Variations of Bacterial Community Composition and Functions in an Estuary Reservoir during Spring and Summer Alternation



Abstract
:1. Introduction
2. Results
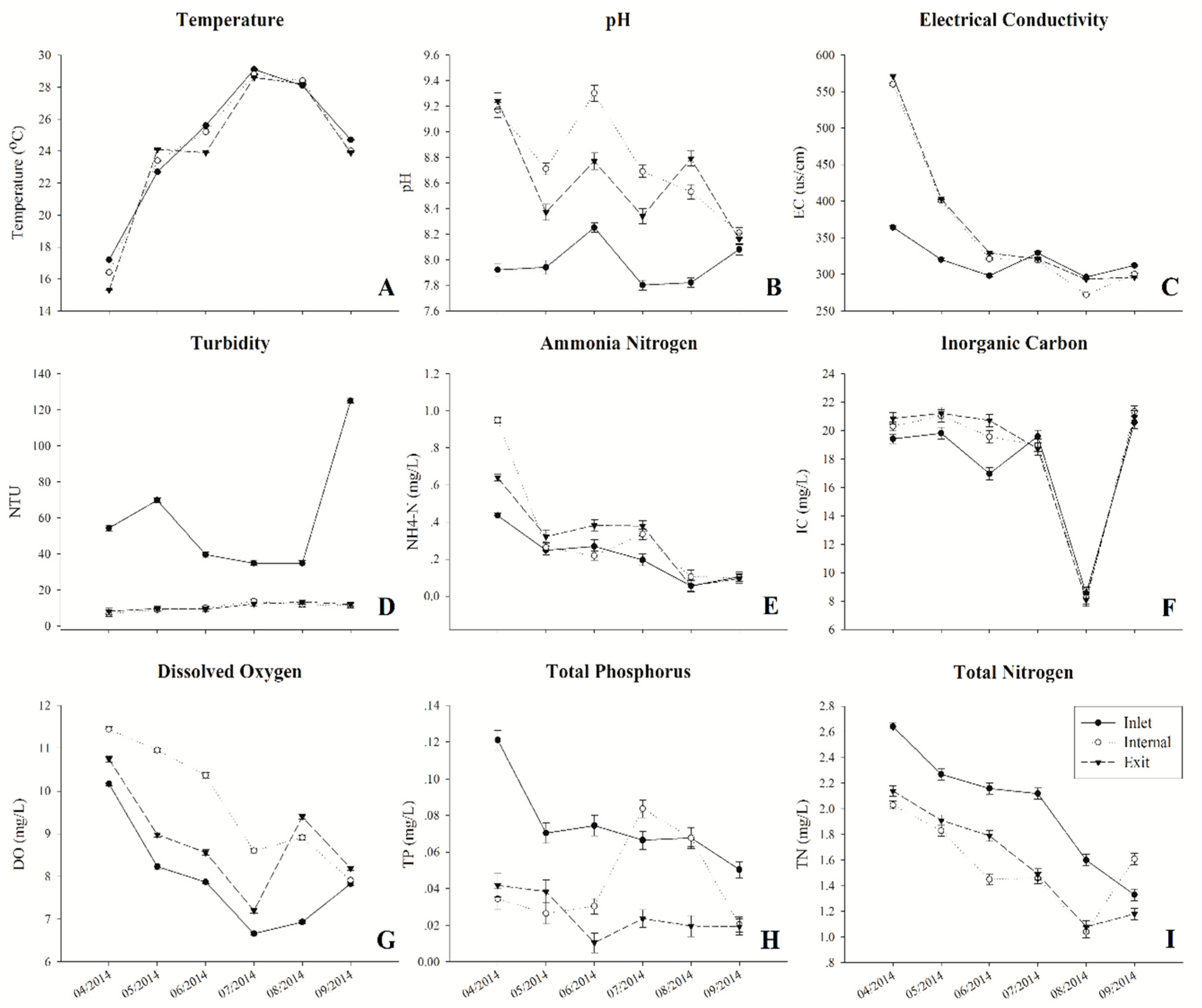
2.1. Physico-Chemical Parameters and Environmental Factor in QCS Reservoir
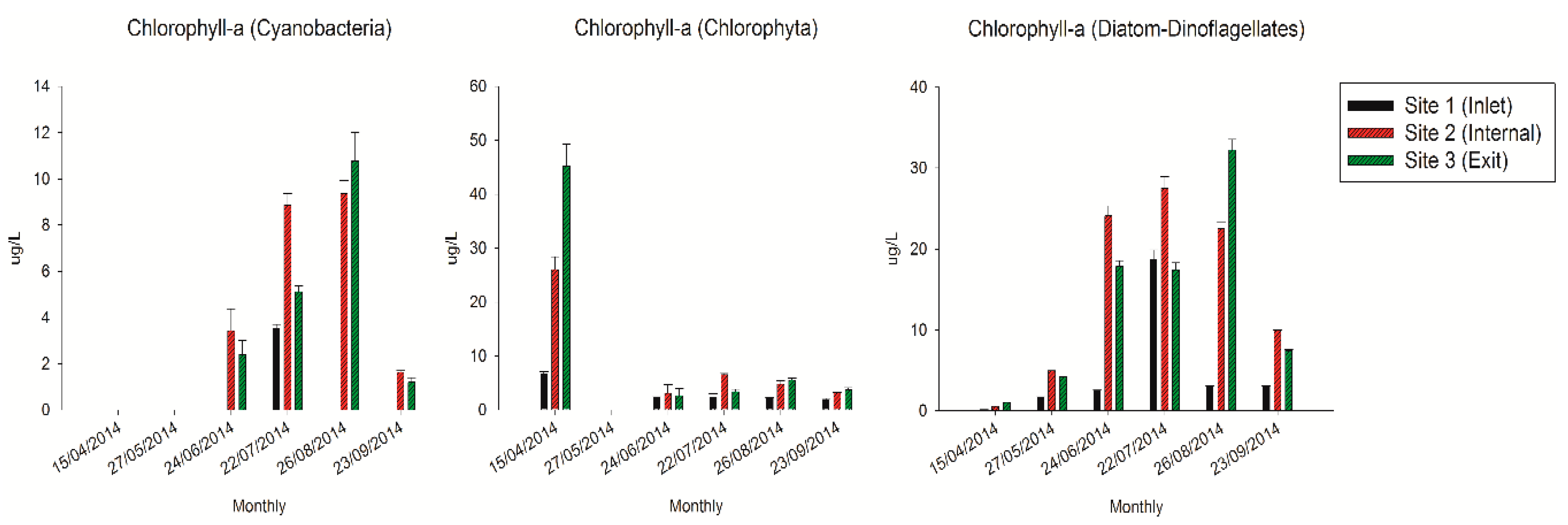
2.2. The Variations of Chlorophyll-α Concentrations in the QCS Reservoir
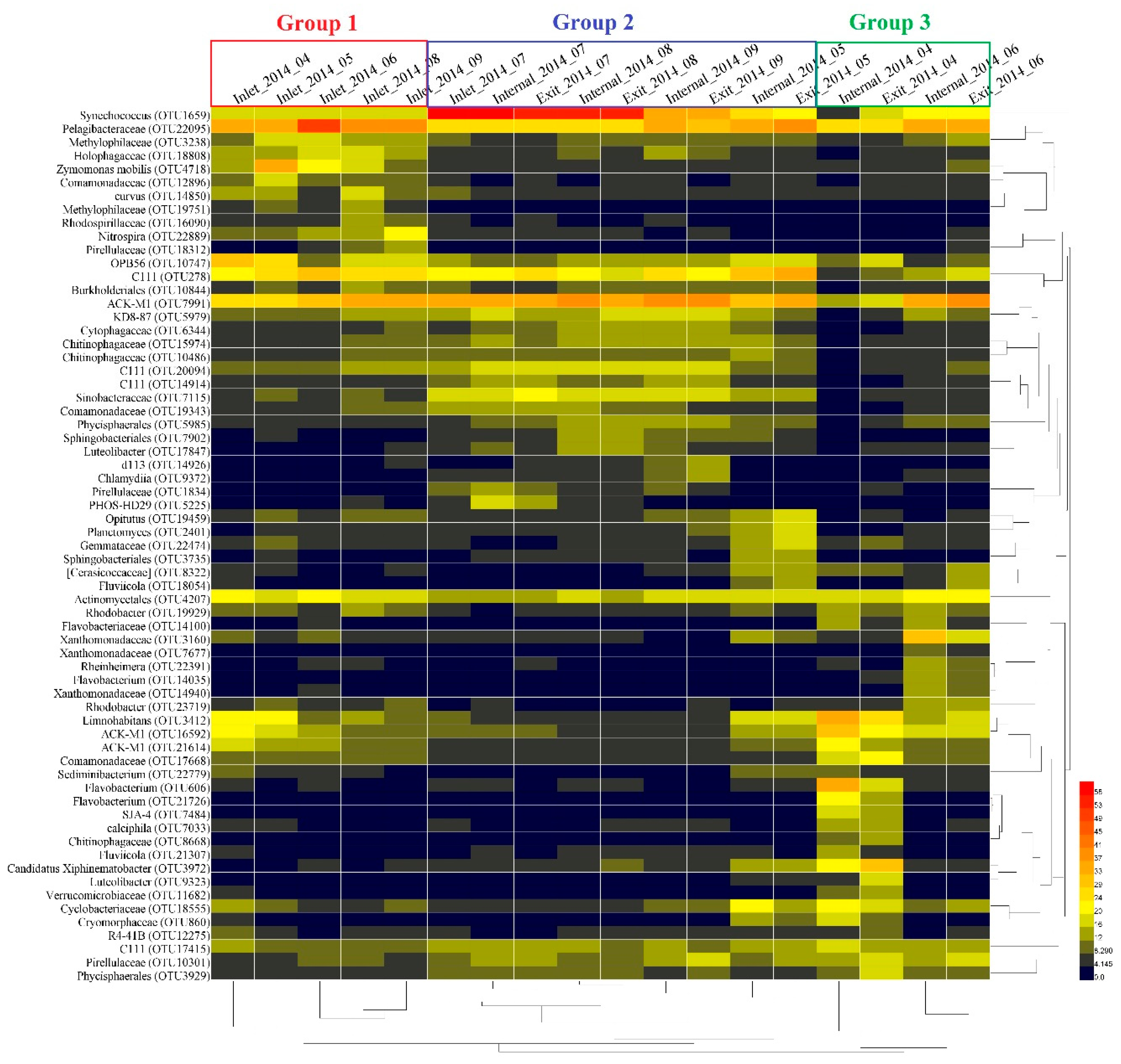
2.3. Dynamic Analysis of Bacterial Community Composition based on the 16S rRNA Sequencing Data
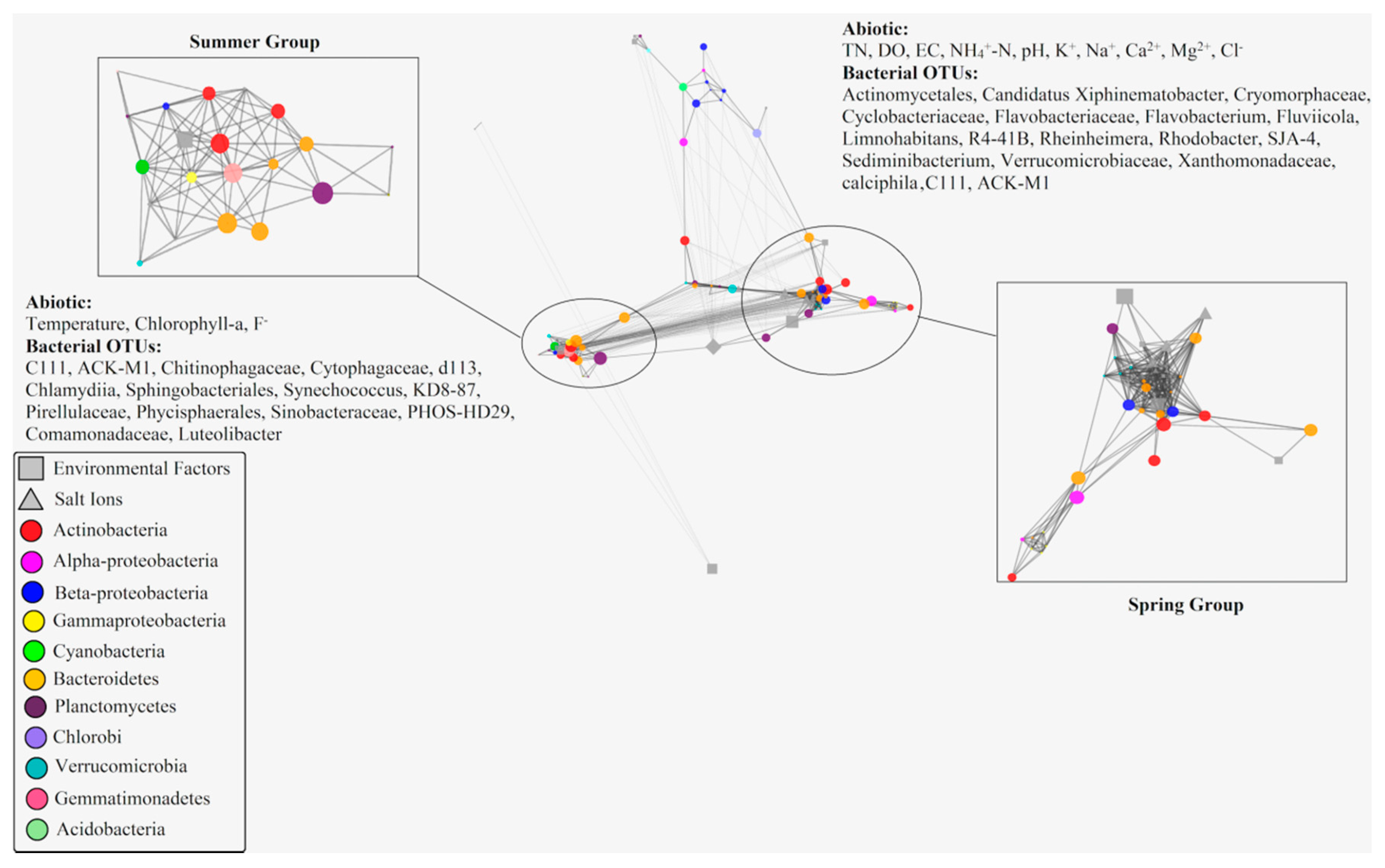
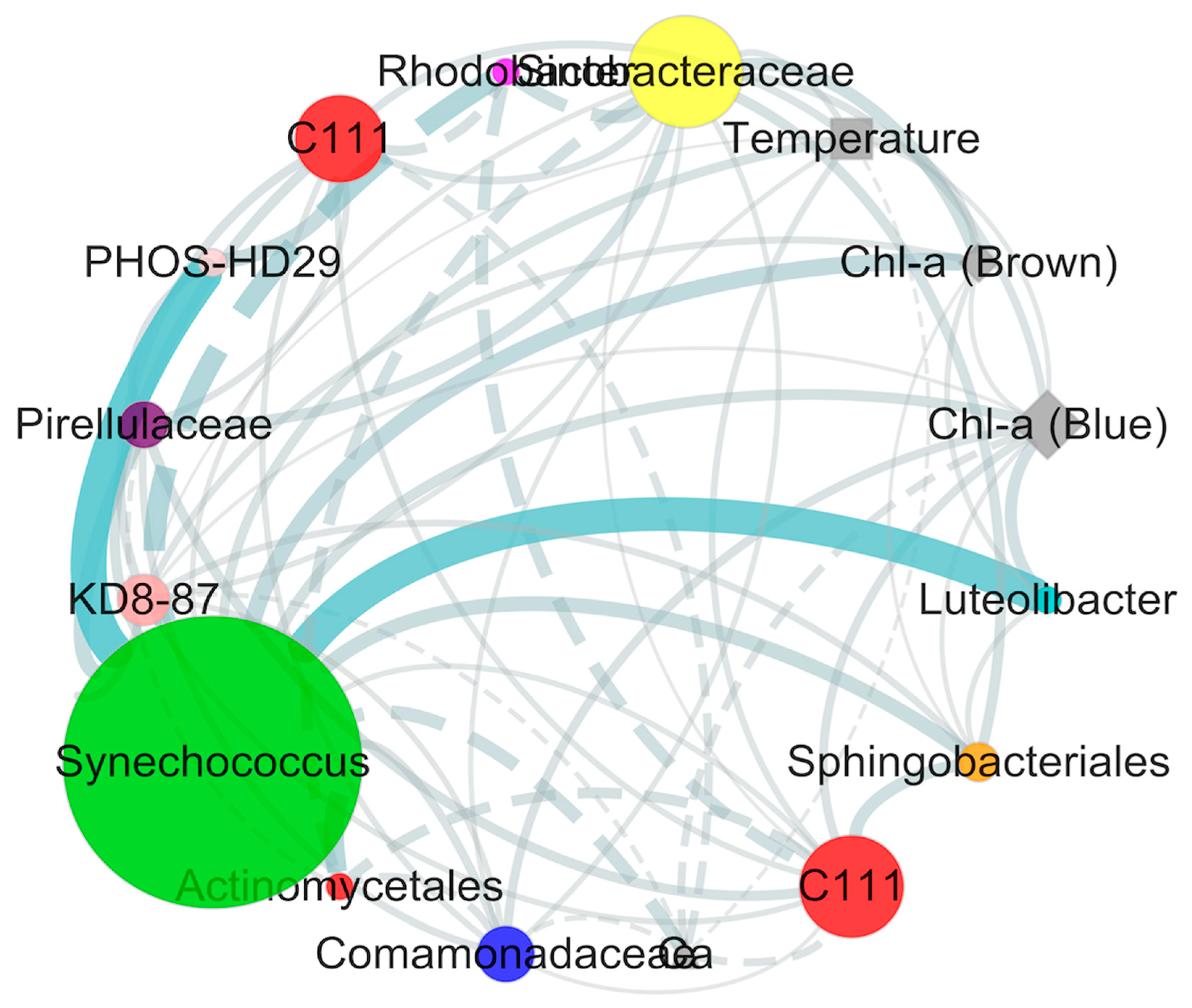
2.4. Covariance Analysis of Bacterial Community Composition and Environmental Variables
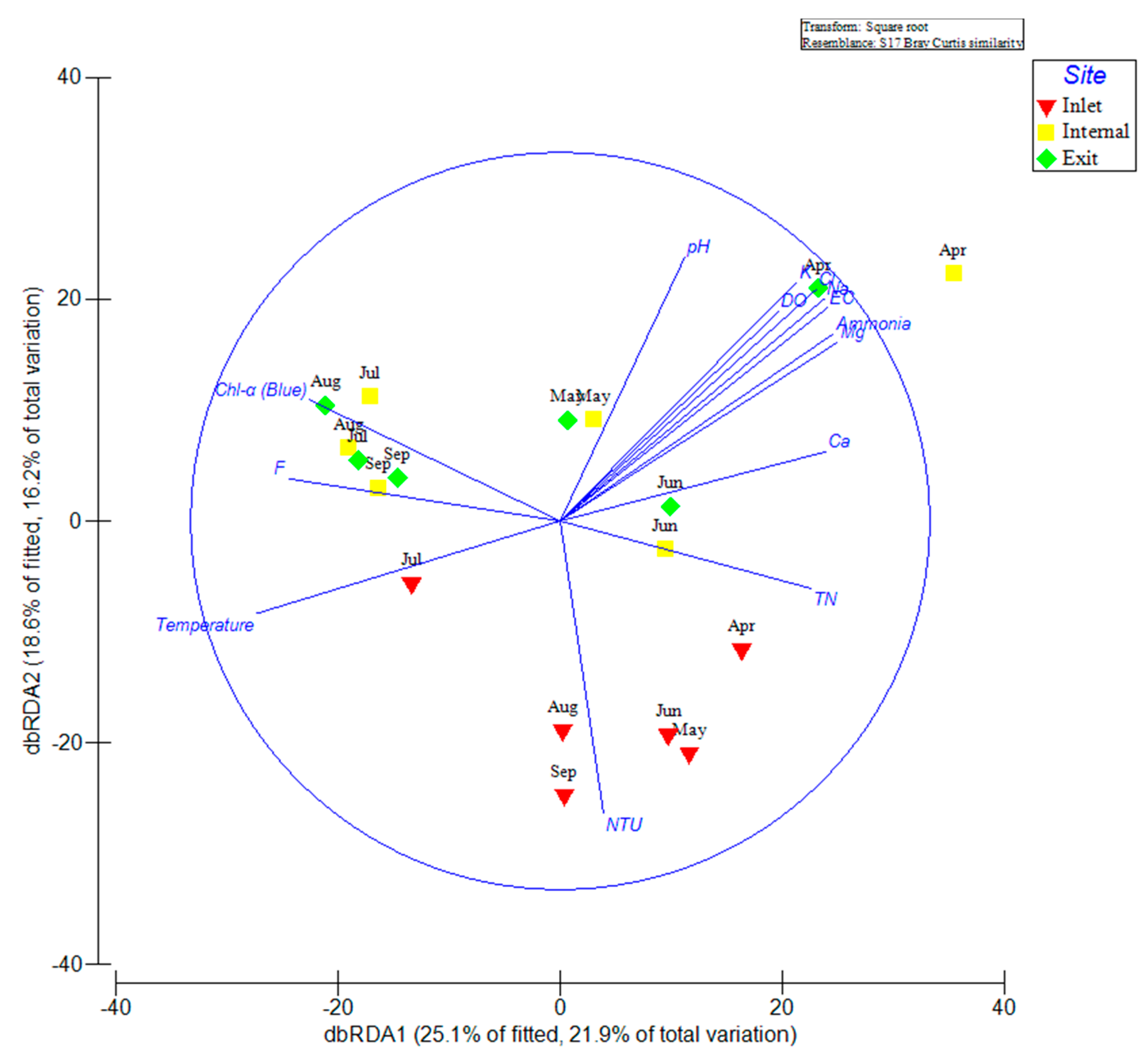
2.5. Multivariate Analysis of Biotic and Abiotic Factors in the QCS Reservoir
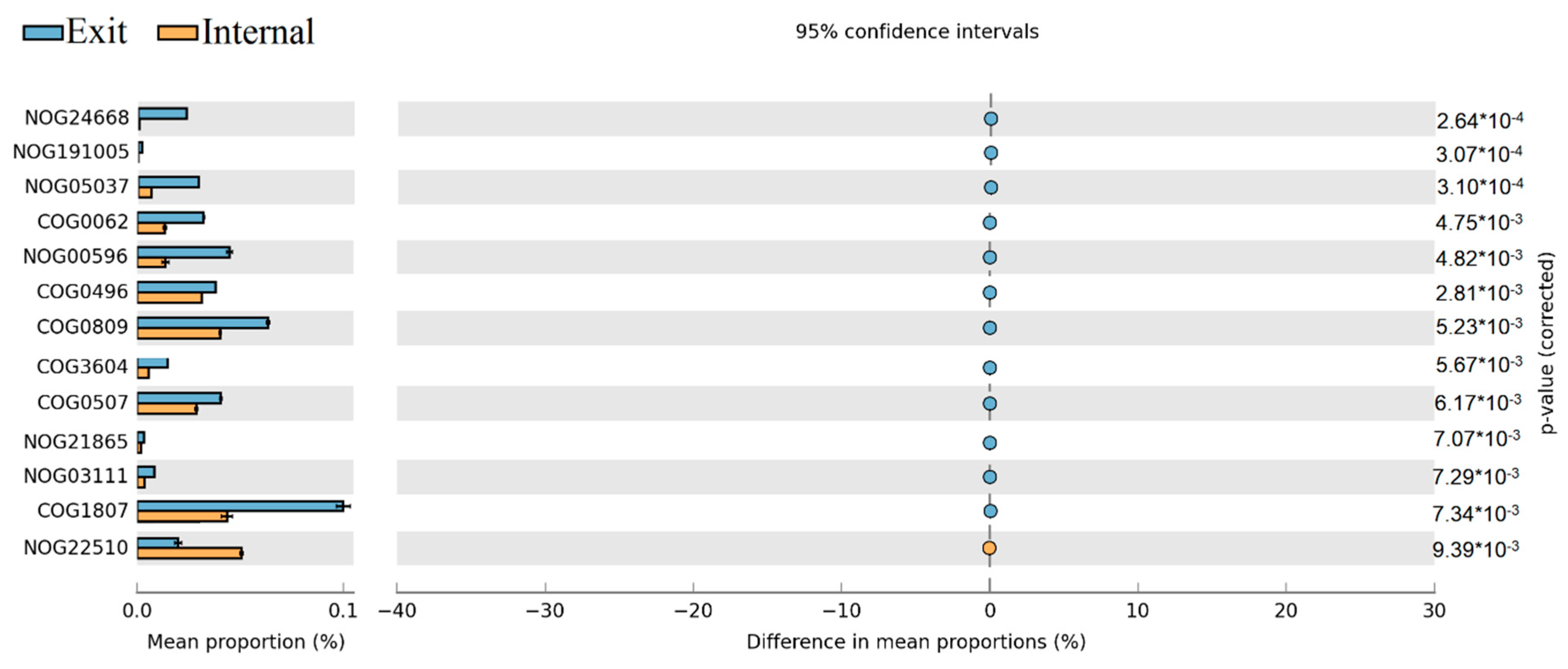
2.6. Shotgun Metagenomic Analysis
3. Discussion
3.1. Temporal and Spatial Dynamics of Microbial Community Composition in the QCS Reservoir
3.2. The Variations of Ecological Functions within the Microbial Community during the Period of Later Spring/Early Summer
4. Conclusions
5. Materials and Methods
5.1. Sampling Sites and In Situ Measurements
5.2. Physic-Chemical Parameters and Environmental Factors
5.3. DNA Extraction
5.4. The 16S rRNA Gene Sequencing via PCR Amplification
5.5. Statistical Analysis of the 16S rRNA Sequencing Data
5.6. Shotgun Metagenomic Analyses
5.7. Sequence Quality Control and Assembly
5.8. Gene Prediction, Taxonomy, and Functional Annotation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shu, H.T.; Gin, Y.H. The dynamics of cyanobacteria and microcystin production in a tropical reservoir of singapore. Harmful Algae 2011, 10, 319–329. [Google Scholar]
- Jin, X.; He, Y.; Kirumba, G.; Hassan, Y.; Li, J. Phosphorus fractions and phosphate sorption-release characteristics of the sediment in the yangtze river estuary reservoir. Ecol. Eng. 2013, 55, 62–66. [Google Scholar] [CrossRef]
- Dong, Y.L.; Owens, M.S.; Doherty, M.; Eggleston, E.M.; Hewson, I.; Crump, B.C.; Cornwell, J.C. The effects of oxygen transition on community respiration and potential chemoautotrophic production in a seasonally stratified anoxic estuary. Estuaries Coasts 2014, 38, 1–14. [Google Scholar]
- Yong, H.J.; Yang, J.S.; Park, K. Changes in water quality after the construction of an estuary dam in the geum river estuary dam system, korea. J. Coast. Res. 2014, 30, 1278–1286. [Google Scholar]
- Chen, W.; Chen, K.; Kuang, C.; Zhu, D.Z.; He, L.; Mao, X.; Liang, H.; Song, H. Influence of sea level rise on saline water intrusion in the yangtze river estuary, china. Appl. Ocean Res. 2016, 54, 12–25. [Google Scholar] [CrossRef]
- Canuel, E.A.; Hardison, A.K. Sources, ages, and alteration of organic matter in estuaries. Ann. Rev. Mar. Sci. 2016, 8, 409–434. [Google Scholar] [CrossRef] [PubMed]
- Osburn, C.L.; Boyd, T.J.; Montgomery, M.T.; Bianchi, T.S.; Coffin, R.B.; Paerl, H.W. Optical proxies for terrestrial dissolved organic matter in estuaries and coastal waters. Front. Mar. Sci. 2016, 2. [Google Scholar] [CrossRef]
- Jin, X.; He, Y.; Zhang, B.; Hassan, Y.; George, K. Impact of sulfate and chloride on sediment phosphorus release in the yangtze estuary reservoir, china. Water Sci. Technol. A J. Int. Assoc. Water Poll. Res. 2013, 67, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Li, G.; Wang, C.; Jing, Y.; Zhu, Y.; Zhang, S.; Liu, Y. Community dynamics of prokaryotic and eukaryotic microbes in an estuary reservoir. Sci. Rep. 2014, 4, 6966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Eom, Y.B. Analysis of microbial composition associated with freshwater and seawater. Biomed. Sci. Lett. 2016, 22, 150–159. [Google Scholar] [CrossRef]
- Crump, B.C.; Baross, J.A.; Simenstad, C.A. Dominance of particle-attached bacteria in the columbia river estuary, USA. Aquat. Microb. Ecol. 1998, 14, 7–18. [Google Scholar] [CrossRef]
- Angelika, R.; Herlemann, D.P.R.; Klaus, J.; Hans-Peter, G. Particle-associated differ from free-living bacteria in surface waters of the baltic sea. Front. Microbiol. 2015, 6, 1297. [Google Scholar]
- Yung, C.M.; Ward, C.S.; Davis, K.M.; Johnson, Z.I.; Hunt, D.E. Insensitivity of diverse and temporally variable particle-associated microbial communities to bulk seawater environmental parameters. Appl. Environ. Microbiol. 2016, 82, 3431. [Google Scholar] [CrossRef] [PubMed]
- Ochsenreiter, T.; Selezi, D.; Quaiser, A.; Bonch-Osmolovskaya, L.; Schleper, C. Diversity and abundance of crenarchaeota in terrestrial habitats studied by 16S rna surveys and real time pcr. Environ. Microbiol. 2010, 5, 787–797. [Google Scholar] [CrossRef]
- Humbert, S.; Tarnawski, S.; Fromin, N.; Mallet, M.P.; Aragno, M.; Zopfi, J. Molecular detection of anammox bacteria in terrestrial ecosystems: Distribution and diversity. J. Emultidiscip. J. Microb. Ecol. 2010, 4, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Caroquintero, A.; Tsementzi, D.; Deleonrodriguez, N.; Luo, C.; Poretsky, R.; Konstantinidis, K.T. Metagenomic insights into the evolution, function, and complexity of the planktonic microbial community of lake lanier, a temperate freshwater ecosystem. Appl. Environ. Microbiol. 2011, 77, 6000–6011. [Google Scholar] [CrossRef] [PubMed]
- Lindh, M.V.; Sjöstedt, J.; Andersson, A.F.; Baltar, F.; Hugerth, L.W.; Lundin, D.; Muthusamy, S.; Legrand, C.; Pinhassi, J. Disentangling seasonal bacterioplankton population dynamics by high-frequency sampling. Environ. Microbiol. 2015, 17, 2459–2476. [Google Scholar] [CrossRef] [PubMed]
- Frazão, B.; Martins, R.; Vasconcelos, V. Are known cyanotoxins involved in the toxicity of picoplanktonic and filamentous north atlantic marine cyanobacteria? Mar. Drugs 2010, 8, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Gantar, M.; Sekar, R.; Richardson, L.L. Cyanotoxins from black band disease of corals and from other coral reef environments. Microb. Ecol. 2009, 58, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Fernandez, N.; Beiras, R.; Vasconcelos, V. Toxicity assessment of crude and partially purified extracts of marine synechocystis and synechococcus cyanobacterial strains in marine invertebrates. Toxicon 2007, 50, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, J.N.; Kinsela, A.S.; Collins, R.N.; Bowling, L.C.; Honeyman, G.L.; Holliday, J.K.; Neilan, B.A. Microbial communities reflect temporal changes in cyanobacterial composition in a shallow ephemeral freshwater lake. ISME J. 2016, 10, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Woodhouse, J.N.; Te, S.H.; Yew-Hoong, G.K.; He, Y.; Xu, C.; Chen, L. Seasonal variation in the bacterial community composition of a large estuarine reservoir and response to cyanobacterial proliferation. Chemosphere 2018, 202, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.J.; Maruya, K.A.; Snyder, S.A.; Zeng, E.Y. China’s water pollution by persistent organic pollutants. Environ. Pollut. 2012, 163, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liang, D.; Ren, L.; Shi, S.; Li, Z.; Zhang, T.; Huang, Y. Concentration and source identification of polycyclic aromatic hydrocarbons and phthalic acid esters in the surface water of the yangtze river delta, china. J. Environ. Sci. (China) 2012, 24, 335–342. [Google Scholar] [CrossRef]
- Floehr, T.; Xiao, H.; Scholz-Starke, B.; Wu, L.; Hou, J.; Yin, D.; Zhang, X.; Ji, R.; Yuan, X.; Ottermanns, R. Solution by dilution?—A review on the pollution status of the yangtze river. Environ. Sci. Pollut. Res. 2013, 20, 6934–6971. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.A.C.; Kalyuzhnaya, M.G.; Malfatti, S.; Tringe, S.G.; Rio, T.G.D.; Ivanova, N.; Lidstrom, M.E.; Chistoserdova, L. A metagenomic insight into freshwater methane-utilizing communities and evidence for cooperation between the methylococcaceae and the methylophilaceae. PeerJ 2013, 1, e23. [Google Scholar] [CrossRef] [PubMed]
- Garrity, G.M.; Bell, J.A.; Lilburn, T. Methylophilaceae fam. Nov; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Hahn, M.W.; Kasalický, V.; Jezbera, J.; Brandt, U.; Jezberová, J.; Simek, K. Limnohabitans curvus gen. Nov., sp. Nov., a planktonic bacterium isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 2010, 60, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Willems, A. The Family Comamonadaceae; Springer: Heidelberg/Berlin, Germany, 2014; pp. 777–851. [Google Scholar]
- Pujalte, M.J.; Lucena, T.; Ruvira, M.A.; Arahal, D.R.; Macián, M.C. The Family Rhodobacteraceae; Springer: Heidelberg/Berlin, Germany, 2014; pp. 439–512. [Google Scholar]
- Huang, Z.; Xie, B.; Yuan, Q.; Xu, W.; Lu, J. Microbial community study in newly established qingcaosha reservoir of shanghai, china. Appl. Microbiol. Biotechnol. 2014, 98, 9849–9858. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.L.; Lovindeer, R.; Xi, Y.; Radway, J.C.; Armstrong, R.A. Differences in growth and physiology of marine synechococcus (cyanobacteria) on nitrate versus ammonium are not determined solely by nitrogen source redox state1. J. Phycol. 2012, 48, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, J.; Liu, L.; Fan, Y.; Li, L.; Yang, Y.; Lu, Z.; Zhang, X. Annual periodicity in planktonic bacterial and archaeal community composition of eutrophic lake taihu. Sci. Rep. 2015, 5, 15488. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.J.; Jones, S.E.; Eiler, A.; Mcmahon, K.D.; Bertilsson, S. A guide to the natural history of freshwater lake bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 14–49. [Google Scholar] [CrossRef] [PubMed]
- Needham, D.M.; Chow, C.E.T.; Cram, J.A.; Sachdeva, R.; Parada, A.; Fuhrman, J.A. Short-term observations of marine bacterial and viral communities: Patterns, connections and resilience. ISME J. 2013, 7, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Currie, D.J.; Kalff, J. The relative importance of bacterioplankton and phytoplankton in phosphorus uptake in freshwater. Limnol. Oceanogr. 1984, 29, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Vadstein, O. Growth and phosporus status of limnetic phytoplankton and bacteria. Limnol. Oceanogr. 1988, 33, 489–503. [Google Scholar] [CrossRef]
- Smith, E.M.; Prairie, Y.T. Bacterial metabolism and growth efficiency in lakes: The importance of phosphorus availability. Limnol. Oceanogr. 2004, 49, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.; Gilbert, J.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, S.; Treuren, W.V.; Lozupone, C.; Faust, K.; Friedman, J.; Ye, D.; Li, C.X.; Xu, Z.Z.; Ursell, L.; Alm, E.J. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016, 10, 1669–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshef, D.N.; Reshef, Y.A.; Finucane, H.K.; Grossman, S.R.; Mcvean, G.; Turnbaugh, P.J.; Lander, E.S.; Mitzenmacher, M.; Sabeti, P.C. Detecting novel associations in large data sets. Science 2011, 334, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.S.; Wei, C.H.; Deng, Y.; Gao, N.Y. Principal component analysis to assess the composition and fate of impurities in a large river-embedded reservoir: Qingcaosha reservoir. Environ. Sci. Process. Impacts 2013, 15, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Gera, A.; Alcoverro, T.; Mascarã, O.; PãRez, M.; Romero, J. Exploring the utility of posidonia oceanica chlorophyll fluorescence as an indicator of water quality within the european water framework directive. Environ. Monit. Assess. 2012, 184, 3675–3686. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berglyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M. Ultra-high-throughput microbial community analysis on the illumina hiseq and miseq platforms. ISME J. Multidiscip. J. Microb. Ecol. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, Z.; Li, H.; Park, H.D.; Wu, Z. Metagenomes reveal microbial structures, functional potentials, and biofouling-related genes in a membrane bioreactor. Appl. Microbiol. Biotechnol. 2016, 100, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A guide from sampling to data analysis. Microbial. Inf. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. Uchime improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Desantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16s rrna gene database and workbench compatible with arb. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; Mcgarrell, D.M.; Marsh, T.; Garrity, G.M. The ribosomal database project: Improved alignments and new tools for rrna analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The silva ribosomal rna gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Assenov, Y.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Albertsen, M.; Hansen, L.B.S.; Saunders, A.M.; Nielsen, P.H.; Nielsen, K.L. A metagenome of a full-scale microbial community carrying out enhanced biological phosphorus removal. ISME J. 2012, 6, 1094–1106. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Inlet | Internal | Exit | |||
|---|---|---|---|---|---|---|
| Pseudo-F | p | Pseudo-F | p | Pseudo-F | p | |
| TC | 0.751 | 0.933 | 0.731 | 0.655 | 1.528 | 0.059 |
| TOC | 0.734 | 0.943 | 0.673 | 0.828 | 0.673 | 0.971 |
| IC | 0.717 | 0.952 | 0.718 | 0.716 | 0.967 | 0.391 |
| TN | 1.352 | 0.059 | 1.669 | 0.090 | 1.923 | 0.020 |
| TP | 1.231 | 0.172 | 0.883 | 0.470 | 1.215 | 0.247 |
| NH4+-N | 1.368 | 0.052 | 2.437 | 0.045 | 1.658 | 0.034 |
| pH | 1.205 | 0.165 | 1.839 | 0.057 | 1.291 | 0.148 |
| DO | 1.426 | 0.023 | 2.119 | 0.040 | 1.262 | 0.193 |
| EC | 1.158 | 0.239 | 2.628 | 0.029 | 1.802 | 0.021 |
| Turbidity | 0.813 | 0.802 | 2.366 | 0.018 | 2.094 | 0.003 |
| Temperature | 1.426 | 0.029 | 2.417 | 0.030 | 1.738 | 0.033 |
| Cl− | 1.164 | 0.199 | 2.674 | 0.018 | 1.797 | 0.008 |
| SO42- | 0.730 | 0.917 | 0.986 | 0.381 | 1.209 | 0.205 |
| F− | 1.337 | 0.089 | 2.357 | 0.011 | 1.441 | 0.134 |
| Ca2+ | 0.733 | 0.950 | 1.637 | 0.097 | 1.807 | 0.033 |
| Mg2+ | 0.970 | 0.528 | 2.524 | 0.024 | 1.849 | 0.015 |
| Na+ | 1.216 | 0.149 | 2.686 | 0.019 | 1.803 | 0.010 |
| Al3+ | 0.910 | 0.642 | 1.234 | 0.191 | 1.516 | 0.080 |
| K+ | 0.962 | 0.540 | 2.438 | 0.023 | 1.889 | 0.012 |
| Si4+ | 0.683 | 0.978 | 0.795 | 0.605 | 0.863 | 0.604 |
| Chl-α | 1.345 | 0.167 | 1.348 | 0.170 | 1.172 | 0.252 |
| Sample | Total Species | Species Richness | Pielou’s Evenness | Shannon | Simpson |
|---|---|---|---|---|---|
| Inlet_2014_04 | 1380 | 144.6 | 0.6997 | 5.059 | 0.9762 |
| Inlet_2014_05 | 1349 | 141.4 | 0.6724 | 4.846 | 0.9708 |
| Inlet_2014_06 | 1601 | 167.8 | 0.6644 | 4.903 | 0.9536 |
| Inlet_2014_07 | 1131 | 118.5 | 0.6019 | 4.232 | 0.9084 |
| Inlet_2014_08 | 1382 | 144.8 | 0.6969 | 5.039 | 0.9739 |
| Inlet_2014_09 | 1744 | 182.8 | 0.6991 | 5.218 | 0.9718 |
| Internal_2014_04 | 701 | 73.41 | 0.6637 | 4.349 | 0.9685 |
| Internal_2014_05 | 1138 | 119.2 | 0.6798 | 4.784 | 0.9724 |
| Internal_2014_06 | 1127 | 118.1 | 0.6946 | 4.881 | 0.9747 |
| Internal_2014_07 | 1015 | 106.3 | 0.5991 | 4.148 | 0.9185 |
| Internal_2014_08 | 1107 | 116 | 0.6271 | 4.395 | 0.9375 |
| Internal_2014_09 | 1163 | 121.9 | 0.6591 | 4.652 | 0.9627 |
| Exit_2014_04 | 990 | 103.7 | 0.7044 | 4.859 | 0.9806 |
| Exit_2014_05 | 1128 | 118.2 | 0.6642 | 4.667 | 0.9661 |
| Exit_2014_06 | 1251 | 131.1 | 0.7031 | 5.014 | 0.9743 |
| Exit_2014_07 | 1133 | 118.7 | 0.6150 | 4.325 | 0.9294 |
| Exit_2014_08 | 1173 | 122.9 | 0.6311 | 4.459 | 0.9361 |
| Exit_2014_09 | 1265 | 132.6 | 0.6752 | 4.823 | 0.9672 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Te, S.H.; Xu, C.; He, Y.; Gin, K.Y.-H. Variations of Bacterial Community Composition and Functions in an Estuary Reservoir during Spring and Summer Alternation. Toxins 2018, 10, 315. https://doi.org/10.3390/toxins10080315
Xu Z, Te SH, Xu C, He Y, Gin KY-H. Variations of Bacterial Community Composition and Functions in an Estuary Reservoir during Spring and Summer Alternation. Toxins. 2018; 10(8):315. https://doi.org/10.3390/toxins10080315
Chicago/Turabian StyleXu, Zheng, Shu Harn Te, Cong Xu, Yiliang He, and Karina Yew-Hoong Gin. 2018. "Variations of Bacterial Community Composition and Functions in an Estuary Reservoir during Spring and Summer Alternation" Toxins 10, no. 8: 315. https://doi.org/10.3390/toxins10080315
APA StyleXu, Z., Te, S. H., Xu, C., He, Y., & Gin, K. Y. -H. (2018). Variations of Bacterial Community Composition and Functions in an Estuary Reservoir during Spring and Summer Alternation. Toxins, 10(8), 315. https://doi.org/10.3390/toxins10080315