Pharmacological Targeting of Pore-Forming Toxins as Adjunctive Therapy for Invasive Bacterial Infection


Abstract
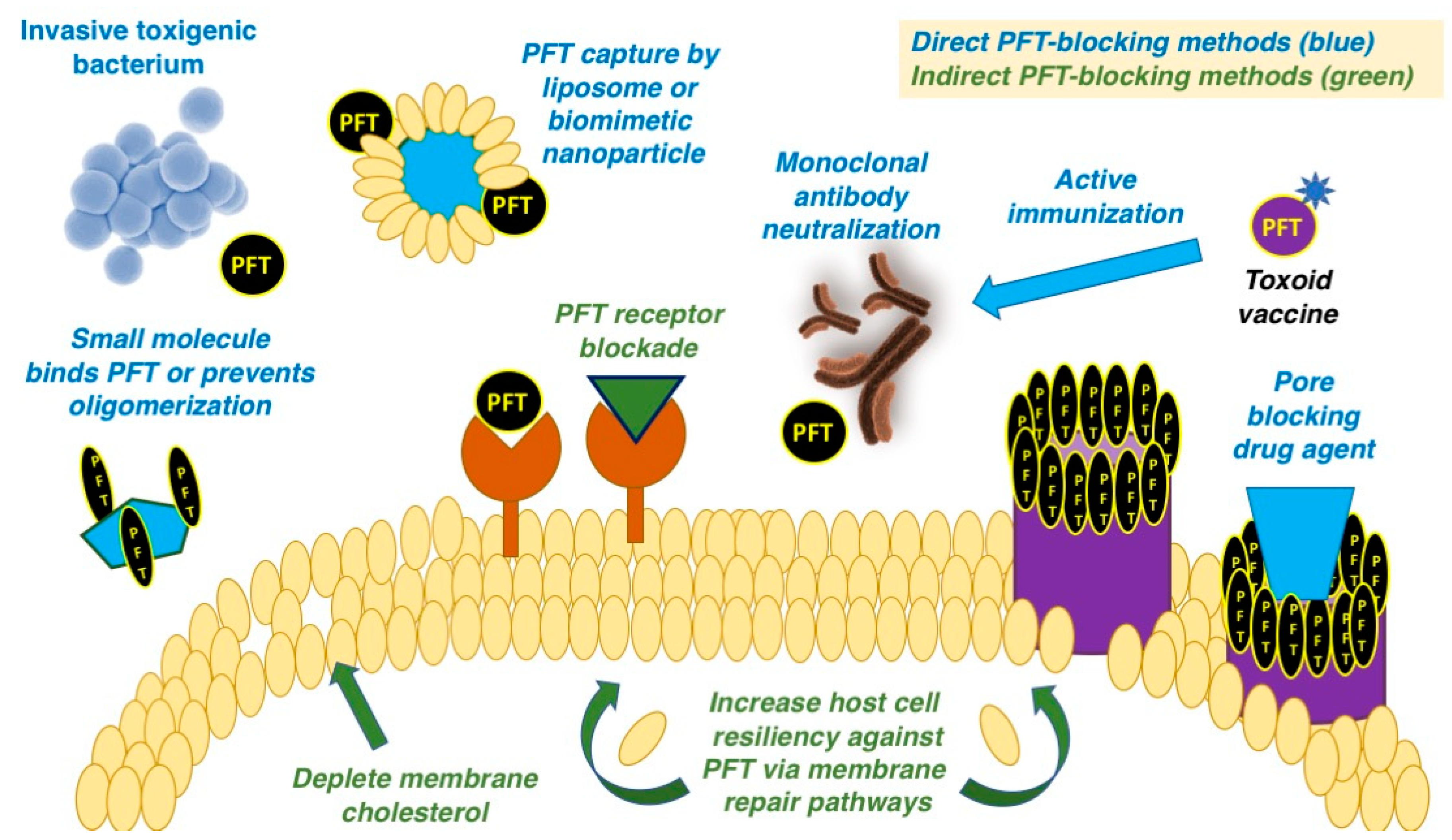
:1. Introduction
2. Classification of Pore-Forming Toxins
3. Overview of Pharmacological Approaches to Pore-Forming Toxin Inhibition
4. Direct Binding or Sequestration of Pore-Forming Toxins
4.1. Passive Antibody Neutralization of Pore-Forming Toxins
4.2. Small Molecules that Bind or Inhibit Toxin Assembly
4.3. Inhibition of Pore-Forming Toxins through Decoy Capture
5. Inhibition of Host Cell Receptors or Uptake of Pore-Forming Toxins
6. Blockade of Pore Formation
7. Increasing Host Cell Resiliency Against Pore-Forming Toxin Action
8. Inactivated PFT (Toxoid) Vaccines for Active Immunization
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alanis, A.J. Resistance to antibiotics: Are we in the post-antibiotic era? Arch. Med. Res. 2005, 36, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Guidos, R.; Gilbert, D.; Bradley, J.; Boucher, H.W.; Scheld, W.M.; Bartlett, J.G.; Edwards, J., Jr. The epidemic of antibiotic-resistant infections: A call to action for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. 2008, 46, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.R.; Bischofberger, M.; Pernot, L.; van der Goot, F.G.; Freche, B. Bacterial pore-forming toxins: The (w)hole story? Cell. Mol. Life Sci. 2008, 65, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Los, F.C.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Kao, C.Y.; Los, F.C.; Huffman, D.L.; Wachi, S.; Kloft, N.; Husmann, M.; Karabrahimi, V.; Schwartz, J.L.; Bellier, A.; Ha, C.; et al. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog. 2011, 7, e1001314. [Google Scholar] [CrossRef]
- Bischofberger, M.; Iacovache, I.; van der Goot, F.G. Pathogenic pore-forming proteins: Function and host response. Cell Host Microbe 2012, 12, 266–275. [Google Scholar] [CrossRef]
- Tweten, R.K. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 2005, 73, 6199–6209. [Google Scholar] [CrossRef]
- Griffitts, J.S.; Haslam, S.M.; Yang, T.; Garczynski, S.F.; Mulloy, B.; Morris, H.; Cremer, P.S.; Dell, A.; Adang, M.J.; Aroian, R.V. Glycolipids as receptors for Bacillus thuringiensis crystal toxin. Science 2005, 307, 922–925. [Google Scholar] [CrossRef]
- Giddings, K.S.; Zhao, J.; Sims, P.J.; Tweten, R.K. Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat. Struct. Mol. Biol. 2004, 11, 1173–1178. [Google Scholar] [CrossRef]
- Diep, D.B.; Nelson, K.L.; Raja, S.M.; Pleshak, E.N.; Buckley, J.T. Glycosylphosphatidylinositol anchors of membrane glycoproteins are binding determinants for the channel-forming toxin aerolysin. J. Biol. Chem. 1998, 273, 2355–2360. [Google Scholar] [CrossRef]
- Muhlen, S.; Dersch, P. Anti-virulence Strategies to Target Bacterial Infections. Curr. Top. Microbiol. Immunol. 2016, 398, 147–183. [Google Scholar]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef]
- Gouaux, E. Channel-forming toxins: Tales of transformation. Curr. Opin. Struct. Biol. 1997, 7, 566–573. [Google Scholar] [CrossRef]
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Prog. Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [CrossRef] [PubMed]
- Rojko, N.; Kristan, K.C.; Viero, G.; Zerovnik, E.; Macek, P.; Dalla Serra, M.; Anderluh, G. Membrane damage by an alpha-helical pore-forming protein, Equinatoxin II, proceeds through a succession of ordered steps. J. Biol. Chem. 2013, 288, 23704–23715. [Google Scholar] [CrossRef] [PubMed]
- Cascales, E.; Buchanan, S.K.; Duche, D.; Kleanthous, C.; Lloubes, R.; Postle, K.; Riley, M.; Slatin, S.; Cavard, D. Colicin biology. Microbiol. Mol. Biol. Rev. 2007, 71, 158–229. [Google Scholar] [CrossRef]
- Lakey, J.H.; van der Goot, F.G.; Pattus, F. All in the family: The toxic activity of pore-forming colicins. Toxicology 1994, 87, 85–108. [Google Scholar] [CrossRef]
- Parker, M.W.; Pattus, F.; Tucker, A.D.; Tsernoglou, D. Structure of the membrane-pore-forming fragment of colicin A. Nature 1989, 337, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Grochulski, P.; Masson, L.; Borisova, S.; Pusztai-Carey, M.; Schwartz, J.L.; Brousseau, R.; Cygler, M. Bacillus thuringiensis CryIA(a) insecticidal toxin: Crystal structure and channel formation. J. Mol. Biol. 1995, 254, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216. [Google Scholar] [CrossRef] [PubMed]
- Allured, V.S.; Collier, R.J.; Carroll, S.F.; McKay, D.B. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc. Natl. Acad. Sci. USA 1986, 83, 1320–1324. [Google Scholar] [CrossRef] [PubMed]
- Ros, U.; Garcia-Saez, A.J. More Than a Pore: The Interplay of Pore-Forming Proteins and Lipid Membranes. J. Membr. Biol. 2015, 248, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Bayley, H. Key residues for membrane binding, oligomerization, and pore forming activity of staphylococcal alpha-hemolysin identified by cysteine scanning mutagenesis and targeted chemical modification. J. Biol. Chem. 1995, 270, 23065–23071. [Google Scholar] [CrossRef] [PubMed]
- Tilley, S.J.; Saibil, H.R. The mechanism of pore formation by bacterial toxins. Curr. Opin. Struct. Biol. 2006, 16, 230–236. [Google Scholar] [CrossRef]
- Tweten, R.K.; Parker, M.W.; Johnson, A.E. The cholesterol-dependent cytolysins. Curr. Top. Microbiol. Immunol. 2001, 257, 15–33. [Google Scholar]
- Palmer, M.; Harris, R.; Freytag, C.; Kehoe, M.; Tranum-Jensen, J.; Bhakdi, S. Assembly mechanism of the oligomeric streptolysin O pore: The early membrane lesion is lined by a free edge of the lipid membrane and is extended gradually during oligomerization. EMBO J. 1998, 17, 1598–1605. [Google Scholar] [CrossRef]
- Sowdhamini, R.; Mitchell, T.J.; Andrew, P.W.; Morgan, P.J. Structural and functional analogy between pneumolysin and proaerolysin. Protein Eng. 1997, 10, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Macey, M.G.; Whiley, R.A.; Miller, L.; Nagamune, H. Effect on polymorphonuclear cell function of a human-specific cytotoxin, intermedilysin, expressed by Streptococcus intermedius. Infect. Immun. 2001, 69, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, C.; Gaillard, J.L.; Alouf, J.E.; Berche, P. Production of thiol-dependent haemolysins by Listeria monocytogenes and related species. J. Gen. Microbiol. 1989, 135, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Mengaud, J.; Vicente, M.F.; Chenevert, J.; Pereira, J.M.; Geoffroy, C.; Gicquel-Sanzey, B.; Baquero, F.; Perez-Diaz, J.C.; Cossart, P. Expression in Escherichia coli and sequence analysis of the listeriolysin O determinant of Listeria monocytogenes. Infect. Immun. 1988, 56, 766–772. [Google Scholar] [PubMed]
- Shannon, J.G.; Ross, C.L.; Koehler, T.M.; Rest, R.F. Characterization of anthrolysin O, the Bacillus anthracis cholesterol-dependent cytolysin. Infect. Immun. 2003, 71, 3183–3189. [Google Scholar] [CrossRef]
- Rossjohn, J.; Feil, S.C.; McKinstry, W.J.; Tweten, R.K.; Parker, M.W. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell 1997, 89, 685–692. [Google Scholar] [CrossRef]
- Parker, M.W.; Buckley, J.T.; Postma, J.P.; Tucker, A.D.; Leonard, K.; Pattus, F.; Tsernoglou, D. Structure of the Aeromonas toxin proaerolysin in its water-soluble and membrane-channel states. Nature 1994, 367, 292–295. [Google Scholar] [CrossRef]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Rojko, N.; Anderluh, G. How lipid membranes affect pore forming toxin activity. Acc. Chem. Res. 2015, 48, 3073–3079. [Google Scholar] [CrossRef]
- Iacovache, I.; van der Goot, F.G.; Pernot, L. Pore formation: An ancient yet complex form of attack. Biochim. Biophys. Acta 2008, 1778, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W. Cryptic clues as to how water-soluble protein toxins form pores in membranes. Toxicon 2003, 42, 1–6. [Google Scholar] [CrossRef]
- Sonnen, A.F.; Henneke, P. Role of pore-forming toxins in neonatal sepsis. Clin. Dev. Immunol. 2013, 2013, 608456. [Google Scholar] [CrossRef]
- Mulvihill, E.; van Pee, K.; Mari, S.A.; Muller, D.J.; Yildiz, O. Directly observing the lipid-dependent self-assembly and pore-forming mechanism of the cytolytic toxin listeriolysin O. Nano Lett. 2015, 15, 6965–6973. [Google Scholar] [CrossRef] [PubMed]
- Van Pee, K.; Neuhaus, A.; D’Imprima, E.; Mills, D.J.; Kuhlbrandt, W.; Yildiz, O. CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of pneumolysin. eLife 2017, 6, e23644. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, N.; Yamaji-Hasegawa, A.; Hullin-Matsuda, F.; Kobayashi, T. Molecular mechanisms of action of sphingomyelin-specific pore-forming toxin, lysenin. Semin. Cell Dev. Biol. 2018, 73, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Bebeacua, C.; Opota, O.; Kellenberger, C.; Klaholz, B.; Orlov, I.; Cambillau, C.; Lemaitre, B.; Roussel, A. X-ray and Cryo-electron microscopy structures of monalysin pore-forming toxin reveal multimerization of the pro-form. J. Biol. Chem. 2015, 290, 13191–13201. [Google Scholar] [CrossRef] [PubMed]
- Iacovache, I.; De Carlo, S.; Cirauqui, N.; Dal Peraro, M.; van der Goot, F.G.; Zuber, B. Cryo-EM structure of aerolysin variants reveals a novel protein fold and the pore-formation process. Nat. Commun. 2016, 7, 12062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, C.M.; Parsons, E.S.; Smith, R.A.; Seddon, J.M.; Ces, O.; Bubeck, D. Disentangling the roles of cholesterol and CD59 in intermedilysin pore formation. Sci. Rep. 2016, 6, 38446. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- DiGiandomenico, A.; Sellman, B.R. Antibacterial monoclonal antibodies: The next generation? Curr. Opin. Microbiol. 2015, 27, 78–85. [Google Scholar] [CrossRef]
- Lang, A.B.; Cryz, S.J., Jr.; Schurch, U.; Ganss, M.T.; Bruderer, U. Immunotherapy with human monoclonal antibodies. Fragment A specificity of polyclonal and monoclonal antibodies is crucial for full protection against tetanus toxin. J. Immunol. 1993, 151, 466–472. [Google Scholar]
- Khazaeli, M.B.; Conry, R.M.; LoBuglio, A.F. Human immune response to monoclonal antibodies. J. Immunother. Emphas. Tumor Immunol. 1994, 15, 42–52. [Google Scholar] [CrossRef]
- Bubeck Wardenburg, J.; Schneewind, O. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 2008, 205, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Bubeck Wardenburg, J.; Gardner, D.J.; Long, D.; Whitney, A.R.; Braughton, K.R.; Schneewind, O.; DeLeo, F.R. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 2010, 202, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Francois, B.; Mercier, E.; Gonzalez, C.; Asehnoune, K.; Nseir, S.; Fiancette, M.; Desachy, A.; Plantefeve, G.; Meziani, F.; de Lame, P.A.; et al. Safety and tolerability of a single administration of AR-301, a human monoclonal antibody, in ICU patients with severe pneumonia caused by Staphylococcus aureus: First-in-human trial. Intensive Care Med. 2018, 44, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Foletti, D.; Strop, P.; Shaughnessy, L.; Hasa-Moreno, A.; Casas, M.G.; Russell, M.; Bee, C.; Wu, S.; Pham, A.; Zeng, Z.; et al. Mechanism of action and in vivo efficacy of a human-derived antibody against Staphylococcus aureus alpha-hemolysin. J. Mol. Biol. 2013, 425, 1641–1654. [Google Scholar] [CrossRef]
- Oganesyan, V.; Peng, L.; Damschroder, M.M.; Cheng, L.; Sadowska, A.; Tkaczyk, C.; Sellman, B.R.; Wu, H.; Dall’Acqua, W.F. Mechanisms of neutralization of a human anti-alpha-toxin antibody. J. Biol. Chem. 2014, 289, 29874–29880. [Google Scholar] [CrossRef]
- Hua, L.; Cohen, T.S.; Shi, Y.; Datta, V.; Hilliard, J.J.; Tkaczyk, C.; Suzich, J.; Stover, C.K.; Sellman, B.R. MEDI4893* Promotes survival and extends the antibiotic treatment window in a Staphylococcus aureus immunocompromised pneumonia model. Antimicrob. Agents Chemother. 2015, 59, 4526–4532. [Google Scholar] [CrossRef]
- Yu, X.Q.; Robbie, G.J.; Wu, Y.; Esser, M.T.; Jensen, K.; Schwartz, H.I.; Bellamy, T.; Hernandez-Illas, M.; Jafri, H.S. Safety, tolerability, and pharmacokinetics of MEDI4893, an investigational, extended-half-life, anti-Staphylococcus aureus alpha-toxin human monoclonal antibody, in healthy adults. Antimicrob. Agents Chemother. 2017, 6, e01020-16. [Google Scholar] [CrossRef]
- Tkaczyk, C.; Hua, L.; Varkey, R.; Shi, Y.; Dettinger, L.; Woods, R.; Barnes, A.; MacGill, R.S.; Wilson, S.; Chowdhury, P.; et al. Identification of anti-alpha toxin monoclonal antibodies that reduce the severity of Staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin. Vaccine Immunol. 2012, 19, 377–385. [Google Scholar] [CrossRef]
- Hua, L.; Hilliard, J.J.; Shi, Y.; Tkaczyk, C.; Cheng, L.I.; Yu, X.; Datta, V.; Ren, S.; Feng, H.; Zinsou, R.; et al. Assessment of an anti-alpha-toxin monoclonal antibody for prevention and treatment of Staphylococcus aureus-induced pneumonia. Antimicrob. Agents Chemother. 2014, 58, 1108–1117. [Google Scholar] [CrossRef]
- Ragle, B.E.; Bubeck Wardenburg, J. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun. 2009, 77, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Seilie, E.S.; Bubeck Wardenburg, J. Staphylococcus aureus pore-forming toxins: The interface of pathogen and host complexity. Semin. Cell Dev. Biol. 2017, 72, 101–116. [Google Scholar] [CrossRef]
- Alonzo, F., 3rd; Torres, V.J. The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol. Mol. Biol. Rev. 2014, 78, 199–230. [Google Scholar] [CrossRef] [PubMed]
- Karauzum, H.; Adhikari, R.P.; Sarwar, J.; Devi, V.S.; Abaandou, L.; Haudenschild, C.; Mahmoudieh, M.; Boroun, A.R.; Vu, H.; Nguyen, T.; et al. Structurally designed attenuated subunit vaccines for S. aureus LukS-PV and LukF-PV confer protection in a mouse bacteremia model. PLoS ONE 2013, 8, e65384. [Google Scholar] [CrossRef] [PubMed]
- Rouha, H.; Badarau, A.; Visram, Z.C.; Battles, M.B.; Prinz, B.; Magyarics, Z.; Nagy, G.; Mirkina, I.; Stulik, L.; Zerbs, M.; et al. Five birds, one stone: Neutralization of alpha-hemolysin and 4 bi-component leukocidins of Staphylococcus aureus with a single human monoclonal antibody. mAbs 2015, 7, 243–254. [Google Scholar] [CrossRef]
- Rouha, H.; Weber, S.; Janesch, P.; Maierhofer, B.; Gross, K.; Dolezilkova, I.; Mirkina, I.; Visram, Z.C.; Malafa, S.; Stulik, L.; et al. Disarming Staphylococcus aureus from destroying human cells by simultaneously neutralizing six cytotoxins with two human monoclonal antibodies. Virulence 2018, 9, 231–247. [Google Scholar] [CrossRef]
- Magyarics, Z.; Leslie, F.; Luperchio, S.A.; Bartko, J.; Schörgenhofer, C.; Schwameis, M.; Derhaschnig, U.; Lagler, H.; Stiebellehner, L.; Jilma, B.; et al. Serum and lung pharmacokinetics of ASN100, a monoclonal antibody combination for the prevention and treatment of Staphylococcus aureus pneumonia. Open Forum Infect. Dis. 2017, 4, S310. [Google Scholar] [CrossRef]
- Laventie, B.J.; Rademaker, H.J.; Saleh, M.; de Boer, E.; Janssens, R.; Bourcier, T.; Subilia, A.; Marcellin, L.; van Haperen, R.; Lebbink, J.H.; et al. Heavy chain-only antibodies and tetravalent bispecific antibody neutralizing Staphylococcus aureus leukotoxins. Proc. Natl. Acad. Sci. USA 2011, 108, 16404–16409. [Google Scholar] [CrossRef]
- Suarez-Alvarez, B.; Garcia-Suarez Mdel, M.; Mendez, F.J.; de los Toyos, J.R. Characterisation of mouse monoclonal antibodies for pneumolysin: Fine epitope mapping and V gene usage. Immunol. Lett. 2003, 88, 227–239. [Google Scholar] [CrossRef]
- Garcia-Suarez Mdel, M.; Cima-Cabal, M.D.; Florez, N.; Garcia, P.; Cernuda-Cernuda, R.; Astudillo, A.; Vazquez, F.; De los Toyos, J.R.; Mendez, F.J. Protection against pneumococcal pneumonia in mice by monoclonal antibodies to pneumolysin. Infect. Immun. 2004, 72, 4534–4540. [Google Scholar] [CrossRef]
- Nakouzi, A.; Rivera, J.; Rest, R.F.; Casadevall, A. Passive administration of monoclonal antibodies to anthrolysin O prolong survival in mice lethally infected with Bacillus anthracis. BMC Microbiol. 2008, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Bokori-Brown, M.; Hall, C.A.; Vance, C.; Fernandes da Costa, S.P.; Savva, C.G.; Naylor, C.E.; Cole, A.R.; Basak, A.K.; Moss, D.S.; Titball, R.W. Clostridium perfringens epsilon toxin mutant Y30A-Y196A as a recombinant vaccine candidate against enterotoxemia. Vaccine 2014, 32, 2682–2687. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Wang, D.; Xiang, H.; Feng, H.; Jiang, Y.; Xia, L.; Dong, J.; Lu, J.; Yu, L.; Deng, X. Subinhibitory concentrations of thymol reduce enterotoxins A and B and alpha-hemolysin production in Staphylococcus aureus isolates. PLoS ONE 2010, 5, e9736. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Stapleton, P.D.; Taylor, P.W. The polyphenol (-)-epicatechin gallate disrupts the secretion of virulence-related proteins by Staphylococcus aureus. Lett. Appl. Microbiol. 2008, 46, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Mooyottu, S.; Yin, H.; Nair, M.S.; Bhattaram, V.; Venkitanarayanan, K. Inhibiting microbial toxins using plant-derived compounds and plant extracts. Medicines 2015, 2, 186–211. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Niu, X.; Dong, J.; Wang, D.; Wang, J.; Li, H.; Luo, M.; Li, S.; Feng, H.; Deng, X. Baicalin protects mice from Staphylococcus aureus pneumonia via inhibition of the cytolytic activity of alpha-hemolysin. J. Infect. Dis. 2012, 206, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Lee, J.H.; Cho, M.H.; Lee, J. Red wines and flavonoids diminish Staphylococcus aureus virulence with anti-biofilm and anti-hemolytic activities. Biofouling 2015, 31, 1–11. [Google Scholar] [CrossRef]
- Dong, J.; Qiu, J.; Zhang, Y.; Lu, C.; Dai, X.; Wang, J.; Li, H.; Wang, X.; Tan, W.; Luo, M.; et al. Oroxylin A inhibits hemolysis via hindering the self-assembly of alpha-hemolysin heptameric transmembrane pore. PLoS Comput. Biol. 2013, 9, e1002869. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, X.; Liu, S.; Li, G.; Shi, L.; Dong, J.; Li, W.; Deng, X.; Niu, X. Morin hydrate attenuates Staphylococcus aureus virulence by inhibiting the self-assembly of alpha-hemolysin. J. Appl. Microbiol. 2015, 118, 753–763. [Google Scholar] [CrossRef]
- Rani, N.; Saravanan, V.; Lakshmi, P.T.V.; Annamalai, A. Inhibition of Pore Formation by Blocking the Assembly of Staphylococcus aureus α-hemolysin through a novel peptide inhibitor: An in silco approach. Int. J. Peptide Res. Therapeut. 2014, 20, 575–583. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, B.; Liu, S.; Wang, L.; Wang, J. Anticytotoxin effects of amentoflavone to pneumolysin. Biol. Pharm. Bull. 2017, 40, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, H.; Wang, J.; Guo, Y.; Liu, B.; Deng, X.; Niu, X. Verbascoside alleviates pneumococcal pneumonia by reducing pneumolysin oligomers. Mol. Pharmacol. 2016, 89, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Li, L.; Li, M.; Cha, Y.; Deng, X.; Wang, J. Apigenin protects mice from pneumococcal pneumonia by inhibiting the cytolytic activity of pneumolysin. Fitoterapia 2016, 115, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, X.; Wang, J.; Dong, Y.; Meng, S.; Li, R.; Niu, X.; Deng, X. beta-sitosterol interacts with pneumolysin to prevent Streptococcus pneumoniae infection. Sci. Rep. 2015, 5, 17668. [Google Scholar] [CrossRef] [PubMed]
- Arzanlou, M.; Bohlooli, S. Inhibition of streptolysin O by allicin—An active component of garlic. J. Med. Microbiol. 2010, 59, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Shewell, L.K.; Harvey, R.M.; Higgins, M.A.; Day, C.J.; Hartley-Tassell, L.E.; Chen, A.Y.; Gillen, C.M.; James, D.B.; Alonzo, F., 3rd; Torres, V.J.; et al. The cholesterol-dependent cytolysins pneumolysin and streptolysin O require binding to red blood cell glycans for hemolytic activity. Proc. Natl. Acad. Sci. USA 2014, 111, E5312–E5320. [Google Scholar] [CrossRef]
- Wang, J.; Qiu, J.; Tan, W.; Zhang, Y.; Wang, H.; Zhou, X.; Liu, S.; Feng, H.; Li, W.; Niu, X.; et al. Fisetin inhibits Listeria monocytogenes virulence by interfering with the oligomerization of listeriolysin O. J. Infect. Dis. 2015, 211, 1376–1387. [Google Scholar] [CrossRef]
- Ghafari, S.; Komeilian, M.; Hashemi, M.S.; Oushani, S.; Rigi, G.; Rashidieh, B.; Yarahmadi, K.; Khoddam, F. Molecular docking based screening of Listeriolysin-O for improved inhibitors. Bioinformation 2017, 13, 160–163. [Google Scholar] [CrossRef]
- Bitounis, D.; Fanciullino, R.; Iliadis, A.; Ciccolini, J. Optimizing druggability through liposomal formulations: New approaches to an old concept. ISRN Pharm. 2012, 2012, 738432. [Google Scholar] [CrossRef]
- Kshirsagar, N.A.; Pandya, S.K.; Kirodian, G.B.; Sanath, S. Liposomal drug delivery system from laboratory to clinic. J. Postgrad. Med. 2005, 51 (Suppl. S1), S5–S15. [Google Scholar]
- Henry, B.D.; Neill, D.R.; Becker, K.A.; Gore, S.; Bricio-Moreno, L.; Ziobro, R.; Edwards, M.J.; Muhlemann, K.; Steinmann, J.; Kleuser, B.; et al. Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat. Biotechnol. 2015, 33, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.W.; Chambers, E.; Mitragotri, S. Factors that control the circulation time of nanoparticles in blood: Challenges, solutions and future prospects. Curr. Pharm. Des. 2010, 16, 2298–2307. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. Engl. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.M.; Fang, R.H.; Copp, J.; Luk, B.T.; Zhang, L. A biomimetic nanosponge that absorbs pore-forming toxins. Nat. Nanotechnol. 2013, 8, 336–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escajadillo, T.; Olson, J.; Luk, B.T.; Zhang, L.; Nizet, V. A red blood cell membrane-camouflaged nanoparticle counteracts streptolysin O-mediated virulence phenotypes of invasive group A Streptococcus. Front. Pharmacol. 2017, 8, 477. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, W.; Chen, Y.; Escajadillo, T.; Ungerleider, J.; Fang, R.H.; Christman, K.; Nizet, V.; Zhang, L. Self-assembled colloidal gel using cell membrane-coated nanosponges as building blocks. ACS Nano 2017, 11, 11923–11930. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, M.; Zhang, Y.; Lee, J.H.; Escajadillo, T.; Gong, H.; Fang, R.H.; Gao, W.; Nizet, V.; Zhang, L. Broad-spectrum neutralization of pore-forming toxins with human erythrocyte membrane-coated nanosponges. Adv. Healthc. Mater. 2018, 7, e1701366. [Google Scholar] [CrossRef]
- Wilke, G.A.; Bubeck Wardenburg, J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef]
- Ludwig, A.; Hundhausen, C.; Lambert, M.H.; Broadway, N.; Andrews, R.C.; Bickett, D.M.; Leesnitzer, M.A.; Becherer, J.D. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb. Chem. High Throughput Screen 2005, 8, 161–171. [Google Scholar] [CrossRef]
- Inoshima, N.; Wang, Y.; Bubeck Wardenburg, J. Genetic requirement for ADAM10 in severe Staphylococcus aureus skin infection. J. Investig. Dermatol. 2012, 132, 1513–1516. [Google Scholar] [CrossRef]
- Sampedro, G.R.; DeDent, A.C.; Becker, R.E.; Berube, B.J.; Gebhardt, M.J.; Cao, H.; Bubeck Wardenburg, J. Targeting Staphylococcus aureus alpha-toxin as a novel approach to reduce severity of recurrent skin and soft-tissue infections. J. Infect. Dis. 2014, 210, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F., 3rd; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; DuMont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [CrossRef]
- Reyes-Robles, T.; Alonzo, F., 3rd; Kozhaya, L.; Lacy, D.B.; Unutmaz, D.; Torres, V.J. Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 2013, 14, 453–459. [Google Scholar] [CrossRef]
- Dowd, K.J.; Farrand, A.J.; Tweten, R.K. The cholesterol-dependent cytolysin signature motif: A critical element in the allosteric pathway that couples membrane binding to pore assembly. PLoS Pathog. 2012, 8, e1002787. [Google Scholar] [CrossRef]
- LaChapelle, S.; Tweten, R.K.; Hotze, E.M. Intermedilysin-receptor interactions during assembly of the pore complex: Assembly intermediates increase host cell susceptibility to complement-mediated lysis. J. Biol. Chem. 2009, 284, 12719–12726. [Google Scholar] [CrossRef]
- Johnson, S.; Brooks, N.J.; Smith, R.A.; Lea, S.M.; Bubeck, D. Structural basis for recognition of the pore-forming toxin intermedilysin by human complement receptor CD59. Cell Rep. 2013, 3, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Xie, S.; Liu, F.; Simone, L.C.; Caplan, S.; Qin, X.; Naslavsky, N. Rapid degradation of the complement regulator, CD59, by a novel inhibitor. J. Biol. Chem. 2014, 289, 12109–12125. [Google Scholar] [CrossRef]
- Wu, Q.; Guo, Z. Glycosylphosphatidylinositols are potential targets for the development of novel inhibitors for aerolysin-type of pore-forming bacterial toxins. Med. Res. Rev. 2010, 30, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Iwashita, Y.; Iwamoto, M.; Ando, S.; Iwashita, S. Effect of lipidic factors on membrane cholesterol topology–mode of binding of theta-toxin to cholesterol in liposomes. Biochim. Biophysica. Scta 1992, 1109, 81–90. [Google Scholar] [CrossRef]
- Maron, D.J.; Fazio, S.; Linton, M.F. Current perspectives on statins. Circulation 2000, 101, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Rosch, J.W.; Boyd, A.R.; Hinojosa, E.; Pestina, T.; Hu, Y.; Persons, D.A.; Orihuela, C.J.; Tuomanen, E.I. Statins protect against fulminant pneumococcal infection and cytolysin toxicity in a mouse model of sickle cell disease. J. Clin. Investig. 2010, 120, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statt, S.; Ruan, J.W.; Hung, L.Y.; Chang, C.Y.; Huang, C.T.; Lim, J.H.; Li, J.D.; Wu, R.; Kao, C.Y. Statin-conferred enhanced cellular resistance against bacterial pore-forming toxins in airway epithelial cells. Am. J. Resrir. Cell Mol. Biol. 2015, 53, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Henning-Knechtel, A.; Knechtel, J.; Magzoub, M. DNA-assisted oligomerization of pore-forming toxin monomers into precisely-controlled protein channels. Nuclear Acids Res. 2017, 45, 12057–12068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alouf, J.E. Cholesterol-binding cytolytic protein toxins. Int. J. Med. Microbiol. 2000, 290, 351–356. [Google Scholar] [CrossRef]
- Majd, S.; Yusko, E.C.; Billeh, Y.N.; Macrae, M.X.; Yang, J.; Mayer, M. Applications of biological pores in nanomedicine, sensing, and nanoelectronics. Curr. Opin. Biotechnol. 2010, 21, 439–476. [Google Scholar] [CrossRef] [Green Version]
- Bischofberger, M.; Gonzalez, M.R.; van der Goot, F.G. Membrane injury by pore-forming proteins. Curr. Opin. Cell Biol. 2009, 21, 589–595. [Google Scholar] [CrossRef]
- Bezrukov, S.M.; Nestorovich, E.M. Inhibiting bacterial toxins by channel blockage. Pathog. Dis. 2016, 74, ftv113. [Google Scholar] [CrossRef]
- Nestorovich, E.M.; Bezrukov, S.M. Obstructing toxin pathways by targeted pore blockage. Chem. Rev. 2012, 112, 6388–6430. [Google Scholar] [CrossRef]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Yannakopoulou, K.; Jicsinszky, L.; Aggelidou, C.; Mourtzis, N.; Robinson, T.M.; Yohannes, A.; Nestorovich, E.M.; Bezrukov, S.M.; Karginov, V.A. Symmetry requirements for effective blocking of pore-forming toxins: Comparative study with alpha-, beta-, and gamma-cyclodextrin derivatives. Antimicrob. Agents Chemother. 2011, 55, 3594–3597. [Google Scholar] [CrossRef]
- Karginov, V.A.; Nestorovich, E.M.; Schmidtmann, F.; Robinson, T.M.; Yohannes, A.; Fahmi, N.E.; Bezrukov, S.M.; Hecht, S.M. Inhibition of S. aureus alpha-hemolysin and B. anthracis lethal toxin by beta-cyclodextrin derivatives. Bioorg. Med. Chem. 2007, 15, 5424–5431. [Google Scholar] [CrossRef] [PubMed]
- Ragle, B.E.; Karginov, V.A.; Bubeck Wardenburg, J. Prevention and treatment of Staphylococcus aureus pneumonia with a beta-cyclodextrin derivative. Antimicrob. Agents Chemother. 2010, 54, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.C.; Teixeira, L.R.; Pol-Fachin, L.; Rodrigues, C.G. Inhibition of the hemolytic activity caused by Staphylococcus aureus alpha-hemolysin through isatin-Schiff copper(II) complexes. FEMS Microbiol. Lett. 2016, 363, fnv207. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.; Weaver, C.D.; McClain, M.S. Identification of small molecule inhibitors of Clostridium perfringens ε-Toxin cytotoxicity using a cell-based high-throughput screen. Toxins 2010, 2, 1825–1847. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.K.; O’Riordan, M.X. More than a pore: The cellular response to cholesterol-dependent cytolysins. Toxins 2013, 5, 618–636. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Brito, C.; Cabanes, D.; Sousa, S. Control of cytoskeletal dynamics during cellular responses to pore forming toxins. Commun. Integr. Biol. 2017, 10, e1349582. [Google Scholar] [CrossRef]
- McNeil, P.L.; Kirchhausen, T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 2005, 6, 499–505. [Google Scholar] [CrossRef]
- Romero, M.; Keyel, M.; Shi, G.; Bhattacharjee, P.; Roth, R.; Heuser, J.E.; Keyel, P.A. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ. 2017, 24, 798–808. [Google Scholar] [CrossRef] [Green Version]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.C.; Yokoyama, W.M.; Heuser, J.E. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; van der Goot, F.G. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 2006, 126, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Yarovinsky, T.O.; Monick, M.M.; Husmann, M.; Hunninghake, G.W. Interferons increase cell resistance to Staphylococcal alpha-toxin. Infect. Immun. 2008, 76, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Rayner, C.F.; Jackson, A.D.; Rutman, A.; Dewar, A.; Mitchell, T.J.; Andrew, P.W.; Cole, P.J.; Wilson, R. Interaction of pneumolysin-sufficient and -deficient isogenic variants of Streptococcus pneumoniae with human respiratory mucosa. Infect. Immun. 1995, 63, 442–447. [Google Scholar] [PubMed]
- Lucas, R.; Czikora, I.; Sridhar, S.; Zemskov, E.; Gorshkov, B.; Siddaramappa, U.; Oseghale, A.; Lawson, J.; Verin, A.; Rick, F.G.; et al. Mini-review: Novel therapeutic strategies to blunt actions of pneumolysin in the lungs. Toxins 2013, 5, 1244–1260. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen homeostasis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 336–361. [Google Scholar] [CrossRef]
- Bellier, A.; Chen, C.S.; Kao, C.Y.; Cinar, H.N.; Aroian, R.V. Hypoxia and the hypoxic response pathway protect against pore-forming toxins in C. elegans. PLoS Pathog. 2009, 5, e1000689. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Investig. 2005, 115, 1806–1815. [Google Scholar] [CrossRef]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef]
- Okumura, C.Y.; Hollands, A.; Tran, D.N.; Olson, J.; Dahesh, S.; von Kockritz-Blickwede, M.; Thienphrapa, W.; Corle, C.; Jeung, S.N.; Kotsakis, A.; et al. A new pharmacological agent (AKB-4924) stabilizes hypoxia inducible factor-1 (HIF-1) and increases skin innate defenses against bacterial infection. J. Mol. Med. 2012, 90, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Banks, D.J.; Bradley, K.A. SILENCE: A new forward genetic technology. Nat. Method 2007, 4, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Guimaraes, C.P.; Varadarajan, M.; Park, A.S.; Wuethrich, I.; Godarova, A.; Kotecki, M.; Cochran, B.H.; Spooner, E.; Ploegh, H.L.; et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 2009, 326, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Popov, L.M.; Marceau, C.D.; Starkl, P.M.; Lumb, J.H.; Shah, J.; Guerrera, D.; Cooper, R.L.; Merakou, C.; Bouley, D.M.; Meng, W.; et al. The adherens junctions control susceptibility to Staphylococcus aureus alpha-toxin. Proc. Natl. Acad. Sci. USA 2015, 112, 14337–14342. [Google Scholar] [CrossRef] [PubMed]
- Ivie, S.E.; Fennessey, C.M.; Sheng, J.; Rubin, D.H.; McClain, M.S. Gene-trap mutagenesis identifies mammalian genes contributing to intoxication by Clostridium perfringens epsilon-toxin. PLoS ONE 2011, 6, e17787. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Autophagy: Regulation and role in disease. Crit. Rev. Clin. Lab. Sci. 2009, 46, 210–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Mostowy, S. Autophagy and bacterial clearance: A not so clear picture. Cell Microbiol. 2013, 15, 395–402. [Google Scholar] [CrossRef]
- Mathieu, J. Interactions between autophagy and bacterial toxins: Targets for therapy? Toxins 2015, 7, 2918–2958. [Google Scholar] [CrossRef]
- Cockeran, R.; Anderson, R.; Feldman, C. Pneumolysin as a vaccine and drug target in the prevention and treatment of invasive pneumococcal disease. Arch. Immunol. Ther. Exp. 2005, 53, 189–198. [Google Scholar]
- Grijalva, C.G.; Nuorti, J.P.; Arbogast, P.G.; Martin, S.W.; Edwards, K.M.; Griffin, M.R. Decline in pneumonia admissions after routine childhood immunisation with pneumococcal conjugate vaccine in the USA: A time-series analysis. Lancet 2007, 369, 1179–1186. [Google Scholar] [CrossRef]
- Crisinel, P.A.; Chevalier, I.; Rallu, F.; Tapiero, B.; Lamarre, V.; Thibault, R.; Ovetchkine, P. Invasive pneumococcal disease after implementation of a reduced three-dose pneumococcal conjugate vaccine program: A pediatric tertiary care center experience. Eur. J. Pediatr. 2010, 169, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Leroux-Roels, G.; Maes, C.; De Boever, F.; Traskine, M.; Ruggeberg, J.U.; Borys, D. Safety, reactogenicity and immunogenicity of a novel pneumococcal protein-based vaccine in adults: A phase I/II randomized clinical study. Vaccine 2014, 32, 6838–6846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.E.; Lock, R.A.; Peeters, C.C.; Poolman, J.T.; Andrew, P.W.; Mitchell, T.J.; Hansman, D.; Paton, J.C. Immunization of mice with pneumolysin toxoid confers a significant degree of protection against at least nine serotypes of Streptococcus pneumoniae. Infect. Immun. 1994, 62, 5683–5688. [Google Scholar]
- Ogunniyi, A.D.; Woodrow, M.C.; Poolman, J.T.; Paton, J.C. Protection against Streptococcus pneumoniae elicited by immunization with pneumolysin and CbpA. Infect. Immun. 2001, 69, 5997–6003. [Google Scholar] [CrossRef] [PubMed]
- Denoel, P.; Philipp, M.T.; Doyle, L.; Martin, D.; Carletti, G.; Poolman, J.T. A protein-based pneumococcal vaccine protects rhesus macaques from pneumonia after experimental infection with Streptococcus pneumoniae. Vaccine 2011, 29, 5495–5501. [Google Scholar] [CrossRef] [PubMed]
- Prymula, R.; Pazdiora, P.; Traskine, M.; Ruggeberg, J.U.; Borys, D. Safety and immunogenicity of an investigational vaccine containing two common pneumococcal proteins in toddlers: A phase II randomized clinical trial. Vaccine 2014, 32, 3025–3034. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, L.A.; Kerr, A.R.; Douce, G.R.; Paterson, G.K.; Dilts, D.A.; Liu, D.F.; Mitchell, T.J. Construction and immunological characterization of a novel nontoxic protective pneumolysin mutant for use in future pneumococcal vaccines. Infect. Immun. 2006, 74, 586–593. [Google Scholar] [CrossRef]
- Cockeran, R.; Steel, H.C.; Theron, A.J.; Mitchell, T.J.; Feldman, C.; Anderson, R. Characterization of the interactions of the pneumolysoid, Delta6 PLY, with human neutrophils in vitro. Vaccine 2011, 29, 8780–8782. [Google Scholar] [CrossRef]
- Farrand, A.J.; LaChapelle, S.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc. Natl. Acad. Sci. USA 2010, 107, 4341–4346. [Google Scholar] [CrossRef] [Green Version]
- Mann, B.; Thornton, J.; Heath, R.; Wade, K.R.; Tweten, R.K.; Gao, G.; El Kasmi, K.; Jordan, J.B.; Mitrea, D.M.; Kriwacki, R.; et al. Broadly protective protein-based pneumococcal vaccine composed of pneumolysin toxoid-CbpA peptide recombinant fusion protein. J. Infect. Dis. 2014, 209, 1116–1125. [Google Scholar] [CrossRef]
- Adhikari, R.P.; Karauzum, H.; Sarwar, J.; Abaandou, L.; Mahmoudieh, M.; Boroun, A.R.; Vu, H.; Nguyen, T.; Devi, V.S.; Shulenin, S.; et al. Novel structurally designed vaccine for S. aureus alpha-hemolysin: Protection against bacteremia and pneumonia. PLoS ONE 2012, 7, e38567. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, R.P.; Thompson, C.D.; Aman, M.J.; Lee, J.C. Protective efficacy of a novel alpha hemolysin subunit vaccine (AT62) against Staphylococcus aureus skin and soft tissue infections. Vaccine 2016, 34, 6402–6407. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.F.; Yang, L.Y.; Feng, Q.; Lu, D.S.; Dong, Y.D.; Cai, C.Z.; Wu, Y.; Guo, Y.; Gu, J.; Zeng, H.; et al. Evaluation of the protective immunity of a novel subunit fusion vaccine in a murine model of systemic MRSA infection. PLoS ONE 2013, 8, e81212. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Flores, K.G.; Vivanco-Cid, H. Biological effects of listeriolysin O: Implications for vaccination. BioMed Res. Int. 2015, 2015, 360741. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.A.; Vivanco-Cid, H.; Unanue, E.R. Listeriolysin o is strongly immunogenic independently of its cytotoxic activity. PLoS ONE 2012, 7, e32310. [Google Scholar] [CrossRef] [PubMed]
- Michel, E.; Reich, K.A.; Favier, R.; Berche, P.; Cossart, P. Attenuated mutants of the intracellular bacterium Listeria monocytogenes obtained by single amino acid substitutions in listeriolysin O. Mol. Microbiol. 1990, 4, 2167–2178. [Google Scholar] [CrossRef] [PubMed]
- Bahey-El-Din, M.; Casey, P.G.; Griffin, B.T.; Gahan, C.G. Expression of two Listeria monocytogenes antigens (P60 and LLO) in Lactococcus lactis and examination for use as live vaccine vectors. J. Med. Microbiol. 2010, 59, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Bokori-Brown, M.; Kokkinidou, M.C.; Savva, C.G.; Fernandes da Costa, S.; Naylor, C.E.; Cole, A.R.; Moss, D.S.; Basak, A.K.; Titball, R.W. Clostridium perfringens epsilon toxin H149A mutant as a platform for receptor binding studies. Protein Sci. 2013, 22, 650–659. [Google Scholar] [CrossRef]
- Chiarot, E.; Faralla, C.; Chiappini, N.; Tuscano, G.; Falugi, F.; Gambellini, G.; Taddei, A.; Capo, S.; Cartocci, E.; Veggi, D.; et al. Targeted amino acid substitutions impair streptolysin O toxicity and group A Streptococcus virulence. mBio 2013, 4, e00387-12. [Google Scholar] [CrossRef]
- Uchiyama, S.; Dohrmann, S.; Timmer, A.M.; Dixit, N.; Ghochani, M.; Bhandari, T.; Timmer, J.C.; Sprague, K.; Bubeck-Wardenburg, J.; Simon, S.I.; et al. Streptolysin O Rapidly Impairs Neutrophil Oxidative Burst and Antibacterial Responses to Group A Streptococcus. Front. Immunol. 2015, 6, 581. [Google Scholar] [CrossRef]
- Hu, C.M.; Fang, R.H.; Luk, B.T.; Zhang, L. Nanoparticle-detained toxins for safe and effective vaccination. Nat. Nanotechnol. 2013, 8, 933–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.M.; Zhang, L. Nanotoxoid Vaccines. Nano Today 2014, 9, 401–404. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| General Mechanism | Subclassification | PFT | Therapeutic Concept or Candidate | References |
|---|---|---|---|---|
| Direct Binding and Inhibition | Passive Immunization with Monoclonal Antibodies | S. aureus α-toxin (Hla) | mAbs MEDI4893, (MedImmune), LTM14. A3 and LC10, 7B8 and 1A9. | [52,53,54,55,56,57,58,59,60,61] |
| LukS-PV, LukF-PV, γ-hemolysin C (HlgC) | AR-301 (Salvecin™) ASN-1 and ASN-2. ASN100 (Arsanis) | [62,63,64,65,66,67] | ||
| S. aureus leukotoxins | Heavy chain-only antibodies | [68] | ||
| Pneumolysin (PLY) | mAbs PLY 4 PLY 7 | [69,70] | ||
| Anthrolysin O (ALO) | mAbs 64F8 and 80C9 | [71] | ||
| Small Molecules that Bind or Inhibit Toxin Assembly | S. aureus α-toxin (Hla) | Baicalin, quercetin, trans-reservertrol, betulinic acid, orolyxins and other flavonoids, peptide IYGSKANRQTDK | [76,77,78,79,80] | |
| Pneumolysin (PLY) | β-sitosterol, apigenin, amentoflavone, verbascoside | [81,82,83,84] | ||
| Streptolysin O (SLO) | Allicin, lacto-N-neotetraose | [85,86] | ||
| Listeriolysin O (LLO) | Finestin, RD-1 | [87,88] | ||
| Decoy Capture | Hla, PLY, and potentially broad-spectrum | (Ch:Sm) liposomes, (Ch:Sm+Sm) liposomes, CAL02 (Combioxin SA) | [91] | |
| HLa, SLO, and potentially broad spectrum | Biomimetic RBC-coated nanoparticles | [95,96,97,98,99] | ||
| Inhibition of Host Cell Receptors or Uptake Mechanisms | Small Molecules, Drug Repurposing, Peptides and Antibodies | S. aureus α-toxin (Hla) | GI254023X (ADAM10 inhibitor) | [101,102,103] |
| Intermediolysin (ILY) | ILY peptide, rILYd4 | [108,109] | ||
| S. aureus LeukED | Maraviroc (CCR5 agonist) | [104,105] | ||
| Pneumolysin (PLY) | Anti-LeX/sLeX antibodies | [86] | ||
| PLY, SLO, tetanolysin | Simvastatin | [113,114] | ||
| Blockade of Pore Formation | Small Molecules | S. aureus α-toxin (Hla) | β-cyclodextrins, isatin-Schiff base copper (II) complex | [122,123,124,125] |
| Clostridium perfringens Etx | Specific quinoline and isoxazol compounds | [126] | ||
| Increase Host Cell Resiliency Against PFT | Stimulate Membrane Repair Pathways | Pneumolysin (PLY) | AP301 TNFα-derived TIP peptide, JI-34 GHRH agonist | [134,135] |
| S. aureus α-toxin (Hla) | IFN-α (increase in lipid metabolism) | [133] | ||
| S. aureus α-toxin (Hla)? | HIF-1 stabilizing PHD inhibitor, AKB4923 | [137,140] | ||
| Active Immunization | Inactivated PFT (Toxoid) Vaccines | Pneumolysin (PLY) | Toxoid dPly, Δ6 PLY, peptide–L460D “pneumolysoid” | [153,154,155,156,157,158,159,160] |
| S. aureus α-toxin (Hla) | HlaH35L, AT62, chimeric bivalent IsdB/Hla | [52,53] [161,162,163] | ||
| Hla, possibly broad-spectrum | RBC “nanotoxoids” with absorbed PFTs | [171,172] | ||
| S. aureus LukS-PV, LukF-PV | LukS-Mut9/LukF-Mut1 | [64] | ||
| Streptolysin O (SLO) | Inactivated W535A toxoid | [169,170] | ||
| Clostridium perfringens Etx | Y30A-Y196A toxoid | [72,168] | ||
| Listeriolysin O (LLO) | LLO W492A, LLO W491-492A toxoid | [164,165,166] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escajadillo, T.; Nizet, V. Pharmacological Targeting of Pore-Forming Toxins as Adjunctive Therapy for Invasive Bacterial Infection. Toxins 2018, 10, 542. https://doi.org/10.3390/toxins10120542
Escajadillo T, Nizet V. Pharmacological Targeting of Pore-Forming Toxins as Adjunctive Therapy for Invasive Bacterial Infection. Toxins. 2018; 10(12):542. https://doi.org/10.3390/toxins10120542
Chicago/Turabian StyleEscajadillo, Tamara, and Victor Nizet. 2018. "Pharmacological Targeting of Pore-Forming Toxins as Adjunctive Therapy for Invasive Bacterial Infection" Toxins 10, no. 12: 542. https://doi.org/10.3390/toxins10120542
APA StyleEscajadillo, T., & Nizet, V. (2018). Pharmacological Targeting of Pore-Forming Toxins as Adjunctive Therapy for Invasive Bacterial Infection. Toxins, 10(12), 542. https://doi.org/10.3390/toxins10120542