Role of the Enterocyte in Fructose-Induced Hypertriglyceridaemia

 , and
, and 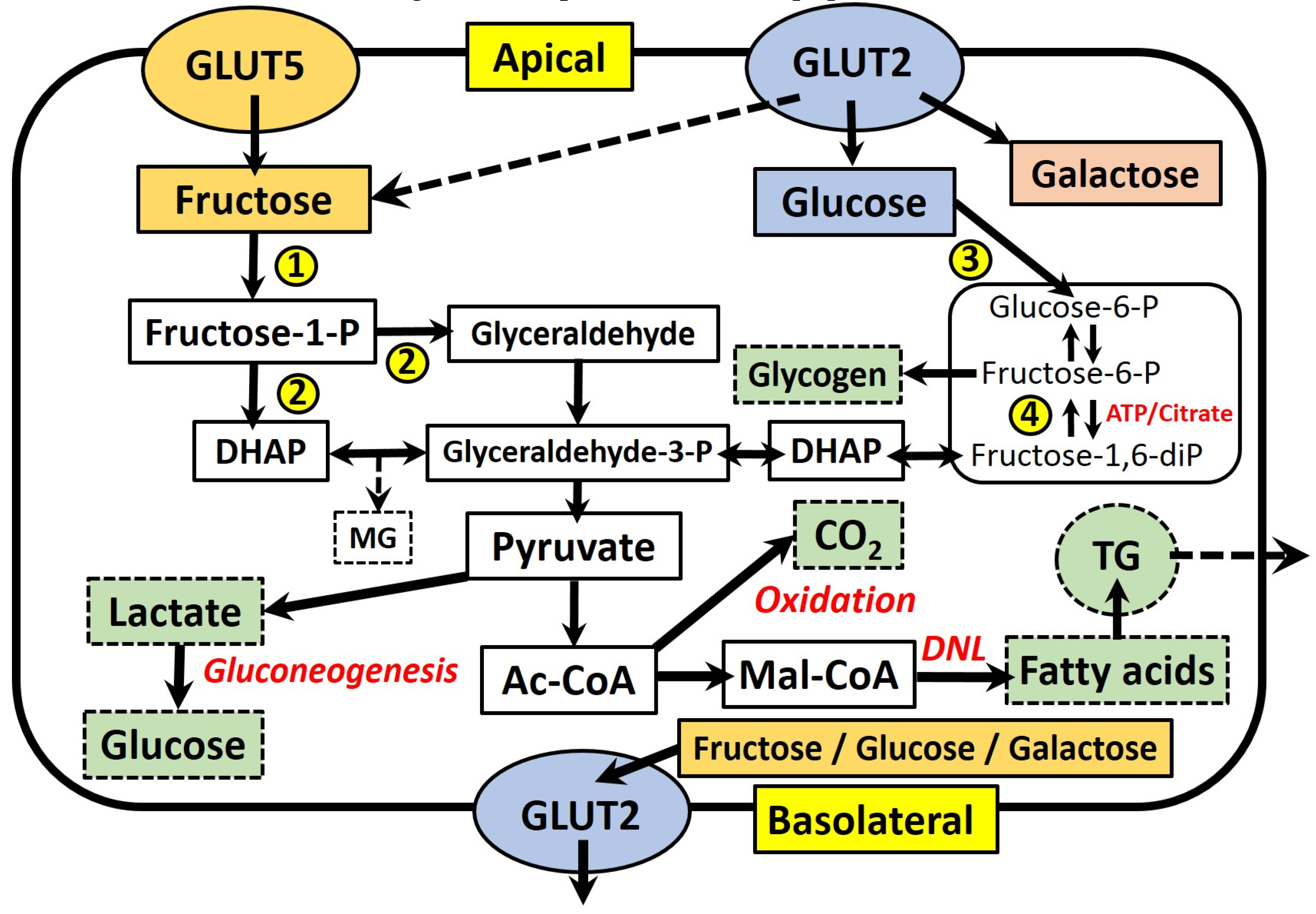{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Fructose Absorption and Metabolism in the Enterocyte
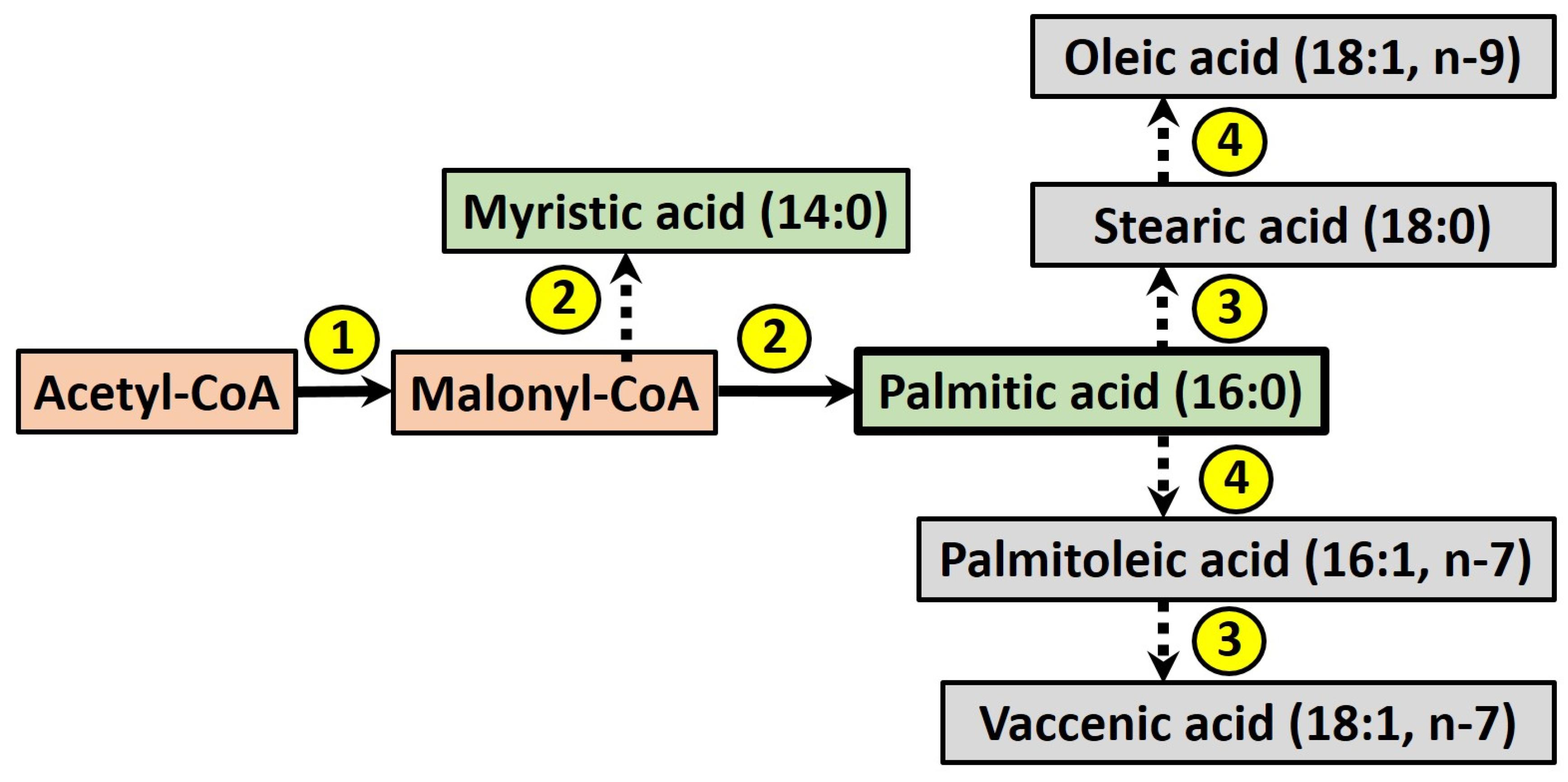
3. Fructose and Intestinal de novo Lipogenesis (DNL)
4. Fructose Effects on Intestinal Lipoprotein Secretion
4.1. Lipid Storage and CM Secretion from the Enterocyte
4.2. Fructose Effects on Apolipoprotein B48 Concentration
4.3. Fructose Effects on Enterocyte CM-TG Secretion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO. Global Status Report on Noncommunicable Diseases 2014; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Eberly, L.E.; Stamler, J.; Neaton, J.D. Relation of triglyceride levels, fasting and nonfasting, to fatal and nonfatal coronary heart disease. Arch. Intern. Med. 2003, 163, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Sullivan, L.; Jacques, P.F.; Wang, T.J.; Fox, C.S.; Meigs, J.B.; D'Agostino, R.B.; Gaziano, J.M.; Vasan, R.S. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation 2007, 116, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Maersk, M.; Belza, A.; Stodkilde-Jorgensen, H.; Ringgaard, S.; Chabanova, E.; Thomsen, H.; Pedersen, S.B.; Astrup, A.; Richelsen, B. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: A 6-mo randomized intervention study. Am. J. Clin. Nutr. 2012, 95, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Bantle, J.P.; Raatz, S.K.; Thomas, W.; Georgopoulos, A. Effects of dietary fructose on plasma lipids in healthy subjects. Am. J. Clin. Nutr. 2000, 72, 1128–1134. [Google Scholar] [PubMed]
- Silbernagel, G.; Machann, J.; Unmuth, S.; Schick, F.; Stefan, N.; Haring, H.U.; Fritsche, A. Effects of 4-week very-high-fructose/glucose diets on insulin sensitivity, visceral fat and intrahepatic lipids: An exploratory trial. Br. J. Nutr. 2011, 106, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Ngo Sock, E.T.; Le, K.A.; Ith, M.; Kreis, R.; Boesch, C.; Tappy, L. Effects of a short-term overfeeding with fructose or glucose in healthy young males. Br. J. Nutr. 2010, 103, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Bremer, A.A.; Medici, V.; Nakajima, K.; Ito, Y.; Nakano, T.; Chen, G.; Fong, T.H.; Lee, V.; Menorca, R.I.; Keim, N.L.; Havel, P.J. Consumption of fructose and high fructose corn syrup increase postprandial triglycerides, LDL-cholesterol, and apolipoprotein-B in young men and women. J. Clin. Endocrinol. Metab. 2011, 96, E1596–E1605. [Google Scholar] [CrossRef] [PubMed]
- Teff, K.L.; Elliott, S.S.; Tschop, M.; Kieffer, T.J.; Rader, D.; Heiman, M.; Townsend, R.R.; Keim, N.L.; D’Alessio, D.; Havel, P.J. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J. Clin. Endocrinol. Metab. 2004, 89, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Teff, K.L.; Grudziak, J.; Townsend, R.R.; Dunn, T.N.; Grant, R.W.; Adams, S.H.; Keim, N.L.; Cummings, B.P.; Stanhope, K.L.; Havel, P.J. Endocrine and metabolic effects of consuming fructose- and glucose-sweetened beverages with meals in obese men and women: Influence of insulin resistance on plasma triglyceride responses. J. Clin. Endocrinol. Metab. 2009, 94, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A.; Tran, C.; Paquot, N. Fructose and metabolic diseases: New findings, new questions. Nutrition 2010, 26, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Sharrett, A.R.; Heiss, G.; Chambless, L.E.; Boerwinkle, E.; Coady, S.A.; Folsom, A.R.; Patsch, W. Metabolic and lifestyle determinants of postprandial lipemia differ from those of fasting triglycerides: The Atherosclerosis Risk In Communities (ARIC) study. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Matikainen, N.; Adiels, M.; Taskinen, M.R. Postprandial hypertriglyceridemia as a coronary risk factor. Clin. Chim. Acta. 2014, 431, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Zavaroni, I.; Chen, Y.D.; Reaven, G.M. Studies of the mechanism of fructose-induced hypertriglyceridemia in the rat. Metabolism 1982, 31, 1077–1083. [Google Scholar] [CrossRef]
- Le, K.A.; Faeh, D.; Stettler, R.; Ith, M.; Kreis, R.; Vermathen, P.; Boesch, C.; Ravussin, E.; Tappy, L. A 4-wk high-fructose diet alters lipid metabolism without affecting insulin sensitivity or ectopic lipids in healthy humans. Am. J. Clin. Nutr. 2006, 84, 1374–1379. [Google Scholar] [PubMed]
- Le, K.A.; Ith, M.; Kreis, R.; Faeh, D.; Bortolotti, M.; Tran, C.; Boesch, C.; Tappy, L. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am. J. Clin. Nutr. 2009, 89, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Campos, V.C.; Tappy, L. Physiological handling of dietary fructose-containing sugars: Implications for health. Int. J. Obes. (Lond.) 2016, 40, S6–S11. [Google Scholar] [CrossRef] [PubMed]
- Haidari, M.; Leung, N.; Mahbub, F.; Uffelman, K.D.; Kohen-Avramoglu, R.; Lewis, G.F.; Adeli, K. Fasting and postprandial overproduction of intestinally derived lipoproteins in an animal model of insulin resistance. Evidence that chronic fructose feeding in the hamster is accompanied by enhanced intestinal de novo lipogenesis and ApoB48-containing lipoprotein overproduction. J. Biol. Chem. 2002, 277, 31646–31655. [Google Scholar] [CrossRef] [PubMed]
- Shojaee-Moradie, F.; Ma, Y.; Lou, S.; Hovorka, R.; Umpleby, A.M. Prandial hypertriglyceridemia in metabolic syndrome is due to an overproduction of both chylomicron and VLDL triacylglycerol. Diabetes 2013, 62, 4063–4069. [Google Scholar] [CrossRef] [PubMed]
- Berliner, J.A.; Navab, M.; Fogelman, A.M.; Frank, J.S.; Demer, L.L.; Edwards, P.A.; Watson, A.D.; Lusis, A.J. Atherosclerosis: Basic mechanisms. Oxidation, inflammation, and genetics. Circulation 1995, 91, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Elsegood, C.L.; Mamo, J.C. An investigation by electron microscopy of chylomicron remnant uptake by human monocyte-derived macrophages. Atherosclerosis 2006, 188, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Nakajima, K.; Niimi, M.; Fujita, M.Q.; Nakajima, Y.; Takeichi, S.; Kinoshita, M.; Matsushima, T.; Teramoto, T.; Tanaka, A. Detection of apolipoproteins B-48 and B-100 carrying particles in lipoprotein fractions extracted from human aortic atherosclerotic plaques in sudden cardiac death cases. Clin. Chim. Acta 2008, 390, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Semorine, K.; Watts, G.F.; Mamo, J. Identification of lipoproteins of intestinal origin in human atherosclerotic plaque. Clin. Chem. Lab. Med. 2003, 41, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.; Avella, M.; Botham, K.M.; Bravo, E. Chylomicron remnant induction of lipid accumulation in J774 macrophages is associated with up-regulation of triacylglycerol synthesis which is not dependent on oxidation of the particles. Biochim. Biophys. Acta 2003, 1631, 255–264. [Google Scholar] [CrossRef]
- Prentice, A. The Scientific Advisory Committee on Nutrition recommendations on carbohydrates, including sugars and fibre. Public. Health. Engl. 2015. Available online: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/489906/Why_5__-_The_Science_Behind_SACN.pdf (accessed on 17 March 2017).
- 2015–2020 Dietary Guidelines for Americans. 2015. Available online: http://health.gov/dietaryguidelines/2015/guidelines/ (accessed on 17 March 2017).
- Bates, B.; Lennox, A.; Prentice, A.; Bates, C.; Page, P.; Nicholson, S.; Swan, G. (Eds.) National Diet and Nutrition Survey - Results from Years 1, 2, 3 and 4 (combined) of the Rolling Programme (2008/2009 – 2011/2012); Public Health England: London, UK, 2014. [Google Scholar]
- White, J.S. Straight talk about high-fructose corn syrup: What it is and what it ain’t. Am. J. Clin. Nutr. 2008, 88, 1716s–1721s. [Google Scholar] [CrossRef] [PubMed]
- Marriott, B.P.; Cole, N.; Lee, E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J. Nutr. 2009, 139, 1228s–1235s. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Kimmons, J.E.; Gillespie, C.; Welsh, J.; Blanck, H.M. Dietary fructose consumption among US children and adults: The Third National Health and Nutrition Examination Survey. Medscape J. Med. 2008, 10, 160. [Google Scholar] [PubMed]
- Welsh, J.A.; Sharma, A.J.; Grellinger, L.; Vos, M.B. Consumption of added sugars is decreasing in the United States. Am. J. Clin. Nutr. 2011, 94, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Whitton, C.; Nicholson, S.K.; Roberts, C.; Prynne, C.J.; Pot, G.K.; Olson, A.; Fitt, E.; Cole, D.; Teucher, B.; Bates, B.; et al. National Diet and Nutrition Survey: UK food consumption and nutrient intakes from the first year of the rolling programme and comparisons with previous surveys. Br. J. Nutr. 2011, 106, 1899–1914. [Google Scholar] [CrossRef] [PubMed]
- Barone, S.; Fussell, S.L.; Singh, A.K.; Lucas, F.; Xu, J.; Kim, C.; Wu, X.; Yu, Y.; Amlal, H.; Seidler, U.; et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J. Biol. Chem. 2009, 284, 5056–5066. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Douard, V.; Yu, S.; Gao, N.; Ferraris, R.P. Transport, metabolism, and endosomal trafficking-dependent regulation of intestinal fructose absorption. Faseb. J. 2015, 29, 4046–4058. [Google Scholar] [CrossRef] [PubMed]
- Kellett, G.L.; Brot-Laroche, E. Apical GLUT2: A major pathway of intestinal sugar absorption. Diabetes 2005, 54, 3056–3062. [Google Scholar] [CrossRef] [PubMed]
- Di Luccia, B.; Crescenzo, R.; Mazzoli, A.; Cigliano, L.; Venditti, P.; Walser, J.C.; Widmer, A.; Baccigalupi, L.; Ricca, E.; Iossa, S. Rescue of Fructose-Induced Metabolic Syndrome by Antibiotics or Faecal Transplantation in a Rat Model of Obesity. PLoS ONE 2015, 10, e0134893. [Google Scholar] [CrossRef]
- Liu, J.P.; Zou, W.L.; Chen, S.J.; Wei, H.Y.; Yin, Y.N.; Zou, Y.Y.; Lu, F.G. Effects of different diets on intestinal microbiota and nonalcoholic fatty liver disease development. World J. Gastroenterol. 2016, 22, 7353–7364. [Google Scholar] [CrossRef] [PubMed]
- Park, D.Y.; Ahn, Y.T.; Huh, C.S.; McGregor, R.A.; Choi, M.S. Dual probiotic strains suppress high fructose-induced metabolic syndrome. World J. Gastroenterol. 2013, 19, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Z.; Empie, M.W. Fructose metabolism in humans—what isotopic tracer studies tell us. Nutr. Metab. (Lond.) 2012, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, O.; Crump, M.; Phillips, R.W. Intestinal metabolism of orally administered glucose and fructose in Yucatan miniature swine. J. Nutr. 1984, 114, 1413–1420. [Google Scholar] [PubMed]
- Patel, C.; Douard, V.; Yu, S.; Tharabenjasin, P.; Gao, N.; Ferraris, R.P. Fructose-induced increases in expression of intestinal fructolytic and gluconeogenic genes are regulated by GLUT5 and KHK. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R499–R509. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Sugimoto, K.; Douard, V.; Shah, A.; Inui, H.; Yamanouchi, T.; Ferraris, R.P. Effect of dietary fructose on portal and systemic serum fructose levels in rats and in KHK-/- and GLUT5-/- mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G779–G790. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Lemaitre, R.N.; Imamura, F.; King, I.B.; Song, X.; Spiegelman, D.; Siscovick, D.S.; Mozaffarian, D. Fatty acids in the de novo lipogenesis pathway and risk of coronary heart disease: The Cardiovascular Health Study. Am. J. Clin. Nutr. 2011, 94, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Hudgins, L.C.; Hellerstein, M.; Seidman, C.; Neese, R.; Diakun, J.; Hirsch, J. Human fatty acid synthesis is stimulated by a eucaloric low fat, high carbohydrate diet. J. Clin. Invest. 1996, 97, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Abate, G.; Marziano, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Nutrition and AGE-ing: Focusing on Alzheimer’s Disease. Oxid. Med. Cell Longev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, L.; Du, R.; Yan, J.; Liu, N.; Yuan, W.; Jiang, Y.; Xu, S.; Ye, F.; Yuan, G.; et al. CML/RAGE signal induces calcification cascade in diabetes. Diabetol. Metab. Syndr. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Hegab, Z.; Gibbons, S.; Neyses, L.; Mamas, M.A. Role of advanced glycation end products in cardiovascular disease. World J. Cardiol. 2012, 4, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox. Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Noworolski, S.M.; Wen, M.J.; Dyachenko, A.; Prior, J.L.; Weinberg, M.E.; Herraiz, L.A.; Tai, V.W.; Bergeron, N.; Bersot, T.P.; et al. Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat. J Clin. Endocrinol. Metab. 2015, 100, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar] [PubMed]
- Egli, L.; Lecoultre, V.; Theytaz, F.; Campos, V.; Hodson, L.; Schneiter, P.; Mittendorfer, B.; Patterson, B.W.; Fielding, B.A.; Gerber, P.A.; et al. Exercise prevents fructose-induced hypertriglyceridemia in healthy young subjects. Diabetes 2013, 62, 2259–2265. [Google Scholar] [CrossRef] [PubMed]
- Theytaz, F.; de Giorgi, S.; Hodson, L.; Stefanoni, N.; Rey, V.; Schneiter, P.; Giusti, V.; Tappy, L. Metabolic fate of fructose ingested with and without glucose in a mixed meal. Nutrients 2014, 6, 2632–2649. [Google Scholar] [CrossRef] [PubMed]
- Surowska, A.; De Giorgi, S.; Theytaz, F.; Campos, V.; Hodson, L.; Stefanoni, N.; Rey, V.; Schneiter, P.; Laville, M.; Giusti, V.; et al. Effects of roux-en-Y gastric bypass surgery on postprandial fructose metabolism. Obes. (Silver Spring) 2016, 24, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Bouchoux, J.; Beilstein, F.; Pauquai, T.; Guerrera, I.C.; Chateau, D.; Ly, N.; Alqub, M.; Klein, C.; Chambaz, J.; Rousset, M.; et al. The proteome of cytosolic lipid droplets isolated from differentiated Caco-2/TC7 enterocytes reveals cell-specific characteristics. Biol. Cell 2011, 103, 499–517. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Lee, B.; Buhman, K.K.; Cheng, J.X. A dynamic, cytoplasmic triacylglycerol pool in enterocytes revealed by ex vivo and in vivo coherent anti-Stokes Raman scattering imaging. J. Lipid. Res. 2009, 50, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Xiao, C.; Morgantini, C.; Lewis, G.F. New Insights into the Regulation of Chylomicron Production. Annu. Rev. Nutr. 2015, 35, 265–294. [Google Scholar] [CrossRef] [PubMed]
- Adeli, K.; Lewis, G.F. Intestinal lipoprotein overproduction in insulin-resistant states. Curr. Opin. Lipidol. 2008, 19, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Demignot, S.; Beilstein, F.; Morel, E. Triglyceride-rich lipoproteins and cytosolic lipid droplets in enterocytes: Key players in intestinal physiology and metabolic disorders. Biochimie 2014, 96, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Giammanco, A.; Cefalu, A.B.; Noto, D.; Averna, M.R. The pathophysiology of intestinal lipoprotein production. Front. Physiol. 2015, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.M. Intestinal lipid absorption and lipoprotein formation. Curr. Opin. Lipidol. 2014, 25, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Julve, J.; Martin-Campos, J.M.; Escola-Gil, J.C.; Blanco-Vaca, F. Chylomicrons: Advances in biology, pathology, laboratory testing, and therapeutics. Clin. Chim. Acta. 2016, 455, 134–148. [Google Scholar] [CrossRef] [PubMed]
- White, D.A.; Bennett, A.J.; Billett, M.A.; Salter, A.M. The assembly of triacylglycerol-rich lipoproteins: An essential role for the microsomal triacylglycerol transfer protein. Br. J. Nutr. 1998, 80, 219–229. [Google Scholar] [PubMed]
- Griffo, E.; Di Marino, L.; Patti, L.; Bozzetto, L.; Annuzzi, G.; Cipriano, P.; Mangione, A.; Della Pepa, G.; Cocozza, S.; Riccardi, G.; et al. Test meals rich in marine long-chain n-3 polyunsaturated fatty acids increase postprandial chylomicron response. Nutr. Res. 2014, 34, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Milan, A.M.; Nuora, A.; Pundir, S.; Pileggi, C.A.; Markworth, J.F.; Linderborg, K.M.; Cameron-Smith, D. Older adults have an altered chylomicron response to a high-fat meal. Br. J. Nutr. 2016, 115, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Knuth, N.D.; Horowitz, J.F. The elevation of ingested lipids within plasma chylomicrons is prolonged in men compared with women. J. Nutr. 2006, 136, 1498–1503. [Google Scholar] [PubMed]
- Desmarchelier, C.; Martin, J.C.; Planells, R.; Gastaldi, M.; Nowicki, M.; Goncalves, A.; Valero, R.; Lairon, D.; Borel, P. The postprandial chylomicron triacylglycerol response to dietary fat in healthy male adults is significantly explained by a combination of single nucleotide polymorphisms in genes involved in triacylglycerol metabolism. J. Clin. Endocrinol. Metab. 2014, 99, E484–E488. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Polansky, M.M.; Sato, Y.; Adeli, K.; Anderson, R.A. Cinnamon extract inhibits the postprandial overproduction of apolipoprotein B48-containing lipoproteins in fructose-fed animals. J. Nutr. Biochem. 2009, 20, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Hein, G.J.; Baker, C.; Hsieh, J.; Farr, S.; Adeli, K. GLP-1 and GLP-2 as yin and yang of intestinal lipoprotein production: Evidence for predominance of GLP-2-stimulated postprandial lipemia in normal and insulin-resistant states. Diabetes 2013, 62, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Pavlic, M.; Xiao, C.; Szeto, L.; Patterson, B.W.; Lewis, G.F. Insulin acutely inhibits intestinal lipoprotein secretion in humans in part by suppressing plasma free fatty acids. Diabetes 2010, 59, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Federico, L.M.; Naples, M.; Taylor, D.; Adeli, K. Intestinal insulin resistance and aberrant production of apolipoprotein B48 lipoproteins in an animal model of insulin resistance and metabolic dyslipidemia: Evidence for activation of protein tyrosine phosphatase-1B, extracellular signal-related kinase, and sterol regulatory element-binding protein-1c in the fructose-fed hamster intestine. Diabetes 2006, 55, 1316–1326. [Google Scholar] [PubMed]
- Lewis, G.F.; Uffelman, K.; Naples, M.; Szeto, L.; Haidari, M.; Adeli, K. Intestinal lipoprotein overproduction, a newly recognized component of insulin resistance, is ameliorated by the insulin sensitizer rosiglitazone: Studies in the fructose-fed Syrian golden hamster. Endocrinology 2005, 146, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Griffiths, B.; Santos, C.R.; Pende, M.; Schulze, A. Regulation of the SREBP transcription factors by mTORC1. Biochem. Soc. Trans. 2011, 39, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Lino, M.; Farr, S.; Baker, C.; Fuller, M.; Trigatti, B.; Adeli, K. Intestinal scavenger receptor class B type I as a novel regulator of chylomicron production in healthy and diet-induced obese states. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G350–G359. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.A.; Webb, J.; Choi, J.; Baker, C.; Lino, M.; Trigatti, B.; Trajcevski, K.E.; Hawke, T.J.; Adeli, K. Intestinal SR-BI is upregulated in insulin-resistant states and is associated with overproduction of intestinal apoB48-containing lipoproteins. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G326–G337. [Google Scholar] [CrossRef] [PubMed]
- Yusta, B.; Holland, D.; Waschek, J.A.; Drucker, D.J. Intestinotrophic glucagon-like peptide-2 (GLP-2) activates intestinal gene expression and growth factor-dependent pathways independent of the vasoactive intestinal peptide gene in mice. Endocrinology 2012, 153, 2623–2632. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.; Taher, J.; Adeli, K. Glucagon-like peptide-1 as a key regulator of lipid and lipoprotein metabolism in fasting and postprandial states. Cardiovasc. Hematol. Disord. Drug. Targets. 2014, 14, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.; Trajcevski, K.E.; Farr, S.L.; Baker, C.L.; Lake, E.J.; Taher, J.; Iqbal, J.; Hussain, M.M.; Adeli, K. Glucagon-Like Peptide 2 (GLP-2) Stimulates Postprandial Chylomicron Production and Postabsorptive Release of Intestinal Triglyceride Storage Pools via Induction of Nitric Oxide Signaling in Male Hamsters and Mice. Endocrinology 2015, 156, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Matikainen, N.; Bjornson, E.; Soderlund, S.; Boren, C.; Eliasson, B.; Pietilainen, K.H.; Bogl, L.H.; Hakkarainen, A.; Lundbom, N.; Rivellese, A.; et al. Minor Contribution of Endogenous GLP-1 and GLP-2 to Postprandial Lipemia in Obese Men. PLoS ONE 2016, 11, e0145890. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Kagaya, M.; Suzuki, M.; Yoshida, A.; Naito, M. Simultaneous ingestion of fructose and fat exacerbates postprandial exogenous lipidemia in young healthy Japanese women. J. Atheroscler. Thromb. 2013, 20, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Kato, M.; Yoshida, A.; Naito, M. The ingestion of a fructose-containing beverage combined with fat cream exacerbates postprandial lipidemia in young healthy women. J. Atheroscler. Thromb. 2015, 22, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Dash, S.; Morgantini, C.; Lewis, G.F. Novel role of enteral monosaccharides in intestinal lipoprotein production in healthy humans. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Mamo, J.C.; Hirano, T.; James, L.; Szeto, L.; Steiner, G. Partial characterization of the fructose-induced defect in very-low-density lipoprotein triglyceride metabolism. Metabolism 1991, 40, 888–893. [Google Scholar] [CrossRef]
- Jackson, K.G.; Robertson, M.D.; Fielding, B.A.; Frayn, K.N.; Williams, C.M. Measurement of apolipoprotein B-48 in the Svedberg flotation rate (S(f))>400, S(f) 60–400 and S(f) 20–60 lipoprotein fractions reveals novel findings with respect to the effects of dietary fatty acids on triacylglycerol-rich lipoproteins in postmenopausal women. Clin. Sci. (Lond) 2002, 103, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.G.; Wolstencroft, E.J.; Bateman, P.A.; Yaqoob, P.; Williams, C.M. Acute effects of meal fatty acids on postprandial NEFA, glucose and apo E response: Implications for insulin sensitivity and lipoprotein regulation? Br. J. Nutr. 2005, 93, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.G.; Wolstencroft, E.J.; Bateman, P.A.; Yaqoob, P.; Williams, C.M. Apolipoprotein E enrichment of immuno-separated chylomicron and chylomicron remnants following saturated fatty acids. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Heath, R.B.; Karpe, F.; Milne, R.W.; Burdge, G.C.; Wootton, S.A.; Frayn, K.N. Selective partitioning of dietary fatty acids into the VLDL TG pool in the early postprandial period. J. Lipid. Res. 2003, 44, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Stolinski, M.; Shojaee-Moradie, F.; Lou, S.; Ma, Y.; Hovorka, R.; Umpleby, A.M. A novel method for measuring intestinal and hepatic triacylglycerol kinetics. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1041–E1047. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit. Rev. Clin. Lab. Sci. 2016, 53, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Van Buul, V.J.; Tappy, L.; Brouns, F.J. Misconceptions about fructose-containing sugars and their role in the obesity epidemic. Nutr. Res. Rev. 2014, 27, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A.; Lustig, R.H.; Caccavello, R.; Erkin-Cakmak, A.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Mulligan, K.; Schwarz, J.M. Short-term isocaloric fructose restriction lowers apoC-III levels and yields less atherogenic lipoprotein profiles in children with obesity and metabolic syndrome. Atherosclerosis 2016, 253, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H.; Mulligan, K.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Erkin-Cakmak, A.; Gugliucci, A.; Schwarz, J.M. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obes. (Silver Spring) 2016, 24, 453–460. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steenson, S.; Umpleby, A.M.; Lovegrove, J.A.; Jackson, K.G.; Fielding, B.A. Role of the Enterocyte in Fructose-Induced Hypertriglyceridaemia. Nutrients 2017, 9, 349. https://doi.org/10.3390/nu9040349
Steenson S, Umpleby AM, Lovegrove JA, Jackson KG, Fielding BA. Role of the Enterocyte in Fructose-Induced Hypertriglyceridaemia. Nutrients. 2017; 9(4):349. https://doi.org/10.3390/nu9040349
Chicago/Turabian StyleSteenson, Simon, A. Margot Umpleby, Julie A. Lovegrove, Kim G. Jackson, and Barbara A. Fielding. 2017. "Role of the Enterocyte in Fructose-Induced Hypertriglyceridaemia" Nutrients 9, no. 4: 349. https://doi.org/10.3390/nu9040349
APA StyleSteenson, S., Umpleby, A. M., Lovegrove, J. A., Jackson, K. G., & Fielding, B. A. (2017). Role of the Enterocyte in Fructose-Induced Hypertriglyceridaemia. Nutrients, 9(4), 349. https://doi.org/10.3390/nu9040349