Domain II of Pseudomonas Exotoxin Is Critical for Efficacy of Bolus Doses in a Xenograft Model of Acute Lymphoblastic Leukemia



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
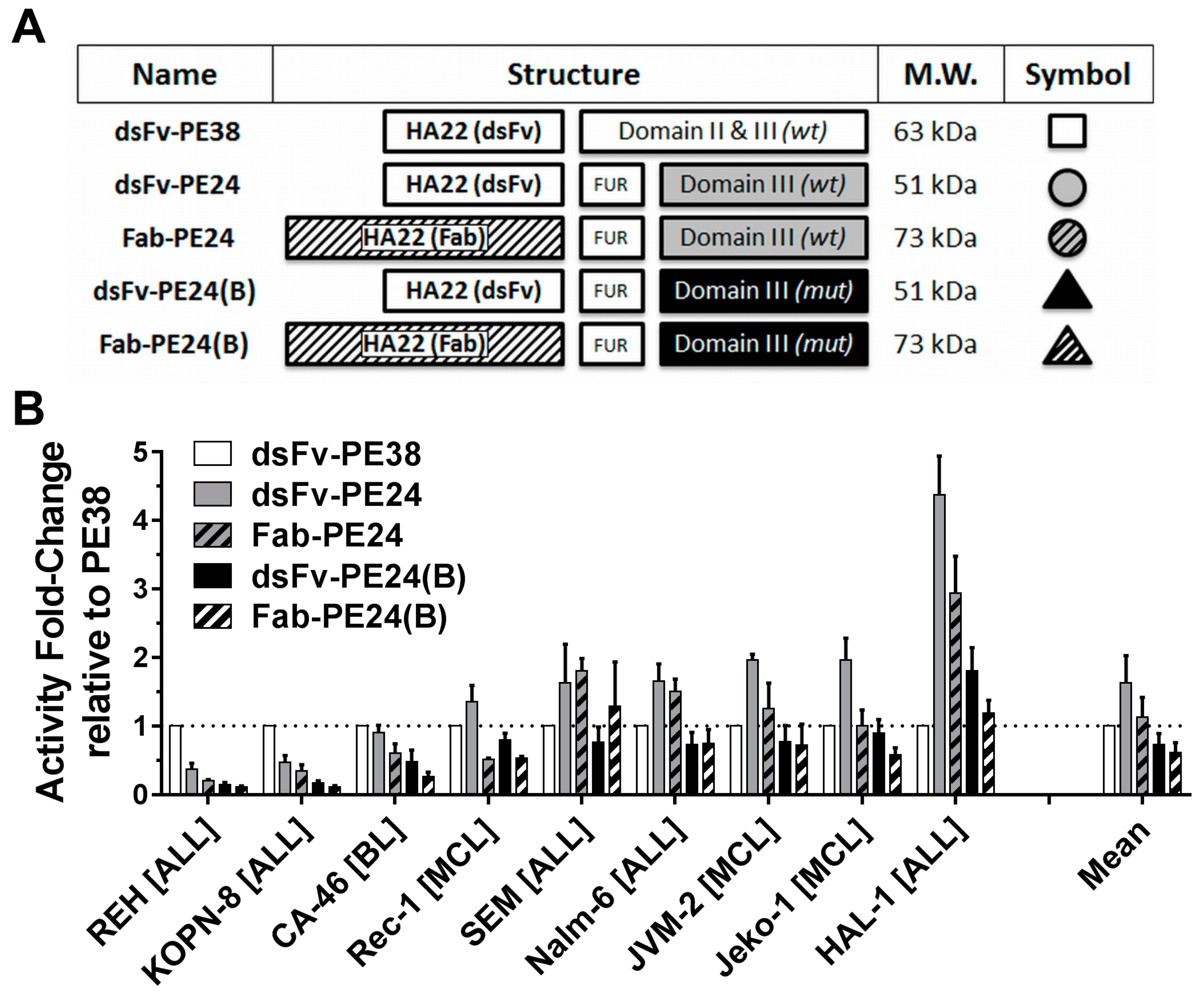
2.1. Wild-Type PE24 Immunotoxin Shows Highest Overall Activity
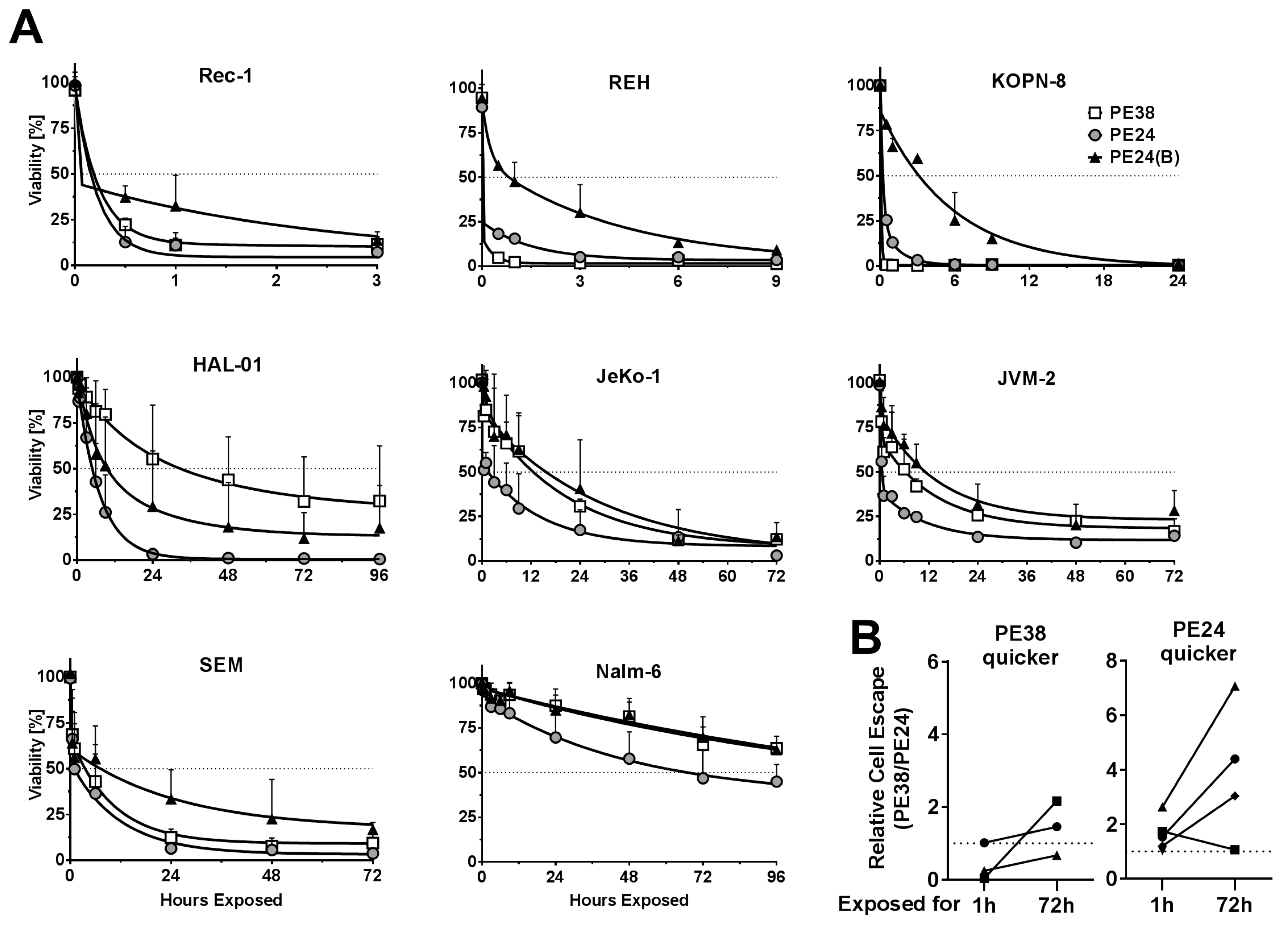
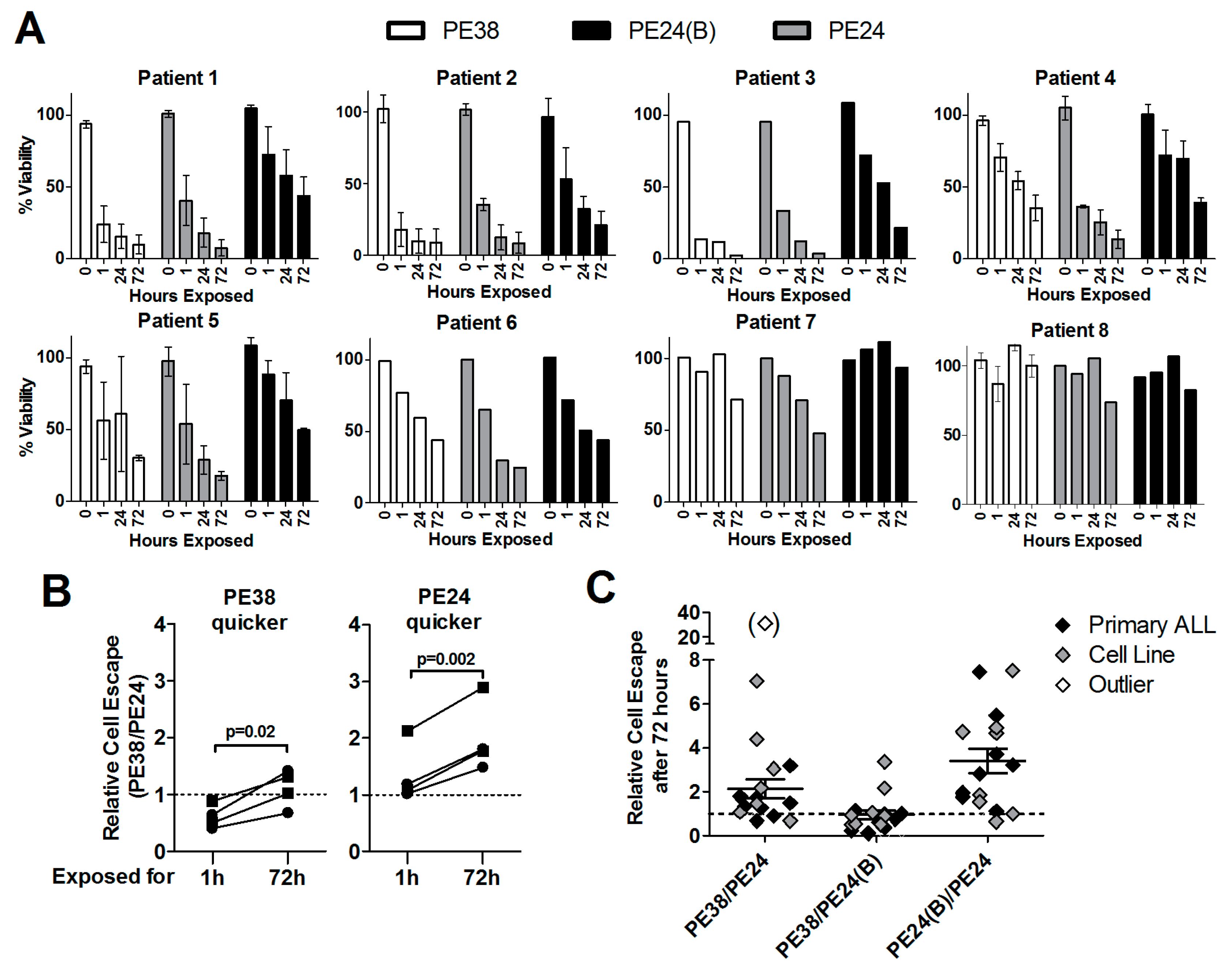
2.2. Immunotoxin Variants Show Highly Variable Time to Reach Maximal Cytotoxicity
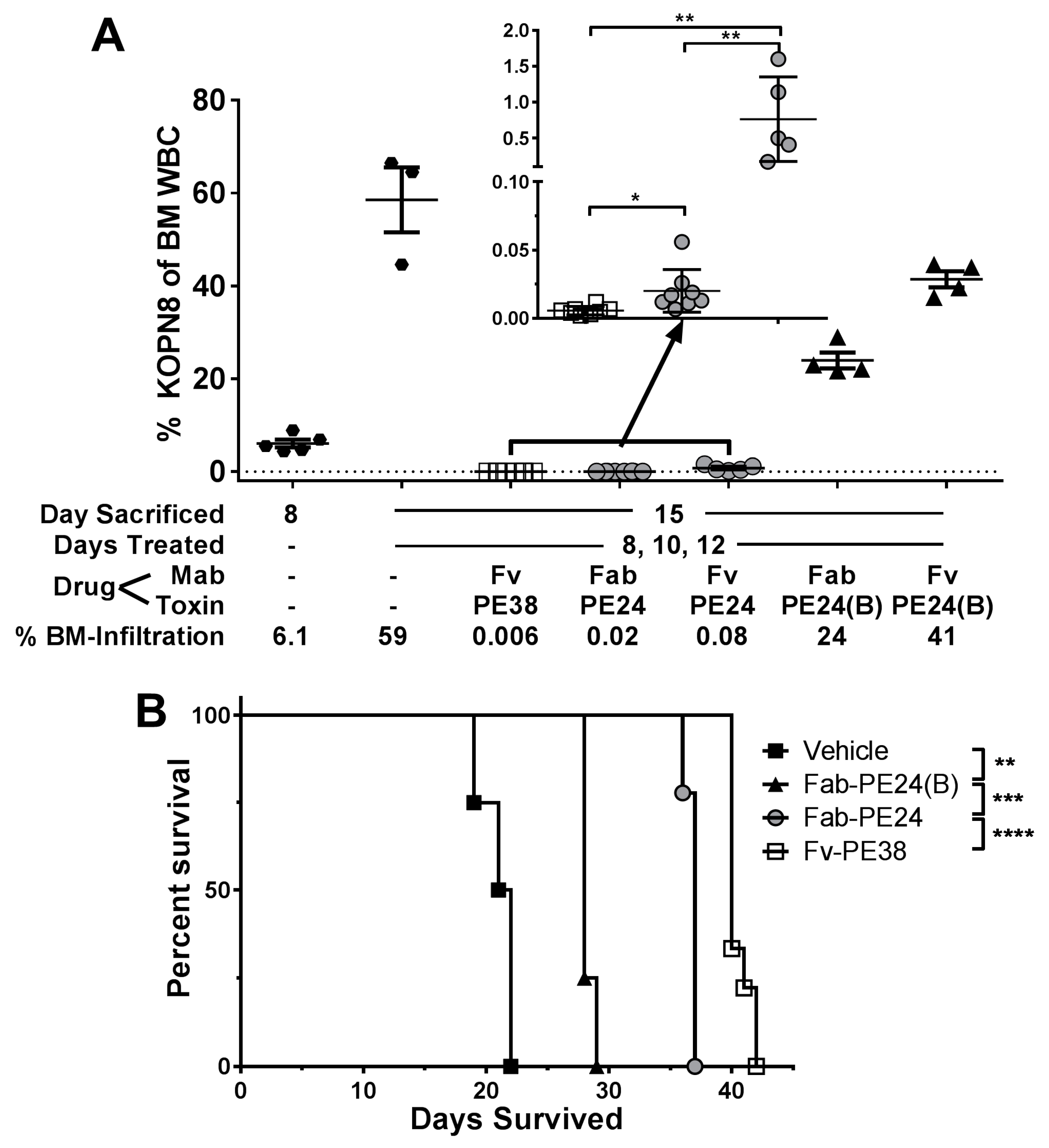
2.3. In Vivo Efficacy Emphasizes Importance of Domain II against KOPN-8
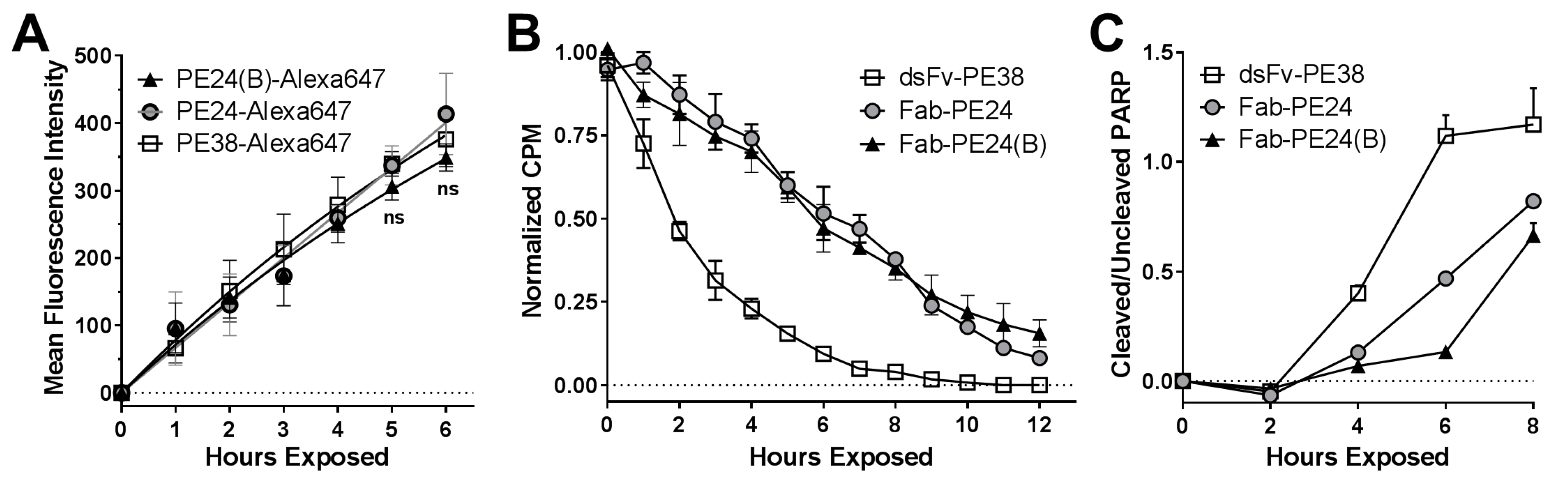
2.4. Biochemical Events Reflect Differences in Immunotoxin Activity
2.5. Lack of Domain II Is Advantageous against Primary ALL Blasts
3. Discussion
3.1. How Domain II Influences Immunotoxin Efficacy
3.2. Efficacy In Vivo Is Influenced by Needed Exposure Time and Half-Life
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines
4.3. Cell Assays
4.4. Patient Samples
4.5. Animal Studies
4.6. Statistics
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Wurch, T.; Bailly, C.; Corvaia, N. Strategies and challenges for the next generation of therapeutic antibodies. Nat. Rev. Immunol. 2010, 10, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, R.M.; Goldenberg, D.M. Cancer radioimmunotherapy. Immunotherapy 2011, 3, 349–370. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Reisfeld, R.A.; Becker, J.C. Antibody targeted drugs as cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, D.J.; Wayne, A.S.; Kreitman, R.J.; Pastan, I. Treatment of hematologic malignancies with immunotoxins and antibody-drug conjugates. Cancer Res. 2011, 71, 6300–6309. [Google Scholar] [CrossRef] [PubMed]
- Wayne, A.S.; Fitzgerald, D.J.; Kreitman, R.J.; Pastan, I. Immunotoxins for leukemia. Blood 2014, 123, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Tallman, M.S.; Robak, T.; Coutre, S.; Wilson, W.H.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Lechleider, R.; Pastan, I. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J. Clin. Oncol. 2012, 30, 1822–1828. [Google Scholar] [CrossRef] [PubMed]
- Wayne, A.S.; Shah, N.N.; Bhojwani, D.; Silverman, L.B.; Whitlock, J.A.; Stetler-Stevenson, M.; Sun, W.; Liang, M.; Yang, J.; Kreitman, R.J.; et al. Phase I study of the anti-CD22 immunotoxin moxetumomab pasudotox for childhood acute lymphoblastic leukemia. Blood 2017, 130, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Sharon, E.; Thomas, A.; Zhang, J.; Ling, A.; Miettinen, M.; Kreitman, R.J.; Steinberg, S.M.; Hollevoet, K.; Pastan, I. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen 125. Cancer 2014, 120, 3311–3319. [Google Scholar] [PubMed]
- Mazor, R.; Vassall, A.N.; Eberle, J.A.; Beers, R.; Weldon, J.E.; Venzon, D.J.; Tsang, K.Y.; Benhar, I.; Pastan, I. Identification and elimination of an immunodominant t-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin a. Proc. Natl. Acad. Sci. USA 2012, 109, E3597–E3603. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Xiang, L.; Chertov, O.; Margulies, I.; Kreitman, R.J.; FitzGerald, D.J.; Pastan, I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 2009, 113, 3792–3800. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Beers, R.; Xiang, L.; Lee, B.; Weldon, J.E.; Kreitman, R.J.; Pastan, I. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc. Natl. Acad. Sci. USA 2011, 108, 5742–5747. [Google Scholar] [CrossRef] [PubMed]
- Bera, T.K.; Onda, M.; Kreitman, R.J.; Pastan, I. An improved recombinant Fab-immunotoxin targeting CD22 expressing malignancies. Leuk. Res. 2014, 38, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.; Cunningham, T.; Liu, X.F.; Wayne, A.S.; Pastan, I. Wide variability in the time required for immunotoxins to kill B lineage acute lymphoblastic leukemia cells: Implications for trial design. Clin. Cancer Res. 2016, 22, 4913–4922. [Google Scholar] [CrossRef] [PubMed]
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Pastan, I. Accumulation of a recombinant immunotoxin in a tumor in vivo: Fewer than 1000 molecules per cell are sufficient for complete responses. Cancer Res. 1998, 58, 968–975. [Google Scholar] [PubMed]
- Johannes, L.; Popoff, V. Tracing the retrograde route in protein trafficking. Cell 2008, 135, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. Multiple routes of protein transport from endosomes to the trans golgi network. FEBS Lett. 2009, 583, 3811–3816. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.; Martin-Belmonte, F. Cargo sorting in the endocytic pathway: A key regulator of cell polarity and tissue dynamics. Cold Spring Harb. Perspect. Biol. 2014, 6, a016899. [Google Scholar] [CrossRef] [PubMed]
- Chiron, M.F.; Fryling, C.M.; FitzGerald, D. Furin-mediated cleavage of Pseudomonas exotoxin-derived chimeric toxins. J. Biol. Chem. 1997, 272, 31707–31711. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.; Lee, F.; Onda, M.; Kolyvas, E.; Bhardwaj, G.; Baker, D.; Pastan, I. Protection of the furin cleavage site in low-toxicity immunotoxins based on Pseudomonas exotoxin A. Toxins (Basel) 2016, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Kaplan, G.; Park, D.; Jang, Y.; Lee, F.; Kreitman, R.; Pastan, I. Rational design of low immunogenic anti CD25 recombinant immunotoxin for T cell malignancies by elimination of T cell epitopes in PE38. Cell. Immunol. 2017, 313, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Xiang, L.; Zhang, J.; Beers, R.; Walker, D.A.; Onda, M.; Hassan, R.; Pastan, I. A recombinant immunotoxin against the tumor-associated antigen mesothelin reengineered for high activity, low off-target toxicity, and reduced antigenicity. Mol. Cancer Ther. 2013, 12, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab gtpases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Moreau, D.; Kumar, P.; Wang, S.C.; Chaumet, A.; Chew, S.Y.; Chevalley, H.; Bard, F. Genome-wide RNAi screens identify genes required for ricin and PE intoxications. Dev. Cell 2011, 21, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Pasetto, M.; Antignani, A.; Ormanoglu, P.; Buehler, E.; Guha, R.; Pastan, I.; Martin, S.E.; FitzGerald, D.J. Whole-genome RNAi screen highlights components of the endoplasmic reticulum/Golgi as a source of resistance to immunotoxin-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, E1135–E1142. [Google Scholar] [CrossRef] [PubMed]
- Verdurmen, W.P.R.; Mazlami, M.; Pluckthun, A. A quantitative comparison of cytosolic delivery via different protein uptake systems. Sci. Rep. 2017, 7, 13194. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Beers, R.; Fitzgerald, D.J.; Pastan, I. Differential cellular internalization of anti-CD19 and -CD22 immunotoxins results in different cytotoxic activity. Cancer Res. 2008, 68, 6300–6305. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.K.; Tian, H.; Paulson, J.C. CD22 is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B cells. J. Immunol. 2011, 186, 1554–1563. [Google Scholar] [CrossRef] [PubMed]
- Michalska, M.; Wolf, P. Pseudomonas exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015, 6, 963. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Kantarjian, H.; Jabbour, E.; Cortes, J.E.; Thomas, D.A.; Rytting, M.E.; Daver, N.; Alvarado, Y.; Konopleva, M.; Kebriaei, P.; et al. A phase I study of moxetumomab pasudotox in adults with relapsed or refractory B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Squires, D.R.; Stetler-Stevenson, M.; Noel, P.; FitzGerald, D.J.; Wilson, W.H.; Pastan, I. Phase I trial of recombinant immunotoxin RFB4(dsfv)-PE38 (BL22) in patients with B-cell malignancies. J. Clin. Oncol. 2005, 23, 6719–6729. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.; Stookey, S.; Cunningham, T.; Pastan, I. Paclitaxel synergizes with exposure time adjusted CD22-targeting immunotoxins against B-cell malignancies. Oncotarget 2017, 8, 30644–30655. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Bera, T.K.; Liu, X.F.; Zhou, Q.; Onda, M.; Ho, M.; Tai, C.H.; Pastan, I. Recombinant immunotoxins with albumin-binding domains have long half-lives and high antitumor activity. Proc. Natl. Acad. Sci. USA 2018, 115, E3501–E3508. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Factors determining antibody distribution in tumors. Trends Pharmacol. Sci. 2008, 29, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, G.; Beers, R.; Margulies, I.; Kreitman, R.J.; Pastan, I. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin. Cancer Res. 2002, 8, 995–1002. [Google Scholar] [PubMed]
- Pastan, I.; Beers, R.; Bera, T.K. Recombinant immunotoxins in the treatment of cancer. Methods Mol. Biol. 2004, 248, 503–518. [Google Scholar] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, F.; Cunningham, T.; Beers, R.; Bera, T.K.; Wayne, A.S.; Pastan, I. Domain II of Pseudomonas Exotoxin Is Critical for Efficacy of Bolus Doses in a Xenograft Model of Acute Lymphoblastic Leukemia. Toxins 2018, 10, 210. https://doi.org/10.3390/toxins10050210
Müller F, Cunningham T, Beers R, Bera TK, Wayne AS, Pastan I. Domain II of Pseudomonas Exotoxin Is Critical for Efficacy of Bolus Doses in a Xenograft Model of Acute Lymphoblastic Leukemia. Toxins. 2018; 10(5):210. https://doi.org/10.3390/toxins10050210
Chicago/Turabian StyleMüller, Fabian, Tyler Cunningham, Richard Beers, Tapan K. Bera, Alan S. Wayne, and Ira Pastan. 2018. "Domain II of Pseudomonas Exotoxin Is Critical for Efficacy of Bolus Doses in a Xenograft Model of Acute Lymphoblastic Leukemia" Toxins 10, no. 5: 210. https://doi.org/10.3390/toxins10050210
APA StyleMüller, F., Cunningham, T., Beers, R., Bera, T. K., Wayne, A. S., & Pastan, I. (2018). Domain II of Pseudomonas Exotoxin Is Critical for Efficacy of Bolus Doses in a Xenograft Model of Acute Lymphoblastic Leukemia. Toxins, 10(5), 210. https://doi.org/10.3390/toxins10050210