A Sensitive LC-MS/MS Method for Palytoxin Using Lithium Cationization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
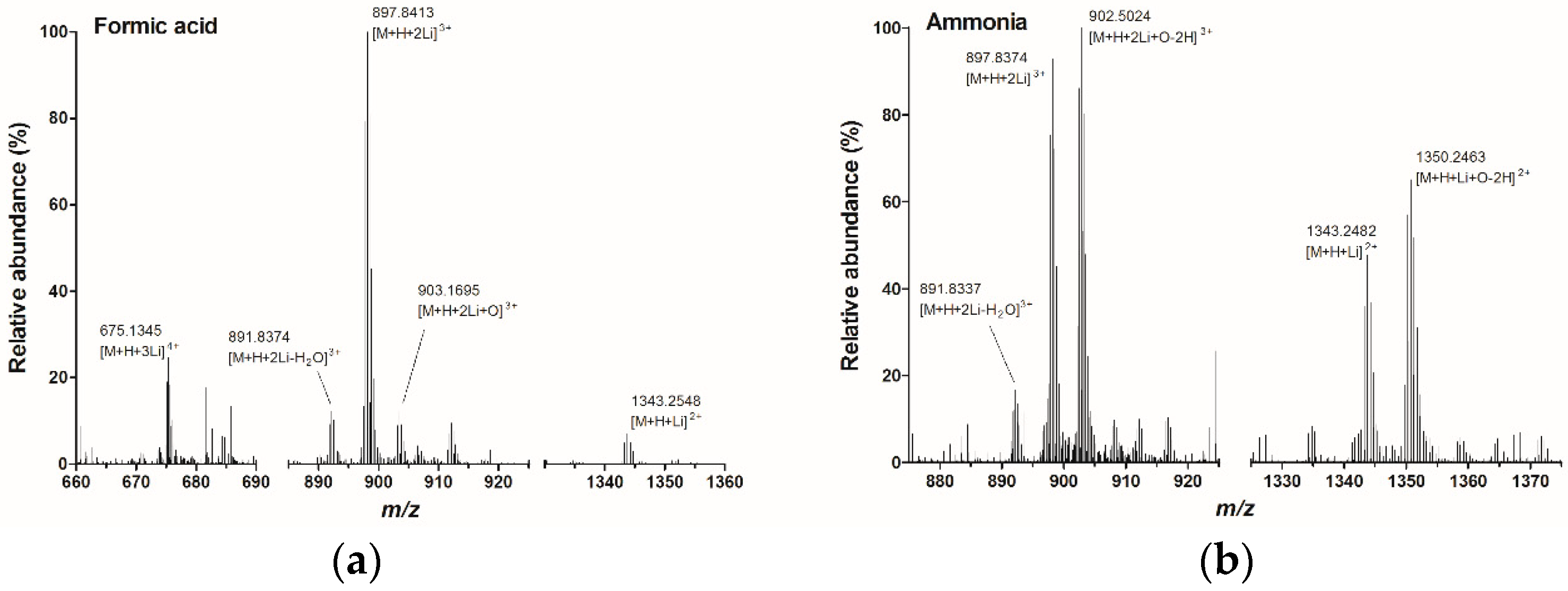
2.1. Infusion Experiments with High Resolution Mass Spectrometry
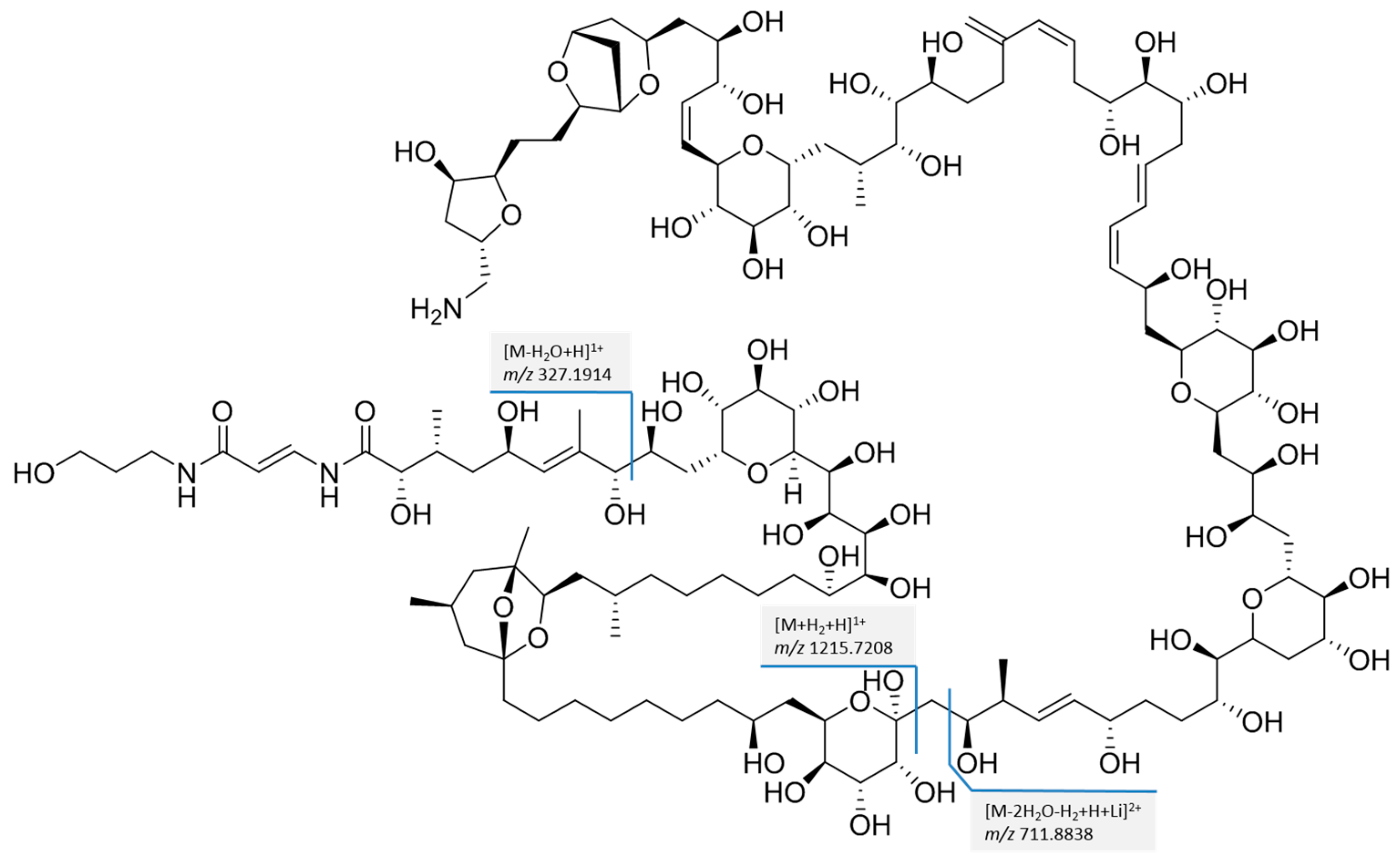
2.2. Fragmentation PlTX with High Resolution Mass Spectrometry
2.3. Chromatography
2.4. Sample Clean-Up
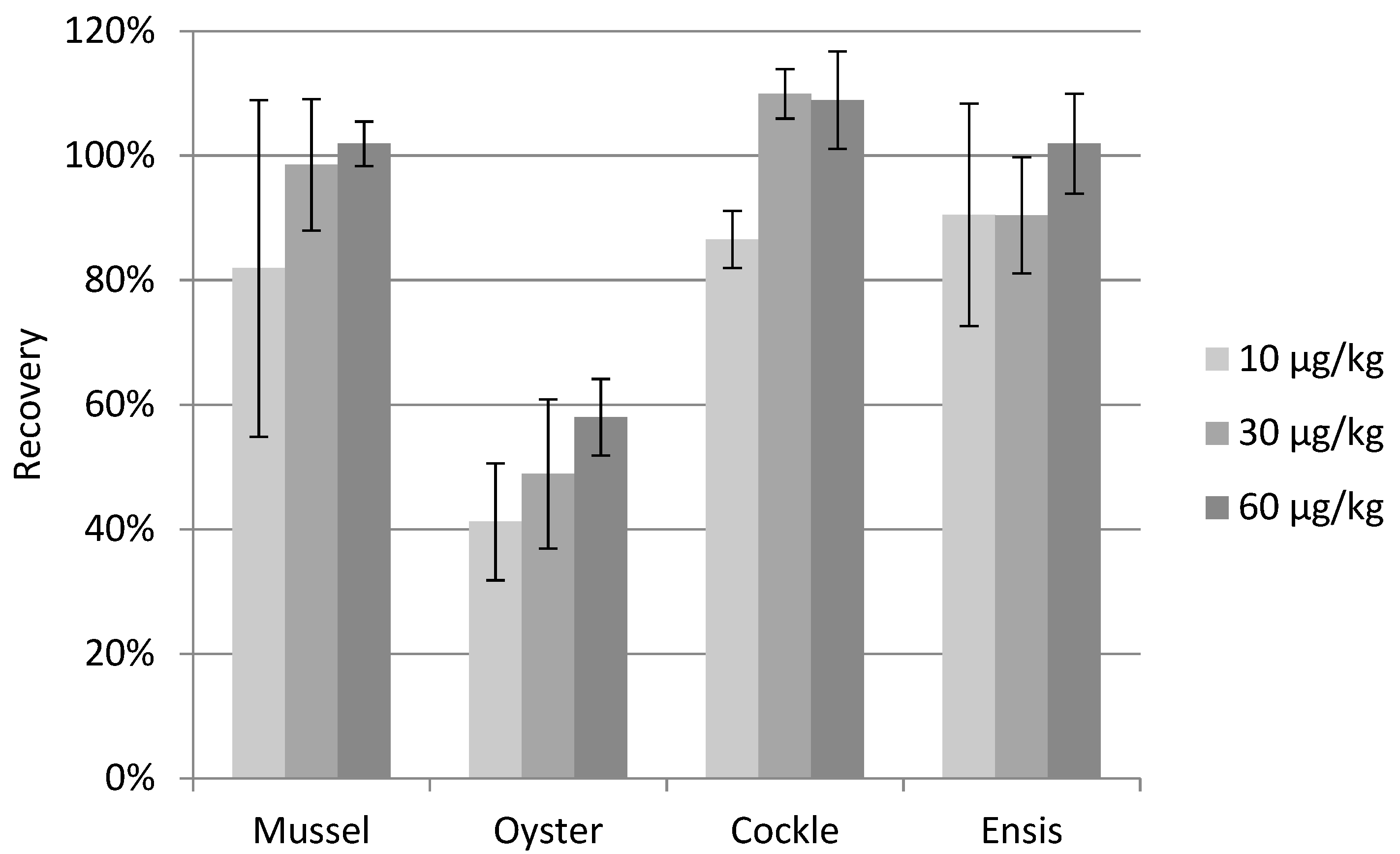
2.5. Method Validation
3. Discussion
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Preparation of Standards
4.3. Liquid Chromatography Coupled with Mass Spectrometry
4.3.1. Sample Clean-up
4.3.2. Liquid Chromatography Methods
4.3.3. High Resolution Mass Spectrometry
4.3.4. Triple Quad Mass Spectrometry
4.4. Method Validation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Usami, M.; Satake, M.; Ishida, S.; Inoue, A.; Kan, Y.; Yasumoto, T. Palytoxin analogs from the dinoflagellate ostreopsis siamensis. J. Am. Chem. Soc. 1995, 117, 5389–5390. [Google Scholar] [CrossRef]
- Ciminiello, P.; Dell’Aversano, C.; Dello Iacovo, E.; Fattorusso, E.; Forino, M.; Grauso, L.; Tartaglione, L.; Guerrini, F.; Pezzolesi, L.; Pistocchi, R.; et al. Isolation and structure elucidation of ovatoxin-a, the major toxin produced by ostreopsis ovata. J. Am. Chem. Soc. 2012, 134, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, L.; Towers, N.; Briggs, L.; Munday, R.; Adamson, J. Uptake of palytoxin-like compounds by shellfish fed ostreopsis siamensis (dinophyceae). N. Z. J. Mar. Freshw. Res. 2002, 36, 631–636. [Google Scholar] [CrossRef]
- Lenoir, S.; Ten-Hage, L.; Turquet, J.; Quod, J.-P.; Bernard, C.; Hennion, M.-C. First evidence of palytoxin analogues from an ostreopsis mascarenensis (dinophyceae) benthic bloom in southwestern indian ocean1. J. Phycol. 2004, 40, 1042–1051. [Google Scholar] [CrossRef]
- Fraga, M.; Vilariño, N.; Louzao, M.C.; Molina, L.; López, Y.; Poli, M.; Botana, L.M. First identification of palytoxin-like molecules in the atlantic coral species palythoa canariensis. Anal. Chem. 2017, 89, 7438–7446. [Google Scholar] [CrossRef]
- Kerbrat, A.S.; Amzil, Z.; Pawlowiez, R.; Golubic, S.; Sibat, M.; Darius, H.T.; Chinain, M.; Laurent, D. First evidence of palytoxin and 42-hydroxy-palytoxin in the marine cyanobacterium trichodesmium. Mar. Drugs 2011, 9, 543. [Google Scholar] [CrossRef]
- Rhodes, L. World-wide occurrence of the toxic dinoflagellate genus ostreopsis schmidt. Toxicon 2011, 57, 400–407. [Google Scholar] [CrossRef]
- Aligizaki, K.; Katikou, P.; Milandri, A.; Diogène, J. Occurrence of palytoxin-group toxins in seafood and future strategies to complement the present state of the art. Toxicon 2011, 57, 390–399. [Google Scholar] [CrossRef]
- Ciminiello, P.; Dell’Aversano, C.; Forino, M. Chemistry of palytoxin and its analogues. In Phycotoxins: Chemistry and Biochemistry, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2015; pp. 85–111. [Google Scholar]
- Tartaglione, L.; Dello Iacovo, E.; Mazzeo, A.; Casabianca, S.; Ciminiello, P.; Penna, A.; Dell’Aversano, C. Variability in toxin profiles of the mediterranean ostreopsis cf. Ovata and in structural features of the produced ovatoxins. Environ. Sci. Technol. 2017, 51, 13920–13928. [Google Scholar] [CrossRef]
- Deeds, J.R.; Schwartz, M.D. Human risk associated with palytoxin exposure. Toxicon 2010, 56, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Murphy, L.T.; Charlton, N.P. Prevalence and characteristics of inhalational and dermal palytoxin exposures reported to the national poison data system in the U.S. Environ. Toxicol. Pharmacol. 2017, 55, 107–109. [Google Scholar] [CrossRef]
- Durando, P.; Ansaldi, F.; Oreste, P.; Moscatelli, P.; Marensi, L.; Grillo, C.; Gasparini, R.; Icardi, G. Ostreopsis ovata and human health: Epidemiological and clinical features of respiratory syndrome outbreaks from a two-year syndromic surveillance, 2005–06, in North-West Italy. Wkly. Releases (1997–2007) 2007, 12, 3212. [Google Scholar]
- Snoeks, L.; Veenstra, J. Family with fever after cleaning a sea aquarium. Ned. Tijdschr. Geneeskd. 2012, 156, A4200. [Google Scholar]
- Patocka, J.; Nepovimova, E.; Wu, Q.; Kuca, K. Palytoxin congeners. Arch. Toxicol. 2017, 92, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Satoh, E.; Ishii, T.; Nishimura, M. Palytoxin-induced increase in cytosolic-free Ca2+ in mouse spleen cells. Eur. J. Pharmacol. 2003, 465, 9–13. [Google Scholar] [CrossRef]
- Wiles, J.S.; Vick, J.A.; Christensen, M.K. Toxicological evaluation of palytoxin in several animal species. Toxicon 1974, 12, 427–433. [Google Scholar] [CrossRef]
- Sosa, S.; Del Favero, G.; De Bortoli, M.; Vita, F.; Soranzo, M.R.; Beltramo, D.; Ardizzone, M.; Tubaro, A. Palytoxin toxicity after acute oral administration in mice. Toxicol. Lett. 2009, 191, 253–259. [Google Scholar] [CrossRef]
- EFSA. Scientific opinion on marine biotoxins in shellfish–palytoxin group. EFSA J. 2009, 1393, 1–40. [Google Scholar]
- Riobó, P.; Franco, J.M. Palytoxins: Biological and chemical determination. Toxicon 2011, 57, 368–375. [Google Scholar] [CrossRef]
- Alfonso, A.; Fernández-Araujo, A.; Alfonso, C.; Caramés, B.; Tobio, A.; Louzao, M.C.; Vieytes, M.R.; Botana, L.M. Palytoxin detection and quantification using the fluorescence polarization technique. Anal. Biochem. 2012, 424, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.; Bovee, T.F.H.; Kamelia, L.; Rietjens, I.M.C.M.; Hendriksen, P.J.M. Exploration of new functional endpoints in neuro-2a cells for the detection of the marine biotoxins saxitoxin, palytoxin and tetrodotoxin. Toxicol. in Vitro 2015, 30, 341–347. [Google Scholar] [CrossRef]
- Brovedani, V.; Sosa, S.; Poli, M.; Forino, M.; Varello, K.; Tubaro, A.; Pelin, M. A revisited hemolytic assay for palytoxin detection: Limitations for its quantitation in mussels. Toxicon 2016, 119, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraga, M.; Vilariño, N.; Louzao, M.C.; Fernández, D.A.; Poli, M.; Botana, L.M. Detection of palytoxin-like compounds by a flow cytometry-based immunoassay supported by functional and analytical methods. Ana. Chim. Acta 2016, 903, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ciminiello, P.; Dell’Aversano, C.; Dello Iacovo, E.; Forino, M.; Tartaglione, L. Liquid chromatography–high-resolution mass spectrometry for palytoxins in mussels. Anal. Bioanal. Chem. 2015, 407, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- García-Altares, M.; Tartaglione, L.; Dell’Aversano, C.; Carnicer, O.; de la Iglesia, P.; Forino, M.; Diogène, J.; Ciminiello, P. The novel ovatoxin-g and isobaric palytoxin (so far referred to as putative palytoxin) from ostreopsis cf. Ovata (nw mediterranean sea): Structural insights by lc-high resolution msn. Anal. Bioanal. Chem. 2015, 407, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Selwood, A.I.; van Ginkel, R.; Harwood, D.T.; McNabb, P.S.; Rhodes, L.R.; Holland, P.T. A sensitive assay for palytoxins, ovatoxins and ostreocins using lc-ms/ms analysis of cleavage fragments from micro-scale oxidation. Toxicon 2012, 60, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Pelin, M.; Forino, M.; Brovedani, V.; Tartaglione, L.; Dell’Aversano, C.; Pistocchi, R.; Poli, M.; Sosa, S.; Florio, C.; Ciminiello, P.; et al. Ovatoxin-a, a palytoxin analogue isolated from ostreopsis cf. Ovata fukuyo: Cytotoxic activity and elisa detection. Environ. Sci. Technol. 2016, 50, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Asam, M.R.; Glish, G.L. Tandem mass spectrometry of alkali cationized polysaccharides in a quadrupole ion trap. J. Am. Soc. Mass Spectrom. 1997, 8, 987–995. [Google Scholar] [CrossRef]
- Ramaley, L.; Herrera, L.C.; Melanson, J.E. Quantitative analysis of tag in oils using lithium cationization and direct-infusion esi tandem mass spectrometry. J. Am. Oil Chem. Soc. 2015, 92, 323–334. [Google Scholar] [CrossRef]
- Wei, J.; Bristow, A.W.T.; O’Connor, P.B. The competitive influence of Li+, Na+, K+, Ag+, and H+ on the fragmentation of a pegylated polymeric excipient. J. Am. Soc. Mass Spectrom. 2015, 26, 166–173. [Google Scholar] [CrossRef]
- Chen, M.; Cook, K.D. Oxidation artifacts in the electrospray mass spectrometry of aβ peptide. Anal. Chem. 2007, 79, 2031–2036. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.D.; Dhanji-Rapkova, M.; Algoet, M.; Suarez-Isla, B.A.; Cordova, M.; Caceres, C.; Murphy, C.J.; Casey, M.; Lees, D.N. Investigations into matrix components affecting the performance of the official bioassay reference method for quantitation of paralytic shellfish poisoning toxins in oysters. Toxicon 2012, 59, 215–230. [Google Scholar] [CrossRef]
- EU Reference Laboratories for Residues of Pesticides. Sante/11945/2015-Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed. Available online: https://www.accredia.it/en/documento/guidance-sante-119452015-guidance-document-on-analytical-quality-control-and-method-validation-procedures-for-pesticides-residues-analysis-in-food-and-feed/ (accessed on 14 December 2018).
- Moreiras, G.; Leão, J.M.; Gago-Martínez, A. Design of experiments for the optimization of electrospray ionization in the lc-ms/ms analysis of ciguatoxins. J. Mass Spectrom. 2018, 53, 1059–1069. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klijnstra, M.D.; Gerssen, A. A Sensitive LC-MS/MS Method for Palytoxin Using Lithium Cationization. Toxins 2018, 10, 537. https://doi.org/10.3390/toxins10120537
Klijnstra MD, Gerssen A. A Sensitive LC-MS/MS Method for Palytoxin Using Lithium Cationization. Toxins. 2018; 10(12):537. https://doi.org/10.3390/toxins10120537
Chicago/Turabian StyleKlijnstra, Mirjam D., and Arjen Gerssen. 2018. "A Sensitive LC-MS/MS Method for Palytoxin Using Lithium Cationization" Toxins 10, no. 12: 537. https://doi.org/10.3390/toxins10120537
APA StyleKlijnstra, M. D., & Gerssen, A. (2018). A Sensitive LC-MS/MS Method for Palytoxin Using Lithium Cationization. Toxins, 10(12), 537. https://doi.org/10.3390/toxins10120537