Meta-Analysis of Fecal Microbiota and Metabolites in Experimental Colitic Mice during the Inflammatory and Healing Phases

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiment
2.2. 16S rRNA Gene-Based Microbiome Profiling
2.3. Fecal Metabolites Analysis Using 1H-NMR Analysis
2.4. Measurements of Lactate and Succinate in Fecal Samples
2.5. Statistical Analysis
3. Results
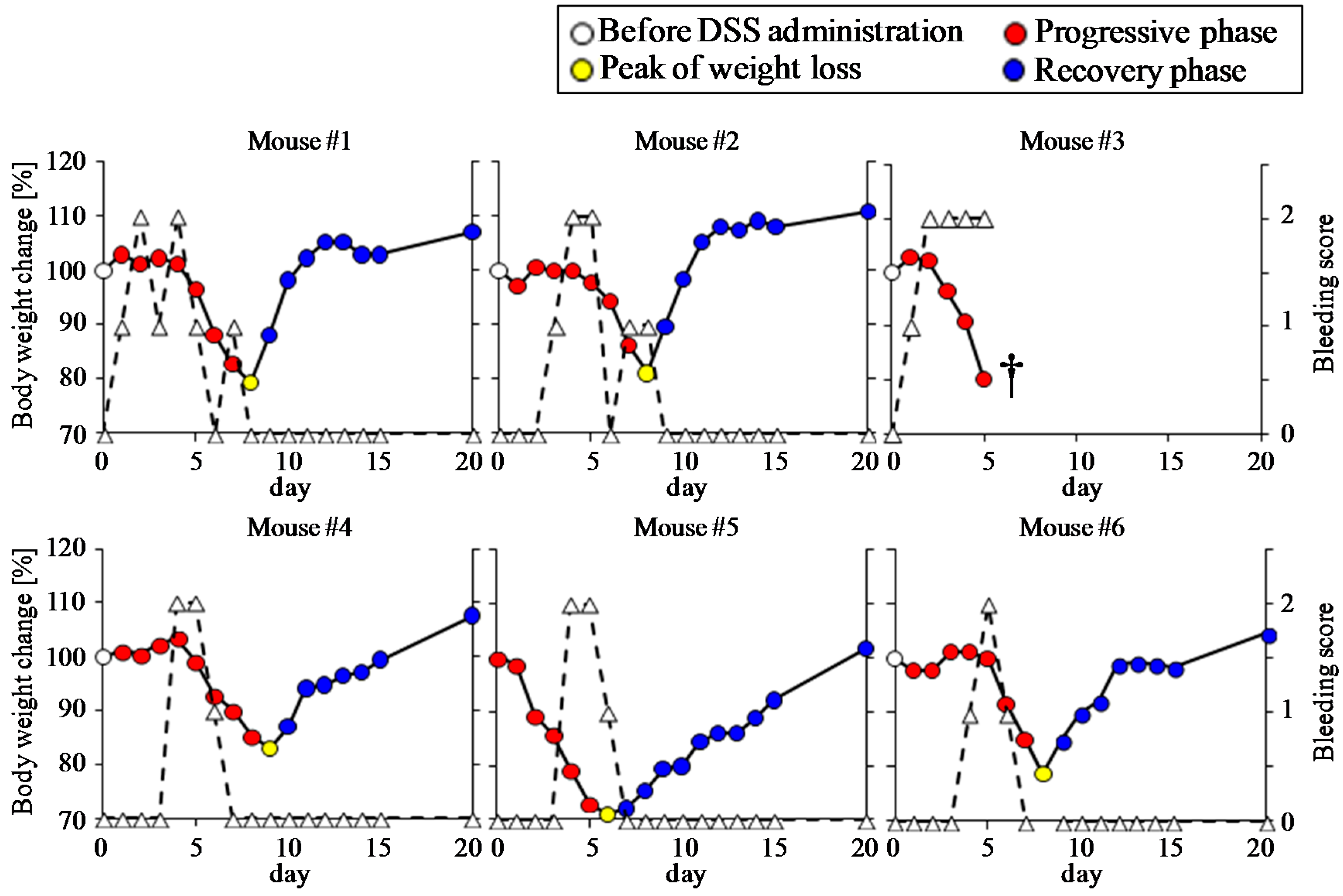
3.1. Clinical Manifestation of DSS-Treated Mice
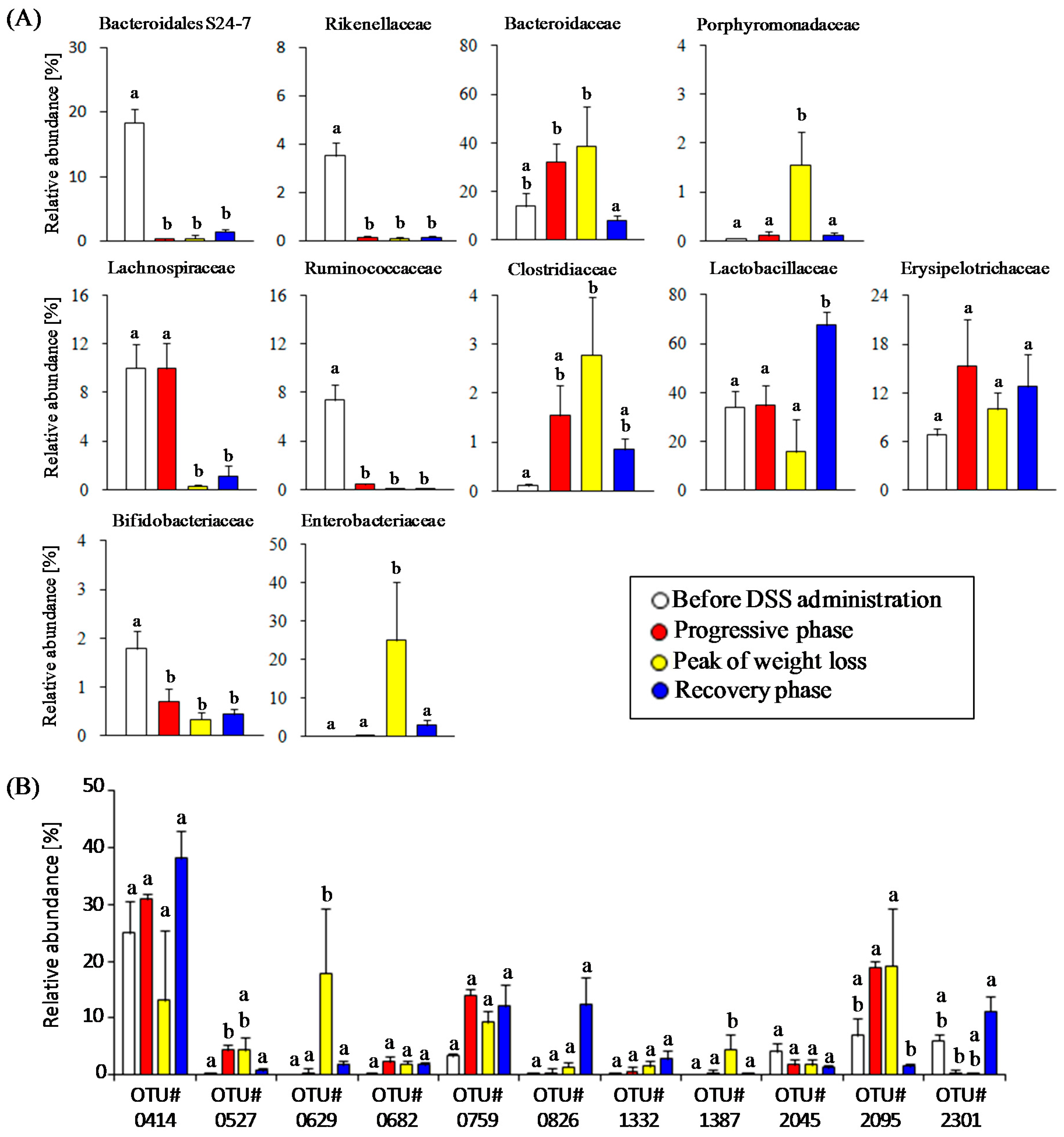
3.2. Time-Course Transitions in Colonic Bacterial Compositions in DSS-Treated Mice
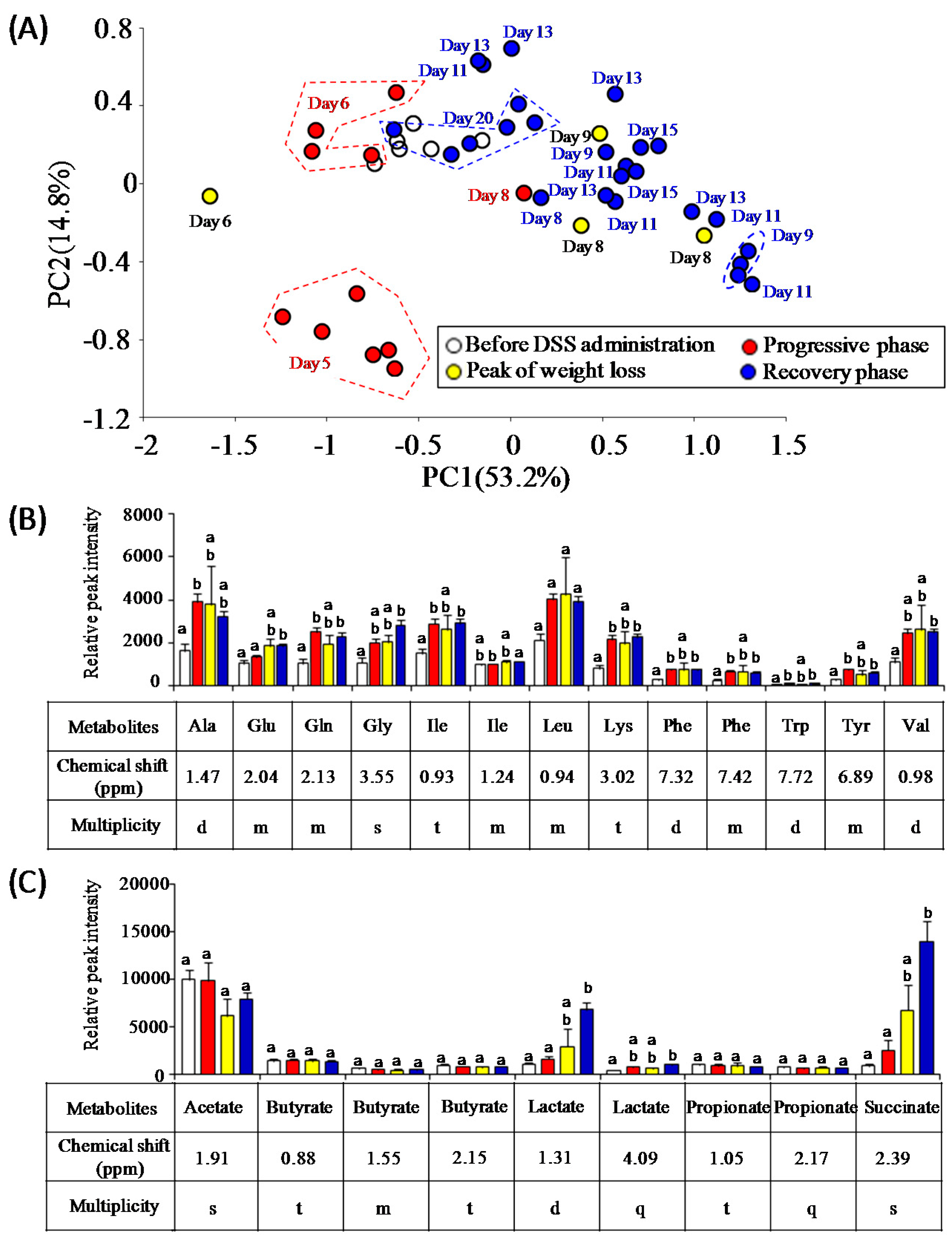
3.3. Characterization of Fecal Metabolites in DSS-Treated Mice
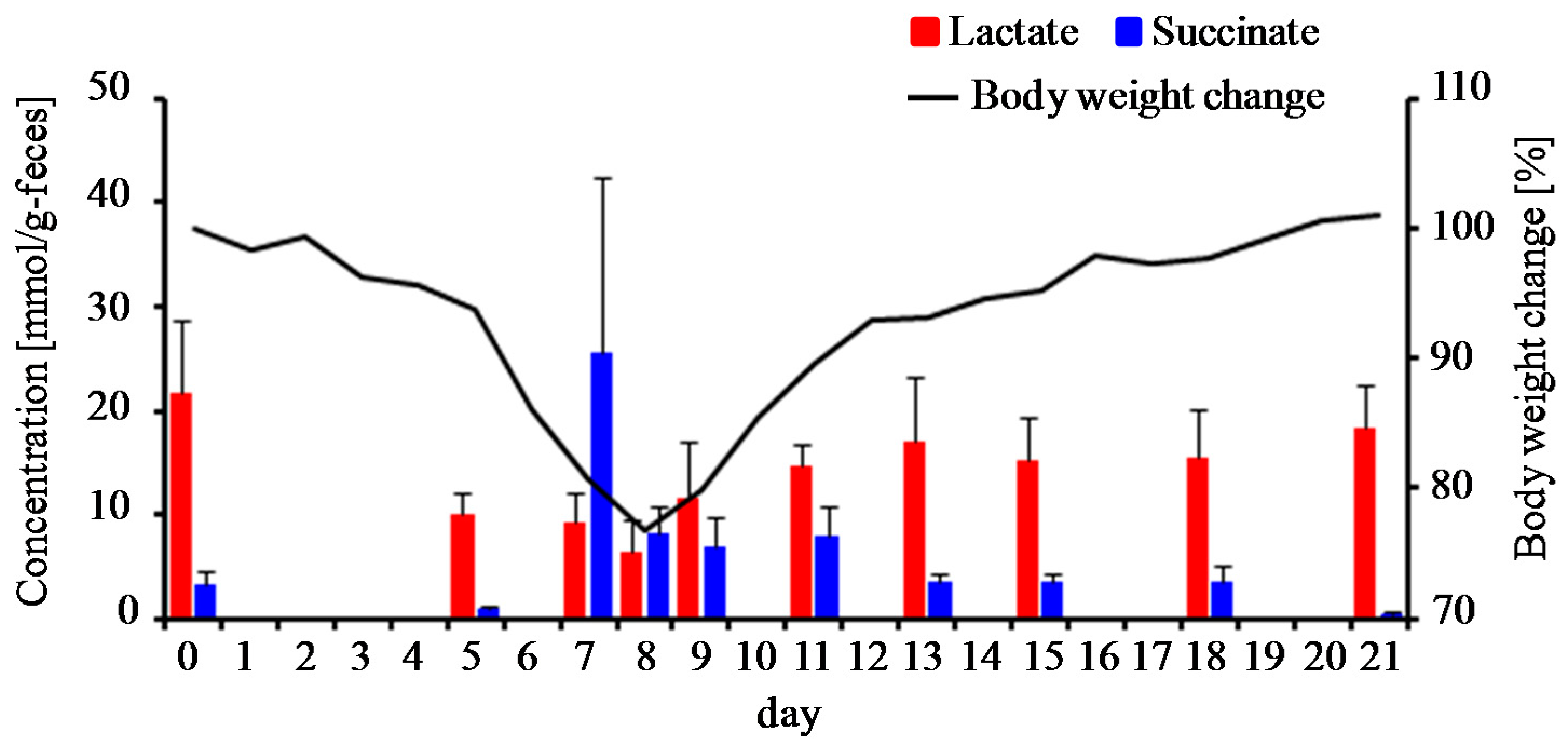
3.4. Estimation of Gut Microbiota Associated with Succinate Production
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contractor, N.V.; Bassiri, H.; Reya, T.; Park, A.Y.; Baumgart, D.C.; Wasik, M.A.; Emerson, S.G.; Carding, S.R. Lymphoid hyperplasia, autoimmunity, and compromised intestinal intraepithelial lymphocyte development in colitis-free gnotobiotic IL-2-deficient mice. J. Immunol. 1998, 160, 385–394. [Google Scholar] [PubMed]
- Dianda, L.; Hanby, A.M.; Wright, N.A.; Sebesteny, A.; Hayday, A.C.; Owen, M.J. T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am. J. Pathol. 1997, 150, 91–97. [Google Scholar] [PubMed]
- Song, F.; Ito, K.; Denning, T.L.; Kuninger, D.; Papaconstantinou, J.; Gourley, W.; Klimpel, G.; Balish, E.; Hokanson, J.; Ernst, P.B. Expression of the neutrophil chemokine KC in the colon of mice with enterocolitis and by intestinal epithelial cell lines: Effects of flora and proinflammatory cytokines. J. Immunol. 1999, 162, 2275–2280. [Google Scholar] [PubMed]
- Frank, D.N.; St. Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.A.; Frank, D.N.; Pace, N.R.; Gordon, J.I. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 2008, 3, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Schwab, C.; Milinovich, G.; Reichert, J.; Mahfoudh, K.B.; Decker, T.; Engel, M.; Hai, B.; Hainzl, E.; Heider, S.; et al. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 2012, 6, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Maharshak, N.; Packey, C.D.; Ellermann, M.; Manick, S.; Siddle, J.P.; Huh, E.Y.; Plevy, S.; Sartor, R.B.; Carroll, I.M. Altered enteric microbiota ecology in interleukin 10-deficient mice during development and progression of intestinal inflammation. Gut Microbes 2013, 4, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; O’Riordan, M.X. Regulation of bacterial pathogenesis by intestinal short-chain fatty acids. Adv. Appl. Microbiol. 2013, 85, 93–118. [Google Scholar] [PubMed]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The role of short-chain fatty acids in health and disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Ohkusa, T.; Nomura, T.; Terai, T.; Miwa, H.; Kobayashi, O.; Hojo, M.; Takei, Y.; Ogihara, T.; Hirai, S.; Okayasu, I.; et al. Effectiveness of antibiotic combination therapy in patients with active ulcerative colitis: A randomized, controlled pilot trial with long-term follow-up. Scand. J. Gastroenterol. 2005, 40, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Rath, H.C.; Schultz, M.; Freitag, R.; Dieleman, L.A.; Li, F.; Linde, H.J.; Schölmerich, J.; Sartor, R.B. Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infect. Immun. 2001, 69, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Bibiloni, R.; Fedorak, R.N.; Tannock, G.W.; Madsen, K.L.; Gionchetti, P.; Campieri, M.; De Simone, C.; Sartor, R.B. VSL#3 probiotic-mixture induces remission in patients with active ulcerative colitis. Am. J. Gastroenterol. 2005, 100, 1539–1546. [Google Scholar] [PubMed]
- Derwa, Y.; Gracie, D.J.; Hamlin, P.J.; Ford, A.C. Systematic review with meta-analysis: The efficacy of probiotics in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2017, 46, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Uronis, J.M.; Arthur, J.C.; Keku, T.; Fodor, A.; Carroll, I.M.; Cruz, M.L.; Appleyard, C.B.; Jobin, C. Gut microbial diversity is reduced by the probiotic VSL#3 and correlates with decreased TNBS-induced colitis. Inflamm. Bowel Dis. 2011, 17, 289–297. [Google Scholar] [PubMed]
- Borody, T.J.; Warren, E.F.; Leis, S.; Surace, R.; Ashman, O. Treatment of ulcerative colitis using fecal bacteriotherapy. J. Clin. Gastroenterol. 2003, 37, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology 2015, 149, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Narula, N.; Kassam, Z.; Yuan, Y.; Colombel, J.F.; Ponsioen, C.; Reinisch, W.; Moayyedi, P. Systematic review and meta-analysis: Fecal microbiota transplantation for treatment of active ulcerative colitis. Inflamm. Bowel Dis. 2017, 23, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, O.; Elias, M.; Peretz, A.; Saliba, W. Role of antibiotics for treatment of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Qazi, T.; Amaratunga, T.; Barnes, E.L.; Fischer, M.; Kassam, Z.; Allegretti, J.R. The risk of inflammatory bowel disease flares after fecal microbiota transplantation: Systematic review and meta-analysis. Gut Microbes 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Y.; Popov, J.; Pai, N. Fecal microbial transplant for the treatment of pediatric inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 10304–10315. [Google Scholar] [CrossRef] [PubMed]
- Alang, N.; Kelly, C.R. Weight gain after fecal microbiota transplantation. Open Forum Infect. Dis. 2015, 2, ofv004. [Google Scholar] [CrossRef] [PubMed]
- Hashash, J.G.; Binion, D.G. Managing clostridium difficile in inflammatory bowel disease (IBD). Curr. Gastroenterol. Rep. 2014, 16, 393. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Loening-Baucke, V.; Bengmark, S.; Scholze, J.; Doerffel, Y. Bacterial biofilm suppression with antibiotics for ulcerative and indeterminate colitis: Consequences of aggressive treatment. Arch. Med. Res. 2008, 39, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lozupone, C.; Hamady, M.; Bushman, F.D.; Knight, R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007, 35, e120. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.Y.; Osaka, T.; Moriyama, E.; Date, Y.; Kikuchi, J.; Tsuneda, S. Strengthening of the intestinal epithelial tight junction by Bifidobacterium bifidum. Physiol. Rep. 2015, 3, e12327. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Fukuda, S.; Fujiwara, A.; Suda, W.; Hattori, M.; Kikuchi, J.; Ohno, H. Multiple omics uncovers host-gut microbial mutualism during prebiotic fructooligosaccharide supplementation. DNA Res. 2014, 21, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Motegi, H.; Tsuboi, Y.; Saga, A.; Kagami, T.; Inoue, M.; Toki, H.; Minowa, O.; Noda, T.; Kikuchi, J. Identification of reliable components in multivariate curve resolution-alternating least squares (MCR-ALS): A data-driven approach across metabolic processes. Sci. Rep. 2015, 5, 15710. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, H.; Odamaki, T.; Fukuda, S.; Kato, T.; Xiao, J.Z.; Abe, F.; Kikuchi, J.; Ohno, H. Probiotic Bifidobacterium longum alters gut luminal metabolism through modification of the gut microbial community. Sci. Rep. 2015, 5, 13548. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Chikayama, E.; Suto, M.; Nishihara, T.; Shinozaki, K.; Kikuchi, J. Systematic NMR analysis of stable isotope labeled metabolite mixtures in plant and animal systems: Coarse grained views of metabolic pathways. PLoS ONE 2008, 3, e3805. [Google Scholar] [CrossRef] [PubMed]
- Chikayama, E.; Sekiyama, Y.; Okamoto, M.; Nakanishi, Y.; Tsuboi, Y.; Akiyama, K.; Saito, K.; Shinozaki, K.; Kikuchi, J. Statistical indices for simultaneous large-scale metabolite detections for a single NMR spectrum. Anal. Chem. 2010, 82, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Nakanishi, Y.; Chikayama, E.; Ohno, H.; Hino, T.; Kikuchi, J. Evaluation and characterization of bacterial metabolic dynamics with a novel profiling technique, real-time metabolotyping. PLoS ONE 2009, 4, e4893. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Tsuboi, Y.; Komatsu, K.; Gomi, M.; Chikayama, E.; Date, Y. SpinCouple: Development of a web tool for analyzing metabolite mixtures via two-dimensional j-resolved NMR database. Anal. Chem. 2016, 88, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Bloom, S.M.; Bijanki, V.N.; Nava, G.M.; Sun, L.; Malvin, N.P.; Donermeyer, D.L.; Dunne, W.M., Jr.; Allen, P.M.; Stappenbeck, T.S. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 2011, 9, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Hans, W.; Schölmerich, J.; Gross, V.; Falk, W. The role of the resident intestinal flora in acute and chronic dextran sulfate sodium-induced colitis in mice. Eur. J. Gastroenterol. Hepatol. 2000, 12, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007, 2, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, S.; Haller, D.; Blaut, M.; Loh, G. Reduced microbial diversity and high numbers of one single Escherichia coli strain in the intestine of colitic mice. Environ. Microbiol. 2009, 11, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Hoentjen, F.; Harmsen, H.J.; Braat, H.; Torrice, C.D.; Mann, B.A.; Sartor, R.B.; Dieleman, L.A. Antibiotics with a selective aerobic or anaerobic spectrum have different therapeutic activities in various regions of the colon in interleukin 10 gene deficient mice. Gut 2003, 52, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Biasi, F.; Leonarduzzi, G.; Oteiza, P.I.; Policorresponding, G. Inflammatory bowel disease: Mechanisms, redox considerations, and therapeutic targets. Antioxid. Redox Signal. 2013, 19, 1711–1747. [Google Scholar] [CrossRef] [PubMed]
- Baughn, A.D.; Malamy, M.H. The strict anaerobe Bacteroides fragilis grows in and benefits from nanomolar concentrations of oxygen. Nature 2004, 427, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, K.L.; Wood, D.L.; Lachner, N.; Gellatly, S.L.; Daly, J.N.; Parsons, J.D.; Dal’Molin, C.G.; Palfreyman, R.W.; Nielsen, L.K.; Cooper, M.A.; et al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 2016, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Choo, J.M.; Kanno, T.; Zain, N.M.; Leong, L.E.; Abell, G.C.; Keeble, J.E.; Bruce, K.D.; Mason, A.J.; Rogers, G.B. Divergent relationships between fecal microbiota and metabolome following distinct antibiotic-induced disruptions. mSphere 2017, 2, e00005–e00017. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Fukuda, S.; Hase, K.; Nishiumi, S.; Izumi, Y.; Yoshida, M.; Hagiwara, T.; Kawashima, R.; Yamazaki, M.; Oshio, T.; et al. Microbiota-derived lactate accelerates colon epithelial cell turnover in starvation-refed mice. Nat. Commun. 2013, 4, 1654. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.N.; Olsen, I.; Bernard, K.; Finegold, S.M.; Gharbia, S.; Gupta, R.S. Approaches to the study of the systematics of anaerobic, gram-negative, non-sporeforming rods: Current status and perspectives. Anaerobe 2009, 15, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Sakamoto, K.; Seo, S.U.; Pickard, J.M.; Gillilland, M.G.; Pudlo, N.A.; Hoostal, M.; Li, X.; Wang, T.D.; Feehley, T.; et al. Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science 2017, 356, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Ariake, K.; Ohkusa, T.; Sakurazawa, T.; Kumagai, J.; Eishi, Y.; Hoshi, S.; Yajima, T. Roles of mucosal bacteria and succinic acid in colitis caused by dextran sulfate sodium in mice. J. Med. Dent. Sci. 2000, 47, 233–241. [Google Scholar] [PubMed]
- Curtis, M.M.; Hu, Z.; Klimko, C.; Narayanan, S.; Deberardinis, R.; Sperandio, V. The gut commensal Bacteroides thetaiotaomicron exacerbates enteric infection through modification of the metabolic landscape. Cell Host Microbe 2014, 16, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Ferreyra, J.A.; Wu, K.J.; Hryckowian, A.J.; Bouley, D.M.; Weimer, B.C.; Sonnenburg, J.L. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 2014, 16, 770–777. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OTU # | Closest Reference Strain (Acsession No.) | Identities | Ranking | |||
|---|---|---|---|---|---|---|
| Phase 1 | Phase 2 | Phase 3 | Phase 4 | |||
| 414 | Lactobacillus johnsonii strain UMNLJ22 (CP021704) | 99.2% | 1 | 1 | 3 | 1 |
| 527 | Bacteroides uniformis strain CECT 7771 (AB021157) | 97.7% | 4 | |||
| 629 | Escherichia coli strain 4060 (FJ405333) | 99.4% | 2 | |||
| 682 | Turicibacter sp. LA62 (AB727349) | 100% | 5 | |||
| 759 | Clostridium sp. ARIAKE1333 (AB809059) | 98.5% | 5 | 3 | 4 | 3 |
| 826 | Lactobacillus murinus (LC159538) | 99.5% | 2 | |||
| 1332 | Bacteroides caecimuris strain I48 (KR364741) | 99.4% | 5 | |||
| 1387 | Escherichia coli strain D1 (CP010134) | 99.7% | 5 | |||
| 2045 | Bacteroides sp. SLC1-38 (AB599946) | 99.7% | 4 | |||
| 2095 | Bacteroides acidifaciens (AB510696) | 99.4% | 2 | 2 | 1 | |
| 2301 | Lactobacillus intestinalis strain ls74 (EU381126) | 99.2% | 3 | 4 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osaka, T.; Moriyama, E.; Arai, S.; Date, Y.; Yagi, J.; Kikuchi, J.; Tsuneda, S. Meta-Analysis of Fecal Microbiota and Metabolites in Experimental Colitic Mice during the Inflammatory and Healing Phases. Nutrients 2017, 9, 1329. https://doi.org/10.3390/nu9121329
Osaka T, Moriyama E, Arai S, Date Y, Yagi J, Kikuchi J, Tsuneda S. Meta-Analysis of Fecal Microbiota and Metabolites in Experimental Colitic Mice during the Inflammatory and Healing Phases. Nutrients. 2017; 9(12):1329. https://doi.org/10.3390/nu9121329
Chicago/Turabian StyleOsaka, Toshifumi, Eri Moriyama, Shunichi Arai, Yasuhiro Date, Junji Yagi, Jun Kikuchi, and Satoshi Tsuneda. 2017. "Meta-Analysis of Fecal Microbiota and Metabolites in Experimental Colitic Mice during the Inflammatory and Healing Phases" Nutrients 9, no. 12: 1329. https://doi.org/10.3390/nu9121329
APA StyleOsaka, T., Moriyama, E., Arai, S., Date, Y., Yagi, J., Kikuchi, J., & Tsuneda, S. (2017). Meta-Analysis of Fecal Microbiota and Metabolites in Experimental Colitic Mice during the Inflammatory and Healing Phases. Nutrients, 9(12), 1329. https://doi.org/10.3390/nu9121329