitage, J.A.; Head, G.A. Exposure to a high-fat diet during development alters leptin and ghrelin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension 2013, 63, 338–345. [Google Scholar]
- Jackson, C.M.; Alexander, B.T.; Roach, L.; Haggerty, D.; Marbury, D.C.; Hutchens, Z.M.; Flynn, E.R.; Maric-Bilkan, C. Exposure to maternal overnutrition and a high-fat diet during early postnatal development increases susceptibility to renal and metabolic injury later in life. Am. J. Physiol. Renal Physiol. 2012, 302, F774–F783. [Google Scholar]
- Armitage, J.A.; Pearce, A.D.; Sinclair, A.J.; Vingrys, A.J.; Weisinger, R.S.; Weisinger, H.S. Increased blood pressure later in life may be associated with perinatal n-3 fatty acid deficiency. Lipids 2003, 38, 459–464. [Google Scholar]
- Schmatz, M.; Madan, J.; Marino, T.; Davis, J. Maternal obesity: The interplay between inflammation, mother and fetus. J. Perinatol. 2010, 30, 441–446. [Google Scholar]
- Lelievre-Pegorier, M.; Vilar, J.; Ferrier, M.-L.; Moreau, E.; Freund, N.; Gilbert, T.; Merlet-Benichou, C. Mild vitamin A deficiency leads to inborn nephron deficit in the rat. Kidney Int. 1998, 54, 1455–1462. [Google Scholar]
- Lisle, S.J.M.; Lewis, R.M.; Petry, C.J.; Ozanne, S.E.; Hales, C.N.; Forhead, A.J. Effect of maternal iron restriction during pregnancy on renal morphology in the adult rat offspring. Br. J. Nutr. 2003, 90, 33–39. [Google Scholar]
- Tomat, A.L.; Costa, M.A.; Girgulsky, L.C.; Veiras, L.; Weisstaub, A.R.; Inserra, F.; Balaszczuk, A.M.; Arranz, C.T. Zinc deficiency during growth: Influence on renal function and morphology. Life Sci. 2007, 80, 1292–1302. [Google Scholar]
- Tomat, A.L.; Veiras, L.C.; Aguirre, S.; Fasoli, H.; Elesgaray, R.; Caniffi, C.; Costa, M.A.; Arranz, C.T. Mild zinc deficiency in male and female rats: Early postnatal alterations in renal nitric oxide system and morphology. Nutrition 2013, 29, 568–573. [Google Scholar]
- Tian, X.; Anthony, K.; Neuberger, T.; Diaz, F.J. Preconception zinc deficiency disrupts postimplantation fetal and placental development in mice. Biol. Reprod. 2014, 90, 83. [Google Scholar]
- Dressler, G.R. The cellular basis of kidney development. Annu. Rev. Cell Dev. Biol. 2006, 22, 509–529. [Google Scholar]
- Caruana, G.; Young, R.J.; Bertram, J.F. Imaging the embryonic kidney. Nephron. Exp. Nephrol. 2006, 103, e62–e68. [Google Scholar]
- Moritz, K.M.; Wintour, E.M. Functional development of the meso- and metanephros. Pediatr. Nephrol. 1999, 13, 171–178. [Google Scholar]
- Ortiz, L.A.; Quan, A.; Weinberg, A.; Baum, M. Effect of prenatal dexamethasone on rat renal development. Kidney Int. 2001, 59, 1663–1669. [Google Scholar]
- Hartman, H.A.; Lai, H.L.; Patterson, L.T. Cessation of renal morphogenesis in mice. Dev. Biol. 2007, 310, 379–387. [Google Scholar]
- Cullen-McEwen, L.A.; Armitage, J.A.; Nyengaard, J.R.; Moritz, K.M.; Bertram, J.F. A design-based method for estimating glomerular number in the developing kidney. Am. J. Physiol. Renal Physiol. 2011, 300, F1448–F1453. [Google Scholar]
- Moritz, K.M.; Wintour, E.M.; Black, M.J.; Bertram, J.F.; Caruana, G. Factors influencing mammalian kidney development: Implications for health in adult life. Adv. Anat. Embryol. Cell Biol. 2008, 196, 1–78. [Google Scholar]
- Zohdi, V.; Sutherland, M.R.; Lim, K.; Gubhaju, L.; Zimanyi, M.A.; Black, M.J. Low birth weight due to intrauterine growth restriction and/or preterm birth: Effects on nephron number and long-term renal health. Int. J. Nephrol. 2012, 2012, 136942. [Google Scholar]
- Davies, J.A.; Fisher, C.E. Genes and proteins in renal development. Exp. Nephrol. 2002, 10, 102–113. [Google Scholar]
- Jain, S. The many faces of ret dysfunction in kidney. Organogenesis 2009, 5, 177–190. [Google Scholar]
- Little, M.; Georgas, K.; Pennisi, D.; Wilkinson, L. Kidney development: Two tales of tubulogenesis. Curr. Top. Dev. Biol. 2010, 90, 193–229. [Google Scholar]
- Michos, O. Kidney development: From ureteric bud formation to branching morphogenesis. Curr. Opin. Genet. Dev. 2009, 19, 484–490. [Google Scholar]
- Popsueva, A.; Poteryaev, D.; Arighi, E.; Meng, X.; Angers-Loustau, A.; Kaplan, D.; Saarma, M.; Sariola, H. GDNF promotes tubulogenesis of GFRalpha1-expressing MDCK cells by Src-mediated phosphorylation of Met receptor tyrosine kinase. J. Cell Biol. 2003, 161, 119–129. [Google Scholar]
- Schedl, A. Renal abnormalities and their developmental origin. Nat. Rev. Genet. 2007, 8, 791–802. [Google Scholar]
- Schwab, K.; Patterson, L.T.; Aronow, B.J.; Luckas, R.; Liang, H.C.; Potter, S.S. A catalogue of gene expression in the developing kidney. Kidney Int. 2003, 64, 1588–1604. [Google Scholar]
- Stuart, R.O.; Bush, K.T.; Nigam, S.K. Changes in global gene expression patterns during development and maturation of the rat kidney. Proc. Natl. Acad. Sci. USA 2001, 98, 5649–5654. [Google Scholar]
- Stuart, R.O.; Bush, K.T.; Nigam, S.K. Changes in gene expression patterns in the ureteric bud and metanephric mesenchyme in models of kidney development. Kidney Int. 2003, 64, 1997–2008. [Google Scholar]
- Sariola, H. Nephron induction revisited: From caps to condensates. Curr. Opin. Nephrol. Hypertens. 2002, 11, 17–21. [Google Scholar]
- Torban, E.; Dziarmaga, A.; Iglesias, D.; Chu, L.L.; Vassilieva, T.; Little, M.; Eccles, M.; Discenza, M.; Pelletier, J.; Goodyer, P. Pax2 activates wnt4 expression during mammalian kidney development. J. Biol. Chem. 2006, 281, 12705–12712. [Google Scholar]
- Kanazawa, T.; Ichii, O.; Otsuka, S.; Namiki, Y.; Hashimoto, Y.; Kon, Y. Hepatocyte nuclear factor 4 alpha is associated with mesenchymal-epithelial transition in developing kidneys of c57bl/6 mice. J. Vet. Medical Sci./Jpn. Soc. Vet. Sci. 2011, 73, 601–607. [Google Scholar]
- Kanazawa, T.; Konno, A.; Hashimoto, Y.; Kon, Y. Hepatocyte nuclear factor 4 alpha is related to survival of the condensed mesenchyme in the developing mouse kidney. Dev. Dyn. 2010, 239, 1145–1154. [Google Scholar]
- Brunskill, E.W.; Aronow, B.J.; Georgas, K.; Rumballe, B.; Valerius, M.T.; Aronow, J.; Kaimal, V.; Jegga, A.G.; Yu, J.; Grimmond, S.; et al. Atlas of gene expression in the developing kidney at microanatomic resolution. Dev. Cell 2008, 15, 781–791. [Google Scholar]
- Goncalves, A.; Zeller, R. Genetic analysis reveals an unexpected role of bmp7 in initiation of ureteric bud outgrowth in mouse embryos. PLoS One 2011, 6, e19370. [Google Scholar]
- Martinez, G.; Bertram, J.F. Organisation of bone morphogenetic proteins in renal development. Nephron. Exp. Nephrol. 2003, 93, e18–e22. [Google Scholar]
- Martinez, G.; Loveland, K.L.; Clark, A.T.; Dziadek, M.; Bertram, J.F. Expression of bone morphogenetic protein receptors in the developing mouse metanephros. Exp. Nephrol. 2001, 9, 372–379. [Google Scholar]
- Hartwig, S.; Hu, M.C.; Cella, C.; Piscione, T.; Filmus, J.; Rosenblum, N.D. Glypican-3 modulates inhibitory Bmp2-Smad signaling to control renal development in vivo. Mech. Dev. 2005, 122, 928–938. [Google Scholar]
- Grieshammer, U.; Cebrian, C.; Ilagan, R.; Meyers, E.; Herzlinger, D.; Martin, G.R. Fgf8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 2005, 132, 3847–3857. [Google Scholar]
- Perantoni, A.O.; Timofeeva, O.; Naillat, F.; Richman, C.; Pajni-Underwood, S.; Wilson, C.; Vainio, S.; Dove, L.F.; Lewandoski, M. Inactivation of fgf8 in early mesoderm reveals an essential role in kidney development. Development 2005, 132, 3859–3871. [Google Scholar]
- Plisov, S.Y.; Yoshino, K.; Dove, L.F.; Higinbotham, K.G.; Rubin, J.S.; Perantoni, A.O. TGF beta 2, LIF and FGF2 cooperate to induce nephrogenesis. Development 2001, 128, 1045–1057. [Google Scholar]
- Dudley, A.T.; Godin, R.E.; Robertson, E.J. Interaction between fgf and bmp signaling pathways regulates development of metanephric mesenchyme. Genes Dev. 1999, 13, 1601–1613. [Google Scholar]
- Kobayashi, A.; Valerius, M.T.; Mugford, J.W.; Carroll, T.J.; Self, M.; Oliver, G.; McMahon, A.P. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 2008, 3, 169–181. [Google Scholar]
- Self, M.; Lagutin, O.V.; Bowling, B.; Hendrix, J.; Cai, Y.; Dressler, G.R.; Oliver, G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006, 25, 5214–5228. [Google Scholar]
- Kobayashi, A.; Mugford, J.W.; Krautzberger, A.M.; Naiman, N.; Liao, J.; McMahon, A.P. Identification of a multipotent self-renewing stromal progenitor population during mammalian kidney organogenesis. Stem Cell Rep. 2014, 3, 650–662. [Google Scholar]
- Charlton, J.R.; Norwood, V.F.; Kiley, S.C.; Gurka, M.J.; Chevalier, R.L. Evolution of the urinary proteome during human renal development and maturation: Variations with gestational and postnatal age. Pediatr Res 2012, 72, 179–185. [Google Scholar]
- Kishore, B.K.; Mandon, B.; Oza, N.B.; DiGiovanni, S.R.; Coleman, R.A.; Ostrowski, N.L.; Wade, J.B.; Knepper, M.A. Rat renal arcade segment expresses vasopressin-regulated water channel and vasopressin v2 receptor. J. Clin. Investig. 1996, 97, 2763–2771. [Google Scholar]
- Zent, R.; Pozzi, A. Connecting the segments. J. Am. Soc. Nephrol. 2012, 23, 1603–1605. [Google Scholar]
- Hughson, M.D.; Douglas-Denton, R.; Bertram, J.F.; Hoy, W.E. Hypertension, glomerular number, and birth weight in african americans and white subjects in the southeastern united states. Kidney Int. 2006, 69, 671–678. [Google Scholar]
- Merlet-Benichou, C. Influence of fetal environment on kidney development. Int. J. Dev. Biol. 1999, 43, 453–456. [Google Scholar]
- Rumballe, B.A.; Georgas, K.M.; Combes, A.N.; Ju, A.L.; Gilbert, T.; Little, M.H. Nephron formation adopts a novel spatial topology at cessation of nephrogenesis. Dev. Biol. 2011, 360, 110–122. [Google Scholar]
- Singh, R.R.; Moritz, K.M.; Bertram, J.F.; Cullen-McEwen, L.A. Effects of dexamethasone exposure on rat metanephric development: In vitro and in vivo studies. Am. J. Physiol. Renal Physiol. 2007, 293, F548–F554. [Google Scholar]
- Welham, S.J.; Riley, P.R.; Wade, A.; Hubank, M.; Woolf, A.S. Maternal diet programs embryonic kidney gene expression. Physiol. Genomics 2005, 22, 48–56. [Google Scholar]
- Douglas-Denton, R.N.; McNamara, B.J.; Hoy, W.E.; Hughson, M.D.; Bertram, J.F. Does nephron number matter in the development of kidney disease? Ethn. Dis. 2006, 16, 40–45. [Google Scholar]
- Hoy, W.E.; Hughson, M.D.; Singh, G.R.; Douglas-Denton, R.; Bertram, J.F. Reduced nephron number and glomerulomegaly in australian aborigines: A group at high risk for renal disease and hypertension. Kidney Int. 2006, 70, 104–110. [Google Scholar]
- Hoy, W.E.; Hughson, M.D.; Bertram, J.F.; Douglas-Denton, R.; Amann, K. Nephron number, hypertension, renal disease, and renal failure. J. Am. Soc. Nephrol. 2005, 16, 2557–2564. [Google Scholar]
- Hoy, W.E.; Samuel, T.; Hughson, M.D.; Nicol, J.L.; Bertram, J.F. How many glomerular profiles must be measured to obtain reliable estimates of mean glomerular areas in human renal biopsies? J. Am. Soc. Nephrol. 2006, 17, 556–563. [Google Scholar]
- Hughson, M.D.; Gobe, G.C.; Hoy, W.E.; Manning, R.D., Jr.; Douglas-Denton, R.; Bertram, J.F. Associations of glomerular number and birth weight with clinicopathological features of African Americans and whites. Am. J. Kidney Dis. 2008, 52, 18–28. [Google Scholar]
- Keller, G.; Zimmer, G.; Mall, G.; Ritz, E.; Amann, K. Nephron number in patients with primary hypertension. N. Engl. J. Med. 2003, 348, 101–108. [Google Scholar]
- Nyengaard, J.R.; Bendsten, T.F. Glomerular number and size in relation to age, kidney weight, and body surface in normal man. Anat. Rec. 1992, 232, 194–201. [Google Scholar]
- Wlodek, M.E.; Mibus, A.; Tan, A.; Siebel, A.L.; Owens, J.A.; Moritz, K.M. Normal lactational environment restores nephron endowment and prevents hypertension after placental restriction in the rat. J. Am. Soc. Nephrol. 2007, 18, 1688–1696. [Google Scholar]
- Wlodek, M.E.; Westcott, K.; Siebel, A.L.; Owens, J.A.; Moritz, K.M. Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int. 2008, 74, 187–195. [Google Scholar]
- Siddique, K.; Guzman, G.L.; Gattineni, J.; Baum, M. Effect of postnatal maternal protein intake on prenatal programming of hypertension. Reprod. Sci. 2014, 21, 1499–1507. [Google Scholar]
- Abdel-Hakeem, A.K.; Henry, T.Q.; Magee, T.R.; Desai, M.; Ross, M.G.; Mansano, R.Z.; Torday, J.S.; Nast, C.C. Mechanisms of impaired nephrogenesis with fetal growth restriction: Altered renal transcription and growth factor expression. Am. J. Obstet. Gynecol. 2008, 199, 252.e1–252.e7. [Google Scholar]
- Henry, T.Q.; Mansano, R.Z.; Nast, C.C.; Lakshmanan, J.; Abdallah, M.; Abdel-Hakeem, A.K.; Desai, M.; Ross, M.G.; Magee, T.R. Gdnf and MAPK-ERK pathway signaling is reduced during nephrogenesis following maternal under-nutrition. J. Dev. Orig. Health Dis. 2010, 1, 67–74. [Google Scholar]
- Sene, L.D.B.; Mesquita, F.F.; de Moraes, L.N.; Santos, D.C.; Carvalho, R.; Gontijo, J.A.; Boer, P.A. Involvement of renal corpuscle microrna expression on epithelial-to-mesenchymal transition in maternal low protein diet in adult programmed rats. PLoS One 2013, 8, e71310. [Google Scholar]
- Welham, S.J.; Wade, A.; Woolf, A.S. Protein restriction in pregnancy is associated with increased apoptosis of mesenchymal cells at the start of rat metanephrogenesis. Kidney Int. 2002, 61, 1231–1242. [Google Scholar]
- Magee, T.R.; Tafti, S.A.; Desai, M.; Liu, Q.; Ross, M.G.; Nast, C.C. Maternal undernourished fetal kidneys exhibit differential regulation of nephrogenic genes including downregulation of the notch signaling pathway. Reprod. Sci. 2011, 18, 563–576. [Google Scholar]
- Tafti, S.A.; Nast, C.C.; Desai, M.; Amaya, K.E.; Ross, M.G.; Magee, T.R. Maternal undernutrition upregulates apoptosis in offspring nephrogenesis. J. Dev. Orig. Health Dis. 2011, 2, 226–235. [Google Scholar]
- Barker, D.J.P.; Bagpy, S.P.; Hanson, M.A. Mechanisms of disease; in utero programming in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2006, 2, 700–707. [Google Scholar]
- Burrow, C.R. Regulatory molecules in kidney development. Pediatr. Nephrol. 2000, 14, 240–253. [Google Scholar]
- Dolinoy, D.C.; Jirtle, R.L. Environmental epigenomics in human health and disease. Environ. Mol. Mutagen. 2008, 49, 4–8. [Google Scholar]
- Hales, B.F.; Grenier, L.; Lalancette, C.; Robaire, B. Epigenetic programming: From gametes to blastocyst. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 652–665. [Google Scholar]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.C.; Boileau, P.; le Bouc, Y.; Deal, C.L.; Lillycrop, K.; et al. Child health, developmental plasticity, and epigenetic programming. Endocr. Rev. 2011, 32, 159–224. [Google Scholar]
- Ikeda, S.; Koyama, H.; Sugimoto, M.; Kume, S. Roles of one-carbon metabolism in preimplantation period—Effects on short-term development and long-term programming. J. Reprod. Dev. 2012, 58, 38–43. [Google Scholar]
- Romagnolo, D.F.; Dashwood, R.; Stover, P.J.; Waterland, R.A.; Ziegler, T.R. Nutritional regulation of epigenetic changes. Adv. Nutr. 2012, 3, 749–750. [Google Scholar]
- Burdge, G.C.; Hanson, M.A.; Slater-Jefferies, J.L.; Lillycrop, K.A. Epigenetic regulation of transcription: A mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? Br. J. Nutr. 2007, 97, 1036–1046. [Google Scholar]
- Cooney, C.A.; Dave, A.A.; Wolff, G.L. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J. Nutr. 2002, 132, 2393S–2400S. [Google Scholar]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. Faseb J. 1998, 12, 949–957. [Google Scholar]
- Waterland, R.A.; Michels, K.B. Epigenetic epidemiology of the developmental origins hypothesis. Annu. Rev. Nutr. 2007, 27, 363–388. [Google Scholar]
- Pham, T.D.; MacLennan, N.K.; Chiu, C.T.; Laksana, G.S.; Hsu, J.L.; Lane, R.H. Uteroplacental insufficiency increases apoptosis and alters p53 gene methylation in the full-term iugr rat kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R962–R970. [Google Scholar]
- Nagalakshmi, V.K.; Ren, Q.; Pugh, M.M.; Valerius, M.T.; McMahon, A.P.; Yu, J. Dicer regulates the development of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int. 2011, 79, 317–330. [Google Scholar]
- Pastorelli, L.M.; Wells, S.; Fray, M.; Smith, A.; Hough, T.; Harfe, B.D.; McManus, M.T.; Smith, L.; Woolf, A.S.; Cheeseman, M.; et al. Genetic analyses reveal a requirement for Dicer1 in the mouse urogenital tract. Mamm. Genome 2009, 20, 140–151. [Google Scholar]
- Lillycrop, K.A.; Phillips, E.S.; Jackson, A.A.; Hanson, M.A.; Burdge, G.C. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J. Nutr. 2005, 135, 1382–1386. [Google Scholar]
- Rees, W.D.; Hay, S.M.; Brown, D.S.; Antipatis, C.; Palmer, R.M. Maternal protein deficiency causes hypermethylation of DNA in the livers of rat fetuses. J. Nutr. 2000, 130, 1821–1826. [Google Scholar]
- Bogdarina, I.; Welham, S.; King, P.J.; Burns, S.P.; Clark, A.J. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ. Res. 2007, 100, 520–526. [Google Scholar]
- Lillycrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restriction diet persistently alters the methylation of specific cytosines in the hepatic PPAR[alpha] promoter of the offspring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar]
- Lillycrop, K.A.; Slater-Jefferies, J.L.; Hanson, M.A.; Godfrey, K.M.; Jackson, A.A.; Burdge, G.C. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br. J. Nutr. 2007, 97, 1064–1073. [Google Scholar]
- Song, R.; van Buren, T.; Yosypiv, I.V. Histone deacetylases are critical regulators of the renin-angiotensin system during ureteric bud branching morphogenesis. Pediatr. Res. 2010, 67, 573–578. [Google Scholar]
- Koleganova, N.; Piecha, G.; Ritz, E.; Becker, L.E.; Muller, A.; Weckbach, M.; Nyengaard, J.R.; Schirmacher, P.; Gross-Weissmann, M.L. Both high and low maternal salt intake in pregnancy alter kidney development in the offspring. Am. J. Physiol. Renal Physiol. 2011, 301, F344–F354. [Google Scholar]
- Cebrian, C.; Asai, N.; D’Agati, V.; Costantini, F. The number of fetal nephron progenitor cells limits ureteric branching and adult nephron endowment. Cell Rep. 2014, 7, 127–137. [Google Scholar]
- Lackland, D.T. Mechanisms and fetal origins of kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2531–2532. [Google Scholar]
- Lackland, D.T.; Bendall, H.E.; Osmond, C.; Egan, B.M.; Barker, D.J. Low birth weights contribute to the high rates of early-onset chronic renal failure in the southeastern United States. Arch. Intern. Med. 2000, 160, 1472–1476. [Google Scholar]
- Hoy, W.E.; Kincaid-Smith, P.; Hughson, M.D.; Fogo, A.B.; Sinniah, R.; Dowling, J.; Samuel, T.; Mott, S.A.; Douglas-Denton, R.N.; Bertram, J.F. Ckd in aboriginal australians. Am. J. Kidney Dis. 2010, 56, 983–993. [Google Scholar]
- Lackland, D.T. Fetal and early life determinants of hypertension in adults: Implications for study. Hypertension 2004, 44, 811–812. [Google Scholar]
- Silver, L.E.; Decamps, P.J.; Korst, L.M.; Platt, L.D.; Castro, L. Intrauterine growth restriction is accompanied by decreased renal volume in the human fetus. Am. J. Obstet. Gynecol. 2003, 188, 1320–1325. [Google Scholar]
- Wang, Y.P.; Chen, X.; Zhang, Z.K.; Cui, H.Y.; Wang, P.; Wang, Y. Effects of a restricted fetal growth environment on human kidney morphology, cell apoptosis and gene expression. J. Renin Angiotensin Aldosterone Syst. 2014. [Google Scholar] [CrossRef]
- Moritz, K.M.; Singh, R.R.; Probyn, M.E.; Denton, K.M. Developmental programming of a reduced nephron endowment: More than just a baby’s birth weight. Am. J. Physiol. Renal Physiol. 2009, 296, F1–F9. [Google Scholar]
- Langley-Evans, S.C.; Welham, S.J.M.; Jackson, A.A. Fetal exposure to a maternal low protein diet impairs nephrogenesis and promotes hypertension in the rat. Life Sci. 1999, 64, 965–974. [Google Scholar]
- Kett, M.M.; Denton, K.M. Renal programming: Cause for concern? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R791–R803. [Google Scholar]
- Black, M.J.; Briscoe, T.A.; Constantinou, M.; Kett, M.M.; Bertram, J.F. Is there an association between level of adult blood pressure and nephron number or renal filtration surface area? Kidney Int. 2004, 65, 582–588. [Google Scholar]
- Skov, K.; Nyengaard, J.R.; Korsgaard, N.; Mulvany, M.J. Number and size of renal glomeruli in spontaneously hypertensive rats. J. Hypertens. 1994, 12, 1373–1376. [Google Scholar]
- Kramer, K.; Kinter, L.; Brockway, B.P.; Voss, H.P.; Remie, R.; van Zutphen, B.L. The use of radiotelemetry in small laboratory animals: Recent advances. Contemp. Top. Lab. Anim. Sci. 2001, 40, 8–16. [Google Scholar]
- Kramer, K.; Remie, R. Measuring blood pressure in small laboratory animals. Methods Mol. Med. 2005, 108, 51–62. [Google Scholar]
- Tonkiss, J.; Trzcinska, M.; Galler, J.R.; Ruiz-Opazo, N.; Herrera, V.L. Prenatal malnutrition-induced changes in blood pressure: Dissociation of stress and nonstress responses using radiotelemetry. Hypertension 1998, 32, 108–114. [Google Scholar]
- Brennan, K.A.; Kaufman, S.; Reynolds, S.W.; McCook, B.T.; Kan, G.; Christiaens, I.; Symonds, M.E.; Olson, D.M. Differential effects of maternal nutrient restriction through pregnancy on kidney development and later blood pressure control in the resulting offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R197–R205. [Google Scholar]
- Lim, K.; Lombardo, P.; Schneider-Kolsky, M.; Hilliard, L.; Denton, K.M.; Black, M.J. Induction of hyperglycemia in adult intrauterine growth-restricted rats: Effects on renal function. Am. J. Physiol. Renal Physiol. 2011, 301, F288–F294. [Google Scholar]
- Alwasel, S.H.; Ashton, N. Prenatal programming of renal sodium handling in the rat. Clin. Sci. 2009, 117, 75–84. [Google Scholar]
- Jennings, B.J.; Ozanne, S.E.; Dorling, M.W.; Hales, C.N. Early growth determines longevity in male rats and may be related to telomere shortening in the kidney. FEBS Lett. 1999, 448, 4–8. [Google Scholar]
- Petry, C.J.; Desai, M.; Ozanne, S.E.; Hales, C.N. Early and late nutritional windows for diabetes susceptibility. Proc. Nutr. Soc. 1997, 56, 233–242. [Google Scholar]
- Snoeck, A.; Remacle, C.; Reusens, B.; Hoet, J.J. Effect of a low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol. Neonate 1990, 57, 107–118. [Google Scholar]
- Langley-Evans, S.C.; Phillips, G.J.; Jackson, A.A. In utero exposure to maternal low protein diets induces hypertension in weanling rats, independently of maternal blood pressure changes. Clin. Nutr. 1994, 13, 319–324. [Google Scholar]
- Langley-Evans, S.C.; Welham, S.J.; Sherman, R.C.; Jackson, A.A. Weanling rats exposed to maternal low-protein diets during discrete periods of gestation exhibit differing severity of hypertension. Clin. Sci. (Lond.) 1996, 91, 607–615. [Google Scholar]
- Gubhaju, L.; Sutherland, M.R.; Horne, R.S.; Medhurst, A.; Kent, A.L.; Ramsden, A.; Moore, L.; Singh, G.; Hoy, W.E.; Black, M.J. Assessment of renal functional maturation and injury in preterm neonates during the first month of life. Am. J. Physiol. Renal. Physiol. 2014, 307, F149–F158. [Google Scholar]
- Sutherland, M.R.; Gubhaju, L.; Moore, L.; Kent, A.L.; Dahlstrom, J.E.; Horne, R.S.; Hoy, W.E.; Bertram, J.F.; Black, M.J. Accelerated maturation and abnormal morphology in the preterm neonatal kidney. J. Am. Soc. Nephrol. 2011, 22, 1365–1374. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wood-Bradley, R.J.; Barrand, S.; Giot, A.; Armitage, J.A. Understanding the Role of Maternal Diet on Kidney Development; an Opportunity to Improve Cardiovascular and Renal Health for Future Generations. Nutrients 2015, 7, 1881-1905. https://doi.org/10.3390/nu7031881
Wood-Bradley RJ, Barrand S, Giot A, Armitage JA. Understanding the Role of Maternal Diet on Kidney Development; an Opportunity to Improve Cardiovascular and Renal Health for Future Generations. Nutrients. 2015; 7(3):1881-1905. https://doi.org/10.3390/nu7031881
Chicago/Turabian StyleWood-Bradley, Ryan James, Sanna Barrand, Anais Giot, and James Andrew Armitage. 2015. "Understanding the Role of Maternal Diet on Kidney Development; an Opportunity to Improve Cardiovascular and Renal Health for Future Generations" Nutrients 7, no. 3: 1881-1905. https://doi.org/10.3390/nu7031881
APA StyleWood-Bradley, R. J., Barrand, S., Giot, A., & Armitage, J. A. (2015). Understanding the Role of Maternal Diet on Kidney Development; an Opportunity to Improve Cardiovascular and Renal Health for Future Generations. Nutrients, 7(3), 1881-1905. https://doi.org/10.3390/nu7031881