Emerging Roles of Aryl Hydrocarbon Receptors in the Altered Clearance of Drugs during Chronic Kidney Disease


Abstract
:1. Chronic Kidney Disease
2. Drug Metabolism
3. Alterations of Drug Metabolism during Chronic Kidney Disease
3.1. What Do We Learn from Patients and Animals Models of Chronic Kidney Disease?
3.2. Proposed Factors Involved in Drug Metabolism Impairments in Chronic Kidney Disease
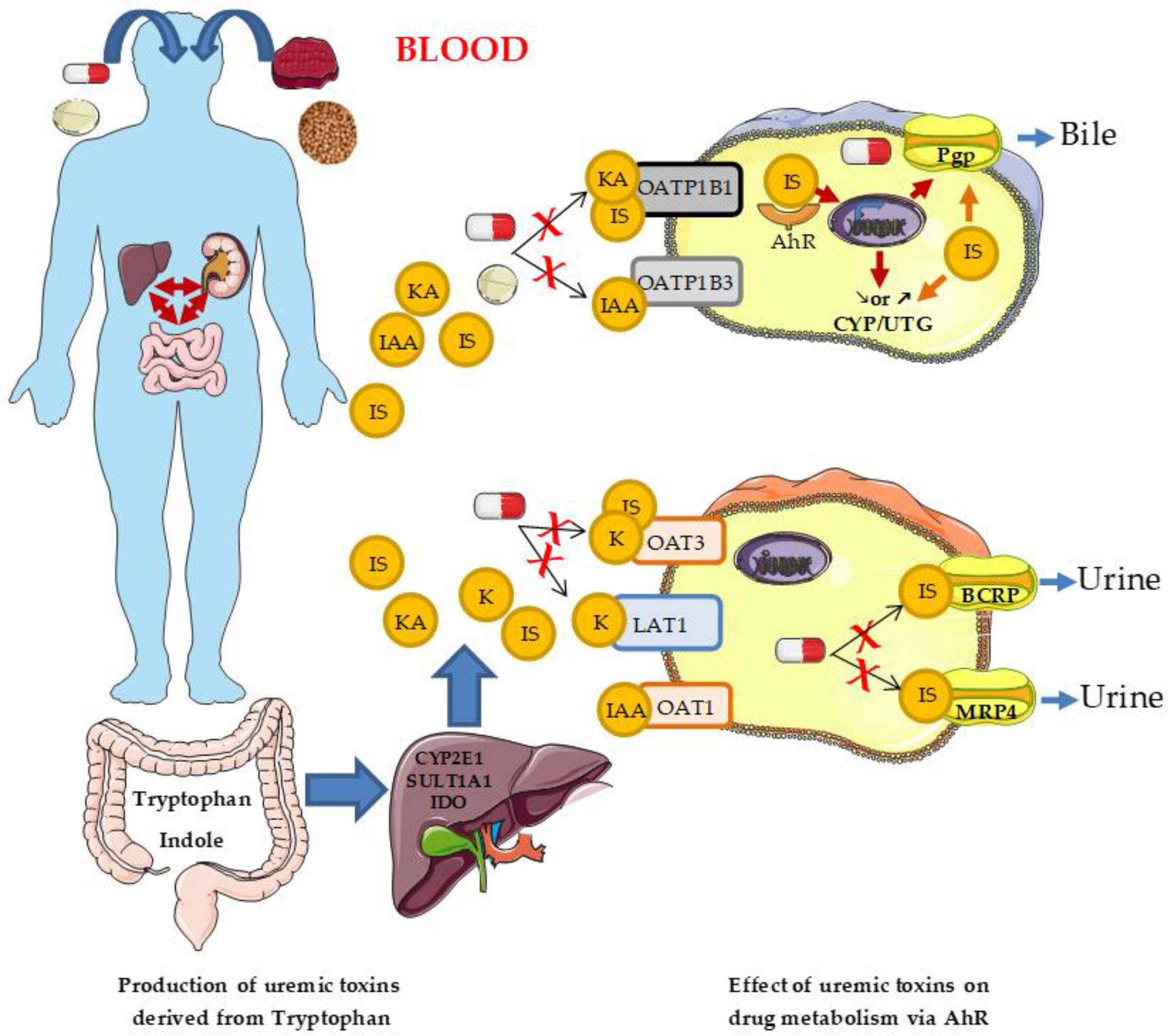
3.2.1. Uremic Toxins
3.2.2. Aryl Hydrocarbon Receptor
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hallan, S.I.; Coresh, J.; Astor, B.C.; Asberg, A.; Powe, N.R.; Romundstad, S.; Hallan, H.A.; Lydersen, S.; Holmen, J. International comparison of the relationship of chronic kidney disease prevalence and ESRD risk. J. Am. Soc. Nephrol. 2006, 17, 2275–2284. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: global dimension and perspectives. Lancet. 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Ojo, A. Addressing the global burden of chronic kidney disease through clinical and translational research. Trans. Am. Clin. Climatol. Assoc. 2014, 125, 229–243. [Google Scholar] [PubMed]
- Steenkamp, R.; Castledine, C.; Feest, T.; Fogarty, D. UK Renal Registry 13th Annual Report [December 2010]: Chapter 2: UK RRT prevalence in 2009: national and centre-specific analyses. Nephron. Clin. Pract. 2011, 2, c27–c52. [Google Scholar] [CrossRef]
- Modi, G.K.; Jha, V. The incidence of end-stage renal disease in India: a population-based study. Kidney Int. 2006, 70, 2131–2133. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.H.; Gullion, C.M.; Nichols, G.; Keith, D.S.; Brown, J.B. Cost of medical care for chronic kidney disease and comorbidity among enrollees in a large HMO population. J. Am. Soc. Nephrol. 2004, 15, 1300–1306. [Google Scholar] [CrossRef]
- Eckardt, K.U.; Coresh, J.; Devuyst, O.; Johnson, R.J.; Köttgen, A.; Levey, A.S.; Levin, A. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet. 2013, 382, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Epidemiology of cardiovascular disease in chronic renal disease. J. Am. Soc. Nephrol. 1998, 9, S16–S23. [Google Scholar] [CrossRef]
- Matsushita, K.; van der Velde, M.; Astor, B.C.; Woodward, M.; Levey, A.S.; de Jong, P.E.; Coresh, J.; Gansevoort, R.T. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative metaanalysis. Lancet. 2010, 375, 2073–2081. [Google Scholar]
- Levin, A.; Singer, J.; Thompson, C.R.; Ross, H.; Lewis, M. Prevalent left ventricular hypertrophy in the predialysis population: identifying opportunities for intervention. Am. J. Kidney Dis. 1996, 27, 347–354. [Google Scholar] [CrossRef]
- Raggi, P.; Bommer, J.; Chertow, G.M. Valvular calcification in hemodialysis patients randomized to calcium-based phosphorus binders or sevelamer. J. Heart. Valve. Dis. 2004, 13, 134–141. [Google Scholar]
- London, G.M.; Guérin, A.P.; Marchais, S.J.; Métivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 2003, 18, 1731–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asami, M.; Tanabe, K.; Ito, S.; Yoshida, E.; Aoki, J.; Tanimoto, S.; Horiuchi, Y.; Yoshida, M. Impact of Indoxyl Sulfate on Coronary Plaques in Patients on Hemodialysis. Int. Heart. J. 2018, 59, 489–4968. [Google Scholar] [CrossRef]
- Karbowska, M.; Kaminski, T.W.; Znorko, B.; Domaniewski, T.; Misztal, T.; Rusak, T.; Pryczynicz, A.; Guzinska-Ustymowicz, K.; Pawlak, K.; Pawlak, D. Indoxyl Sulfate Promotes ArterialThrombosis in Rat Model via Increased Levels of Complex TF/VII, PAI-1, Platelet Activation as Well as Decreased Contents ofSIRT1 and SIRT3. Front. Physiol. 2018, 9, 1623. [Google Scholar] [CrossRef]
- Kaminski, T.W.; Pawlak, K.; Karbowska, M.; Myśliwiec, M.; Pawlak, D. Indoxyl sulfate - the uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrology 2017, 18(1), 35. [Google Scholar] [CrossRef] [PubMed]
- Maciel, R.A.P.; Cunha, R.S.; Busato, V.; Franco, C.R.C.; Gregório, P.C.; Dolenga, C.J.R.; Nakao, L.S.; Massy, Z.A.; Boullier, A.; Pecoits-Filho, R.; et al. Uremia Impacts VE-Cadherin and ZO-1 Expression in Human Endothelial Cell-to-Cell Junctions. Toxins 2018, 10, 404. [Google Scholar] [CrossRef]
- Blacher, J.; Safar, M.E.; Guerin, A.P.; Pannier, B.; Marchais, S.J.; London, G.M. Aortic pulse wave velocity index and mortality in end-stage renal disease. Kidney Int. 2003, 63, 1852–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef]
- Namikoshi, T.; Tomita, N.; Satoh, M.; Sakuta, T.; Kuwabara, A.; Kobayashi, S.; Higuchi, Y.; Nishijima, F.; Kashihara, N. Oral adsorbent AST-120 ameliorates endothelial dysfunction independent of renal function in rats with subtotal nephrectomy. Hypertens. Res. 2009, 32, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Valenti, V.; Tocci, G.; Pontremoli, R.; Rosei, E.A.; Ambrosioni, E.; Costa, V.; Leonetti, G.; Pessina, A.C.; Trimarco, B.; et al. Association of renal damage with cardiovascular diseases is independent of individual cardiovascular risk profile in hypertension: data from the Italy -Developing Education and awareness on MicroAlbuminuria in patients with hypertensive Disease study. J. Hypertens. 2010, 28, 251–258. [Google Scholar] [CrossRef]
- Barreto, F.C.; Stinghen, A.E.; de Oliveira, R.B.; Franco, A.T.; Moreno, A.N.; Barreto, D.V.; Pecoits-Filho, R.; Drüeke, T.B.; Massy, Z.A. The quest for a better understanding of chronic kidney disease complications: an update on uremic toxins. J. Bras. Nefrol. 2014, 36, 221–235. [Google Scholar] [CrossRef]
- Mason, N.A. Polypharmacy and medication-related complications in the chronic kidney disease patient. Curr. Opin. Nephrol. Hypertens. 2011, 20, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Verbeeck, R.K.; Musuamba, F.T. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur. J. Clin. Pharmacol. 2009, 65, 757–773. [Google Scholar] [CrossRef]
- Nolin, T.D. A Synopsis of Clinical Pharmacokinetic Alterations in Advanced CKD. Semin. Dial. 2015, 28, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Haufroid, V.; Mourad, M.; Van Kerckhove, V.; Wawrzyniak, J.; De Meyer, M.; Eddour, D.C.; Malaise, J.; Lison, D.; Squifflet, J.P.; Wallemacq, P. The effect of CYP3A5 and MDR1 [ABCB1] polymorphisms on cyclosporine and tacrolimus dose requirements and trough blood levels in stable renal transplant patients. Pharmacogenetics. 2004, 14, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Gomez, A. The past, present and future of pharmacoepigenomics. Pharmacogenomics. 2010, 11, 625–627. [Google Scholar] [CrossRef]
- Cotreau, M.M.; von Moltke, L.L.; Greenblatt, D.J. The influence of age and sex on the clearance of cytochrome P450 3A substrates. Clin. Pharmacokinet. 2005, 44, 33–60. [Google Scholar] [CrossRef]
- Kinirons, M.T.; O’Mahony, M.S. Drug metabolism and ageing. Br. J. Clin. Pharmacol. 2004, 57, 540–544. [Google Scholar] [CrossRef] [Green Version]
- Zanger, U.M.; Raimundo, S.; Eichelbaum, M. Cytochrome P450 2D6: Overview and updateon pharmacology, genetics, biochemistry. Naunyn. Schmiedebergs. Arch. Pharmacol. 2004, 369, 23–37. [Google Scholar] [CrossRef]
- Dettli, L.; Spring, P.; Habersang, R. Drug dosage in patients with impaired renal function. Postgrad. Med. J. 1970, 10, 32–35. [Google Scholar]
- Dettli, L. Elimination of drugs in patients with renal insufficiency. Schweiz. Rundsch. Med. Prax. 1973, 62, 476–478. [Google Scholar]
- Dreisbach, A.W.; Lertora, J.J. The effect of chronic renal failure on drug metabolism and transport. Expert. Opin. Drug. Metab. Toxicol. 2008, 4, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Abraham, S.; Apparaju, S.; Wu, T.C.; Strong, J.M.; Xiao, S.; Atkinson, A.J., Jr.; Thummel, K.E.; Leeder, J.S.; et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug applications. Clin. Pharmacol. Ther. 2009, 85, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Iyanagi, T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: implications for detoxification. Int. Rev. Cytol. 2007, 260, 35–112. [Google Scholar]
- Köhle, C.; Bock, K.W. Coordinate regulation of human drug-metabolizing enzymes, and conjugate transporters by the Ah receptor, pregnane X receptor and constitutive androstane receptor. Biochem. Pharmacol. 2009, 77, 689–699. [Google Scholar] [CrossRef]
- Xu, C.; Li, C.Y.; Kong, A.N. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 2005, 28, 249–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kot, M.; Daniel, W.A. Caffeine as a marker substrate for testing cytochrome P450 activity in human and rat. Pharmacol Rep. 2008, 60, 789–797. [Google Scholar]
- Ohnishi, A.; Kato, M.; Kojima, J.; Ushiama, H.; Yoneko, M.; Kawai, H. Differential pharmacokinetics of theophylline in elderly patients. Drugs Aging 2003, 20, 71–84. [Google Scholar] [CrossRef]
- Uno, T.; Nakano, R.; Kitagawa, R.; Okada, M.; Kanamaru, K.; Takenaka, S.; Uno, Y.; Imaishi, H. Metabolism of steroids by cytochrome P450 2C9 variants. Biopharm Drug Dispos. 2018, 39, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Seden, K.; Dickinson, L.; Khoo, S.; Back, D. Grapefruit-drug interactions. Drugs 2010, 70, 2373–2407. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Tukey, R.H. Chapter 3. Drug Metabolism. In Goodman & Gilman’s. The Pharmacological Basis of Therapeutics. Brunton, L.; Lazo, J., Parker, K., Eds.; McGraw-Hill: New York, NY, USA, 1996. [Google Scholar]
- Bolton, J.L.; Chang, M. Quinoids as reactive intermediates in estrogen carcinogenesis. Adv. Exp. Med. Biol. 2001, 500, 497–507. [Google Scholar]
- Köhle, C.; Bock, K.W. Coordinate regulation of Phase I and II xenobiotic metabolisms by the Ah receptor and Nrf2. Biochem. Pharmacol. 2007, 73, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics. 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug. Discov. 2015, 14, 29–44. [Google Scholar] [CrossRef]
- Nigam, S.K.; Wu, W.; Bush, K.T.; Hoenig, M.P.; Blantz, R.C.; Bhatnagar, V. Handling of Drugs, Metabolites, and Uremic Toxins by Kidney Proximal Tubule Drug Transporters. Clin. J. Am. Soc. Nephrol. 2015, 10, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.R. Repaglinide-prandial glucose regulator: a new class of oral antidiabetic drugs. Diabet. Med. 1998, 4, S28–S36. [Google Scholar] [CrossRef]
- Schumacher, S.; Abbasi, I.; Weise, D.; Hatorp, V.; Sattler, K.; Sieber, J.; Hasslacher, C. Single- and multiple-dose pharmacokinetics of repaglinide in patients with type 2 diabetes and renal impairment. Eur. J. Clin. Pharmacol. 2001, 57, 147–152. [Google Scholar] [CrossRef]
- Coulomb, F.; Ducret, F.; Laneury, J.P.; Fiorentini, F.; Poggesi, I.; Jannuzzo, M.G.; Fleishaker, J.C.; Houin, G.; Duchêne, P. Pharmacokinetics of single-dose reboxetine involunteers with renal insufficiency. J. Clin. Pharmacol. 2000, 40, 482–487. [Google Scholar] [CrossRef]
- Yuan, R.; Venitz, J. Effect of chronic renal failure on the disposition of highly hepatically metabolized drugs. Int. J. Clin. Pharmacol. Ther. 2000, 38, 245–253. [Google Scholar] [CrossRef]
- Limdi, N.A.; Limdi, M.A.; Cavallari, L.; Anderson, A.M.; Crowley, M.R.; Baird, M.F.; Allon, M.; Beasley, T.M. Warfarin dosing in patients with impaired kidney function. Am. J. Kidney Dis. 2010, 56, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Park, H.W.; Cho, J.G.; Yoon, N.S.; Kim, S.S.; Kim, M.R.; Kim, M.C.; Cho, K.H.; Kim, H.K.; Kim, C.H.; et al. Comparison of non-vitamin K antagonist oral anticoagulants and warfarin on clinical outcomes in atrial fibrillation patients with renal dysfunction. Europace 2015, 10, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Nolin, T.D.; Frye, R.F.; Le, P.; Sadr, H.; Naud, J.; Leblond, F.A.; Pichette, V.; Himmelfarb, J. ESRD impairs nonrenal clearance of fexofenadine but not midazolam. J. Am. Soc. Nephrol. 2009, 20, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Dowling, T.C.; Briglia, A.E.; Fink, J.C.; Hanes, D.S.; Light, P.D.; Stackiewicz, L.; Karyekar, C.S.; Eddington, N.D.; Weir, M.R.; Henrich, W.L. Characterization of hepatic cytochrome p4503A activity in patients with end-stage renal disease. Clin. Pharmacol. Ther. 2003, 73, 427–434. [Google Scholar] [CrossRef]
- Frassetto, L.A.; Poon, S.; Tsourounis, C.; Valera, C.; Benet, L.Z. Effects of uptake and efflux transporter inhibition on erythromycin breath test results. Clin. Pharmacol. Ther. 2007, 81, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Leblond, F.A.; Petrucci, M.; Dubé, P.; Bernier, G.; Bonnardeaux, A.; Pichette, V. Downregulation of intestinal cytochrome p450 in chronic renal failure. J. Am. Soc. Nephrol. 2002, 13, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Leblond, F.A.; Giroux, L.; Villeneuve, J.P.; Pichette, V. Decreased in vivo metabolismof drugs in chronic renal failure. Drug. Metab. Dispos. 2000, 28, 1317–1320. [Google Scholar]
- Leblond, F.A.; Guévin, C.; Demers, C.; Pellerin, I.; Gascon-Barré, M.; Pichette, V. Downregulation of hepatic cytochrome P450 in chronic renal failure. J. Am. Soc. Nephrol. 2001, 12, 326–332. [Google Scholar]
- Uchida, N.; Kurata, N.; Shimada, K.; Nishimura, Y.; Yasuda, K.; Hashimoto, M.; Uchida, E.; Yasuhara, H. Changes of hepatic microsomal oxidative drug metabolizing enzymes inchronic renal failure [CRF] rats by partial nephrectomy. Jpn. J. Pharmacol. 1995, 68, 431–439. [Google Scholar] [CrossRef]
- Velenosi, T.J.; Fu, A.Y.; Luo, S.; Wang, H.; Urquhart, B.L. Down-regulation of hepatic CYP3A and CYP2C mediated metabolism in rats with moderate chronic kidney disease. Drug. Metab. Dispos. 2012, 40, 1508–1514. [Google Scholar] [CrossRef]
- Sindhu, R.K.; Vaziri, N.D. Upregulation of cytochrome P450 1A2 in chronic renal failure: does oxidized tryptophan play a role? Adv. Exp. Med. Biol. 2003, 527, 401–407. [Google Scholar]
- Simard, E.; Naud, J.; Michaud, J.; Leblond, F.A.; Bonnardeaux, A.; Guillemette, C.; Sim, E.; Pichette, V. Downregulation of hepatic acetylation of drugs in chronic renal failure. J. Am. Soc. Nephrol. 2008, 19, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Naud, J.; Michaud, J.; Boisvert, C.; Desbiens, K.; Leblond, F.A.; Mitchell, A.; Jones, C.; Bonnardeaux, A.; Pichette, V. Down-regulation of intestinal drug transporters in chronic renal failure in rats. J. Pharmacol. Exp. Ther. 2007, 320, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Naud, J.; Michaud, J.; Leblond, F.A.; Lefrancois, S.; Bonnardeaux, A.; Pichette, V. Effects of chronic renal failure on liver drug transporters. Drug. Meta. Dispos. 2008, 36, 124–128. [Google Scholar] [CrossRef]
- Laouari, D.; Yang, R.; Veau, C.; Blanke, I.; Friedlander, G. Two apical multidrug transporters, P-gp and MRP2, are differently altered in chronic renal failure. Am. J. Physiol. Renal. Physiol. 2001, 280, F636–F645. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. European Uremic Toxin Work Group [EUTox]. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney. Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- Deltombe, O.; Van Biesen, W.; Glorieux, G.; Massy, Z.; Dhondt, A.; Eloot, S. Exploring Protein Binding of Uremic Toxins in Patients with Different Stages of Chronic Kidney Disease and during Hemodialysis. Toxins 2015, 710, 3933–3946. [Google Scholar] [CrossRef]
- Rhee, E.P.; Souza, A.; Farrell, L.; Pollak, M.R.; Lewis, G.D.; Steele, D.J.; Thadhani, R.; Clish, C.B.; Greka, A.; Gerszten, R.E. Metabolite profiling identifies markers of uremia. J. Am. Soc. Nephrol. 2010, 6, 1041–1051. [Google Scholar] [CrossRef]
- Mischak, H.; Delles, C.; Vlahou, A.; Vanholder, R. Proteomic biomarkers in kidney disease: issues in development and implementation. Nat. Rev. Nephrol. 2015, 11(4), 221–232. [Google Scholar] [CrossRef]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 1, 33. [Google Scholar] [CrossRef]
- Ortiz, A.; Covic, A.; Fliser, D.; Fouque, D.; Goldsmith, D.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Rossignol, P.; Vanholder, R.; et al. Board of theEURECA-m Working Group of ERA-EDTA. Epidemiology, contributors to, and clinical trials of mortality risk in chronic kidney failure. Lancet. 2014, 383, 1831–1843. [Google Scholar] [CrossRef]
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J.; European Uremic Toxin Work Group. A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef]
- Bammens, B.; Evenepoel, P.; Keuleers, H.; Verbeke, K.; Vanrenterghem, Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006, 69, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. European Uraemic Toxin Work Group [EUTox]. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group [EUTox]. Serum indoxyl sulfate is associated with vascular disease and mortality in chronickidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; Mallet, B.; Dignat-George, F.; Burtey, S. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. SocNephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: a new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar]
- Jourde-Chiche, N.; Dou, L.; Sabatier, F.; Calaf, R.; Cerini, C.; Robert, S.; Camoin-Jau, L.; Charpiot, P.; Argiles, A.; Dignat-George, F.; et al. Levels of circulating endothelial progenitor cells are related to uremic toxins and vascular injury in hemodialysis patients. J. Thromb. Haemost. 2009, 7, 1576–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Ji, S.; Dong, W.; Qi, Y.; Song, W.; Cui, D.; Shi, J. Indolic uremic solutes enhance procoagulant activity of red blood cells through phosphatidylserine exposure and microparticle release. Toxins 2015, 7, 4390–4403. [Google Scholar] [CrossRef]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef]
- Chitalia, V.C.; Shivanna, S.; Martorell, J.; Balcells, M.; Bosch, I.; Kolandaivelu, K.; Edelman, E.R. Uremic serum and solutes increase post-vascular interventional thrombotic risk through altered stability of smooth muscle cell tissue factor. Circulation. 2013, 127, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, D.; Pawlak, K.; Malyszko, J.; Mysliwiec, M.; Buczko, W. Accumulation of toxic products degradation of kynurenine in hemodialyzed patients. Int. Urol. Nephrol. 2001, 33, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, D.; Tankiewicz, A.; Buczko, W. Kynurenine and its metabolites in the rat with experimental renal insufficiency. J. Physiol. Pharmacol. 2001, 52, 755–766. [Google Scholar] [PubMed]
- Pawlak, K.; Kowalewska, A.; Mysliwiec, M.; Pawlak, D. Kynurenine and its metabolites -kynurenic acid and anthranilic acid- are associated with soluble endothelial adhesion molecules and oxidative status in patients with chronic kidney disease. Am. J. Med.Sci. 2009, 338, 293–300. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 5, 197–203. [Google Scholar] [CrossRef]
- Reyes, M.; Benet, L.Z. Effects of uremic toxins on transport and metabolism of different biopharmaceutics drug disposition classification system xenobiotics. J. Pharm. Sci. 2011, 100, 3831–3842. [Google Scholar] [CrossRef]
- Sun, H.; Frassetto, L.; Benet, L.Z. Effects of renal failure on drug transport and metabolism. Pharmacol. Ther. 2006, 109, 1–11. [Google Scholar] [CrossRef]
- Sato, T.; Yamaguchi, H.; Kogawa, T.; Abe, T.; Mano, N. Organic anion transporting polypeptides 1B1 and 1B3 play an important role in uremic toxin handling and drug-uremic toxin interactions in the liver. J. Pharm. Pharm Sci. 2014, 17, 475–484. [Google Scholar] [CrossRef]
- Sun, H.; Huang, Y.; Frassetto, L.; Benet, L.Z. Effects of uremic toxins on hepatic uptake and metabolism of erythromycin. Drug. Metab. Dispos. 2004, 32, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, H.A.; Caetano-Pinto, P.; Seegers, A.E.; Dankers, A.C.; van den Broek, P.H.; Wetzels, J.F.; van den Brand, J.A.; van den Heuvel, L.P.; Hoenderop, J.G.; Wilmer, M.J.; et al. Proximal tubular efflux transporters involved in renal excretion of p-cresyl sulfate and p-cresyl glucuronide: Implications for chronic kidney disease pathophysiology. Toxicol. In. Vitro. 2015, 29, 1868–1877. [Google Scholar] [CrossRef]
- Sinclair, L.V.; Neyens, D.; Ramsay, G.; Taylor, P.M.; Cantrell, D.A. Single cell analysis of kynurenine and System L amino acid transport in T cells. Nat. Commun. 2018, 9(1), 1981–1992. [Google Scholar] [CrossRef]
- Michaud, J.; Dubé, P.; Naud, J.; Leblond, F.A.; Desbiens, K.; Bonnardeaux, A.; Pichette, V. Effects of serum from patients with chronic renal failure on rat hepatic cytochrome P450. Br. J. Pharmacol. 2005, 144, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Guévin, C.; Michaud, J.; Naud, J.; Leblond, F.A.; Pichette, V. Down-regulation of hepatic cytochrome p450 in chronic renal failure: Role of uremic mediators. Br. J. Pharmacol. 2002, 137, 1039–1046. [Google Scholar] [CrossRef]
- Liu, H.; Narayanan, R.; Hoffmann, M.; Surapaneni, S. The Uremic Toxin Indoxyl-3-Sulfate Induces CYP1A2 In Primary Human Hepatocytes. Drug. Metab. Lett. 2016, 10, 195–199. [Google Scholar] [CrossRef]
- Weigand, K.M.; Schirris, T.J.J.; Houweling, M.; van den Heuvel, J.J.M.W.; Koenderink, J.B.; Dankers, A.C.A.; Russel, F.G.M.; Greupink, R. Uremic solutes modulate hepatic bile acid handling and induce mitochondrial toxicity. Toxicol. In Vitro. 2019, 56, 52–56. [Google Scholar] [CrossRef]
- Mutsaers, H.A.; van den Heuvel, L.P.; Ringens, L.H.; Dankers, A.C.; Russel, F.G.; Wetzels, J.F.; Hoenderop, J.G.; Masereeuw, R. Uremic toxins inhibit transport by breast cancer resistance protein and multidrug resistance protein 4 at clinically relevant concentrations. PLoS. One 2011, 6, e18438. [Google Scholar] [CrossRef]
- Dankers, A.C.; Mutsaers, H.A.; Dijkman, H.B.; van den Heuvel, L.P.; Hoenderop, J.G.; Sweep, F.C.; Russel, F.G.; Masereeuw, R. Hyperuricemia influences tryptophan metabolism via inhibition of multidrug resistance protein 4 (MRP4) and breast cancer resistanceprotein (BCRP). Biochim. Biophys. Acta. 2013, 1832, 1715–1722. [Google Scholar] [CrossRef]
- Matsuo, K.; Yamamoto, S.; Wakamatsu, T.; Takahashi, Y.; Kawamura, K.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Narita, I. Increased Proinflammatory Cytokine Production and DecreasedCholesterol Efflux Due to Downregulation of ABCG1 in Macrophages Exposed to Indoxyl Sulfate. Toxins 2015, 7, 3155–3166. [Google Scholar] [CrossRef]
- Akiyama, Y.; Kikuchi, K.; Saigusa, D.; Suzuki, T.; Takeuchi, Y.; Mishima, E.; Yamamoto, Y.; Ishida, A.; Sugawara, D.; Jinno, D.; et al. Indoxyl sulfate down-regulates SLCO4C1 transporter through up-regulation of GATA3. PLoS ONE 2013, 8, e66518. [Google Scholar] [CrossRef]
- Enomoto, A.; Takeda, M.; Tak, K.; Takayama, F.; Noshiro, R.; Niwa, T.; Endou, H. Interactions of human organic anion as well as cation transporters with indoxyl sulfate. Eur. J. Pharmacol. 2003, 466, 13–20. [Google Scholar] [CrossRef]
- Deguchi, T.; Kusuhara, H.; Takadate, A.; Endou, H.; Otagiri, M.; Sugiyama, Y. Characterization of uremic toxin transport by organic anion transporters in the kidney. Kidney Int. 2004, 65, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Itoh, Y.; Tateoka, R.; Ezawa, A.; Murakami, K.; Niwa, T. Metabolomic searchfor uremic toxins as indicators of the effect of an oral sorbent AST-120 by liquid chromatography/tandem mass spectrometry. J. Chromatogr. B. Analyt. Technol. Biomed. Life. Sci. 2010, 878, 2997–3002. [Google Scholar] [CrossRef]
- Fujii, H.; Nishijima, F.; Goto, S.; Sugano, M.; Yamato, H.; Kitazawa, R.; Kitazawa, S.; Fukagawa, M. Oral charcoal adsorbent [AST-120] prevents progression of cardiac damage in chronic kidney disease through suppression of oxidative stress. Nephrol. Dial. Transplant. 2009, 24, 2089–2095. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kazama, J.J.; Omori, K.; Matsuo, K.; Takahashi, Y.; Kawamura, K.; Matsuto, T.; Watanabe, H.; Maruyama, T.; Narita, I. Continuous Reduction of Protein-Bound Uraemic Toxins with Improved Oxidative Stress by Using the Oral Charcoal Adsorbent AST-120 in Haemodialysis Patients. Sci. Rep 2015, 5, 1438. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef]
- Koya, Y.; Uchida, S.; Machi, Y.; Shobu, Y.; Namiki, N.; Kotegawa, T. Prediction of drug interaction between oral adsorbent AST-120 and concomitant drugs based on the invitro dissolution and in vivo absorption behavior of the drugs. Eur. J. Clin. Pharmacol. 2016, 72, 1353–1361. [Google Scholar] [CrossRef]
- Aoyama, I.; Enomoto, A.; Niwa, T. Effects of oral adsorbent on gene expression profile in uremic rat kidney: cDNA array analysis. Am. J. Kidney. Dis. 2003, 41, S8–S14. [Google Scholar] [CrossRef] [PubMed]
- Nolin, T.D.; Appiah, K.; Kendrick, S.A.; Le, P.; McMonagle, E.; Himmelfarb, J. Hemodialysis acutely improves hepatic CYP3A4 metabolic activity. J. Am. Soc. Nephrol. 2006, 17, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.; Nolin, T.D.; Naud, J.; Dani, M.; Lafrance, J.P.; Leblond, F.A.; Himmelfarb, J.; Pichette, V. Effect of hemodialysis on hepatic cytochrome P450 functional expression. J. Pharmacol. Sci. 2008, 108, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Van Landschoot, N.; De Smet, R.; Schoots, A.; Ringoir, S. Drug protein binding in chronic renal failure: evaluation of nine drugs. Kidney Int. 1988, 33, 996–1004. [Google Scholar] [CrossRef]
- Takamura, N.; Maruyama, T.; Otagiri, M. Effects of uremic toxins and fatty acids on serum protein binding of furosemide: possible mechanism of the binding defect in uremia. Clin. Chem. 1997, 43, 2274–2280. [Google Scholar]
- Watanabe, H.; Noguchi, T.; Miyamoto, Y.; Kadowaki, D.; Kotani, S.; Nakajima, M.; Miyamura, S.; Ishima, Y.; Otagiri, M.; Maruyama, T. Interaction between two sulfate-conjugated uremic toxins, p-cresyl sulfate and indoxyl sulfate, during binding with human serum albumin. Drug. Metab. Dispos. 2012, 40, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Xu, X.; Cao, W.; Shen, Z.; Wang, N.; Leng, J.; Zou, N.; Shang, E.; Zhu, Z.; et al. Improved dialysis removal of protein-bound uremic toxins by salvianolic acids. Phytomedicine 2018, 57, 166–173. [Google Scholar] [CrossRef]
- Schroeder, J.C.; Dinatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug. Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Poitevin, S.; Sallée, M.; Addi, T.; Gondouin, B.; McKay, N.; Denison, M.S.; Jourde-Chiche, N.; Duval-Sabatier, A.; Cerini, C.; et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 2018, 93, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon [dioxin] receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef]
- Abel, J.; Haarmann-Stemmann, T. An introduction to the molecular basics of aryl hydrocarbon receptor biology. Biol. Chem. 2010, 391, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, T.; Eguchi, H.; Tachibana, T.; Yoneda, Y.; Kawajiri, K. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J. Biol. Chem. 1998, 273, 2895–2904. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 2005, 433, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta. 2003, 1619, 263–268. [Google Scholar] [CrossRef]
- Quattrochi, L.C.; Vu, T.; Tukey, R. The human CYP1A2 gene and induction by 3-methylcholanthrene.A region of DNA that supports Ah-receptor binding and promoter-specific induction. J. Biol. Chem. 1994, 269, 6949–6954. [Google Scholar]
- Erichsen, T.J.; Ehmer, U.; Kalthoff, S.; Lankisch, T.O.; Müller, T.M.; Munzel, P.A.; Manns, M.P.; Strassburg, C.P. Genetic variability of aryl hydrocarbon receptor [AhR]-mediated regulation of the human UDP glucuronosyltransferase [UGT] 1A4 gene. Toxicol. Appl. Pharmacol. 2008, 230, 252–260. [Google Scholar] [CrossRef]
- Emi, Y.; Ikushiro, S.; Iyanagi, T. Xenobiotic responsive element-mediated transcriptional activation in the UDP-glucuronosyltransferase family 1 gene complex. J. Biol. Chem. 1996, 271, 3952–3958. [Google Scholar] [CrossRef]
- Li, W.; Harper, P.A.; Tang, B.K.; Okey, A.B. Regulation of cytochrome P450 enzymes by aryl hydrocarbon receptor in human cells: CYP1A2 expression in the LS180 colon carcinoma cell line after treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin or 3-methylcholanthrene. Biochem. Pharmacol. 1998, 56, 599–612. [Google Scholar] [CrossRef]
- Lankisch, T.O.; Gillman, T.C.; Erichsen, T.J.; Ehmer, U.; Kalthoff, S.; Freiberg, N.; Munzel, P.A.; Manns, M.P.; Strassburg, C.P. Aryl hydrocarbon receptor-mediated regulation of the human estrogen and bile acid UDP-glucuronosyltransferase 1A3 gene. Arch. Toxicol. 2008, 82, 573–582. [Google Scholar]
- Kalthoff, S.; Ehmer, U.; Freiberg, N.; Manns, M.P.; Strassburg, C.P. Interaction between oxidative stress sensor Nrf2 and xenobiotic-activated aryl hydrocarbon receptor in the regulation of the human phase II detoxifying UDP-glucuronosyl-transferase 1A10. J. Biol. Chem. 2010, 285, 5993–6002. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.; Xie, W. Xenobiotic nuclear receptor-mediated regulation of UDP-glucuronosyl-transferases. Curr. Drug. Metab. 2005, 6, 289–298. [Google Scholar] [CrossRef]
- Urquhart, B.L.; Tirona, R.G.; Kim, R.B. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for interindividual variability in response to drugs. J. Clin. Pharmacol. 2007, 47, 566–578. [Google Scholar] [CrossRef]
- Vasiliou, V.; Buetler, T.; Eaton, D.L.; Nebert, D.W. Comparison of oxidative stress response parameters in newborn mouse liver versus simian virus 40 [SV40]-transformed hepatocyte cell lines. Biochem. Pharmacol. 2000, 59, 703–712. [Google Scholar] [CrossRef]
- Gao, X.; Xie, C.; Wang, Y.; Luo, Y.; Yagai, T.; Sun, D.; Qin, X.; Krausz, K.W.; Gonzalez, F.J. The antiandrogen flutamide is a novel aryl hydrocarbon receptor ligand that disrupts bile acid homeostasis in mice through induction of Abcc4. Biochem. Pharmacol. 2016, 119, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Le Vee, M.; Jouan, E.; Stieger, B.; Lecureur, V.; Fardel, O. Regulation of human hepatic drug transporter activity and expression by diesel exhaust particle extract. PLoS One 2015, 10, e0121232. [Google Scholar] [CrossRef]
- Le Vee, M.; Jouan, E.; Lecureur, V.; Fardel, O. Aryl hydrocarbon receptor-dependent up-regulation of the heterodimeric amino acid transporter LAT1 [SLC7A5]/CD98hc [SLC3A2] by diesel exhaust particle extract in human bronchial epithelial cells. Toxicol. Appl. Pharmacol. 2016, 290, 74–85. [Google Scholar] [CrossRef]
- Santana Machado, T.; Poitevin, S.; Paul, P.; McKay, N.; Jourde-Chiche, N.; Legris, T.; Mouly-Bandini, A.; Dignat-George, F.; Brunet, P.; Masereeuw, R.; et al. Indoxyl Sulfate Upregulates Liver P-Glycoprotein Expression and Activity through Aryl Hydrocarbon Receptor Signaling. J. Am. Soc. Nephrol. 2018, 29, 906–918. [Google Scholar]
- Zhou, S.F.; Chan, E.; Zhou, Z.W.; Xue, C.C.; Lai, X.; Duan, W. Insights into the structure, function, and regulation of human cytochrome P450 1A2. Curr. Drug. Metab. 2009, 10, 713–729. [Google Scholar] [CrossRef]
- Zhou, S.F.; Wang, B.; Yang, L.P.; Liu, J.P. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug. Metab. Rev. 2010, 42, 268–354. [Google Scholar] [CrossRef]
- Al Za’abi, M.; Shalaby, A.; Manoj, P.; Ali, B.H. The in vivo effects of adenine-induced chronic kidney disease on some renal and hepatic function and CYP450 metabolizing enzymes. Physiol. Res. 2017, 66, 263–271. [Google Scholar]
- Smith, G.; Wolf, C.R.; Deeni, Y.Y.; Dawe, R.S.; Evans, A.T.; Comrie, M.M.; Ferguson, J.; Ibbotson, S.H. Cutaneous expression of cytochrome P450 CYP2S1: individuality in regulation by therapeutic agents for psoriasis and other skin diseases. Lancet. 2003, 361, 1336–1343. [Google Scholar] [CrossRef]
- Rivera, S.P.; Wang, F.; Saarikoski, S.T.; Taylor, R.T.; Chapman, B.; Zhang, R.; Hankinson, O. A novel promoter element containing multiple overlapping xenobiotic and hypoxia response elements mediates induction of cytochrome P4502S1 by both dioxin and hypoxia. J. Biol. Chem. 2007, 282, 10881–10893. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. Lancet. 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Oh, E.T.; Park, H.J. Implications of NQO1 in cancer therapy. BMB. Rep. 2015, 48, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Ládek, N.E.; Kollander, R.; Sreerama, L.; Kiang, D.T. Cellular levels of aldehyde dehydrogenases [ALDH1A1 and ALDH3A1] as predictors of therapeutic responses to cyclophosphamide-based chemotherapy of breast cancer: a retrospective study. Rational individualization of oxazaphosphorine-based cancer chemotherapeutic regimens. Cancer. Chemother. Pharmacol. 2002, 49, 309–321. [Google Scholar]
- Parajuli, B.; Georgiadis, T.M.; Fishel, M.L.; Hurley, T.D. Development of selective inhibitors for human aldehyde dehydrogenase 3A1 [ALDH3A1] for the enhancement of cyclophosphamide cytotoxicity. Chembio. Chem. 2014, 15, 701–712. [Google Scholar] [CrossRef]
- Ghosh, C.; Hossain, M.; Puvenna, V.; Martinez-Gonzalez, J.; Alexopolous, A.; Janigro, D.; Marchi, N. Expression and functional relevance of UGT1A4 in a cohort of human drug-resistant epileptic brains. Epilepsia. 2013, 54, 1562–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazerska, Z.; Mróz, A.; Pawłowska, M.; Augustin, E. The role of glucuronidation in drug resistance. Pharmacol. Ther. 2016, 159, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Erickson-Ridout, K.K.; Sun, D.; Lazarus, P. Glucuronidation of the secondgeneration antipsychotic clozapine and its active metabolite N desmethylclozapine. Potential importance of the UGT1A1 A[TA]7TAA and UGT1A4 L48V polymorphisms. Pharmacogenet. Genomics. 2012, 22, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Kerdpin, O.; Knights, K.M.; Elliot, D.J.; Miners, J.O. In vitro characterisation of human renal and hepatic frusemide glucuronidation and identification of the UDP-glucuronosyltransferase enzymes involved in this pathway. Biochem. Pharmacol. 2008, 76, 249–257. [Google Scholar] [CrossRef]
- Yamada, A.; Maeda, K.; Ishiguro, N.; Tsuda, Y.; Igarashi, T.; Ebner, T. The impact of pharmacogenetics of metabolic enzymes and transporters on the pharmacokineticsof telmisartan in healthy volunteers. Pharmacogenet. Genomics. 2011, 21, 523–530. [Google Scholar] [CrossRef]
- Jeong, E.S.; Kim, Y.W.; Kim, H.J.; Shin, H.J.; Shin, J.G.; Kim, K.H. Glucuronidation of fimasartan, a new angiotensin receptor antagonist, is mainly mediated by UGT1A3. Xenobiotica 2015, 45, 10–18. [Google Scholar] [CrossRef]
- Cho, S.K.; Oh, E.S.; Park, K.; Park, M.S.; Chung, J.Y. The UGT1A3*2 polymorphism affects atorvastatin lactonization and lipid lowering effect in healthy volunteers. Pharmacogenet. Genomics 2012, 22, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Fujino, H.; Yamada, I.; Shimada, S.; Yoneda, M.; Kojima, J. Metabolic fate of pitavastatin, a new inhibitor of HMG-CoA reductase: Human UDP-glucuronosyltransferase enzymes involved in lactonization. Xenobiotica 2003, 33, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Subramanian, R.; Fang, X.; Ma, B.; Qiu, Y.; Lin, J.H.; Pearson, P.G.; Baillie, T.A. Glucuronidation of statins in animals and humans: a novel mechanism of statin lactonization. Drug. Metab. Dispos. 2002, 30, 505–512. [Google Scholar] [CrossRef]
- Schirris, T.J.; Ritschel, T.; Bilos, A.; Smeitink, J.A.; Russel, F.G. Statin Lactonization by Uridine 5’-Diphospho-glucuronosyltransferases [UGTs]. Mol. Pharm. 2015, 2, 4048–4055. [Google Scholar] [CrossRef]
- Vanlersberghe, C.; Camu, F. Propofol. Handb. Exp. Pharmacol. 2008, 182, 227–252. [Google Scholar]
- Sharma, A.; Patrick, B.; Li, J.; Sharma, R.; Jeyabal, P.V.; Reddy, P.M.; Awasthi, S.; Awasthi, Y.C. Glutathione S-transferases as antioxidant enzymes: Small cell lung cancer [H69] cells transfected with hGSTA1 resist doxorubicin-induced apoptosis. Arch. Biochem. Biophys. 2006, 45, 165–173. [Google Scholar] [CrossRef]
- Locher, K.P. Structure and mechanism of ATP-binding cassette transporters. Philos.Trans. R. Soc. Lond. B. Biol. Sci. 2009, 364, 239–245. [Google Scholar] [CrossRef]
- Chang, G. Multidrug resistance ABC transporters. FEBS. Lett. 2003, 555, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer. Cell. Inter. 2005, 5, 1–13. [Google Scholar] [CrossRef]
- He, S.M.; Li, R.; Kanwar, J.R.; Zhou, S.F. Structural and functional properties of human multidrug resistance protein 1 [MRP1/ABCC1]. Curr. Med. Chem. 2011, 18, 439–481. [Google Scholar] [CrossRef]
- Gynther, M.; Laine, K.; Ropponen, J.; Leppänen, J.; Mannila, A.; Nevalainen, T.; Savolainen, J.; Järvinen, T.; Rautio, J. Large neutral amino acid transporter enables brain drug delivery via prodrugs. J. Med. Chem. 2008, 51, 932–936. [Google Scholar] [CrossRef]
- Del Amo, E.M.; Urtti, A.; Yliperttula, M. Pharmacokinetic role of L-type amino acidtransporters LAT1 and LAT2. Eur. J. Pharm. Sci. 2008, 35, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.; Soares-da-Silva, P. Interaction between L-DOPA and 3-O-methyl-L-DOPA for transport in immortalised rat capillary cerebral endothelial cells. Neuropharmacology 1999, 38, 1371–1380. [Google Scholar] [CrossRef]
- Kageyama, T.; Nakamura, M.; Matsuo, A.; Yamasaki, Y.; Takakura, Y.; Hashida, M.; Kanai, Y.; Naito, M.; Tsuruo, T.; Minato, N.; et al. The 4F2hc/LAT1 complex transports L-DOPA across the blood-brain barrier. Brain. Res. 2000, 879, 115–121. [Google Scholar] [CrossRef]
- Kido, Y.; Tamai, I.; Uchino, H.; Suzuki, F.; Sai, Y.; Tsuji, A. Molecular and functional identification of large neutral amino acid transporters LAT1 and LAT2 and their pharmacological relevance at the blood-brain barrier. J. Pharm. Pharmacol. 2001, 53, 497–503. [Google Scholar] [CrossRef]
- Sampaio-Maia, B.; Serrão, M.P.; Soares-da-Silva, P. Regulatory pathways and uptake of L-DOPA by capillary cerebral endothelial cells, astrocytes, and neuronal cells. Am. J. Physiol. Cell. Physiol. 2001, 280, C333–C342. [Google Scholar] [CrossRef]
- Soares-da-Silva, P.; Serrão, P.; Fraga, S.; Pinho, M.J. Expression and function of LAT1, a neutral amino acid exchanger, in renal porcine epithelial cell line LLC-PK. Acta. Physiol. Scand. 2005, 185, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Fraga, S.; Sampaio-Maia, B.; Serrão, M.P.; Soares-da-Silva, P. Regulation of apical transporter of L-DOPA in human intestinal Caco-2 cells. Acta. Physiol. Scand. 2002, 175, 103–111. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cardiovascular Drugs | Metabolism |
| β-receptor blockers | insulin |
| calcium channel blockers | antidiabetics (sulfonylureas, biguanides/metformin, thiazolidinediones, insulin) |
| antianginals | thyroid hormones |
| angiotensin-converting-enzyme inhibitors | diphosphonate |
| angiotensin receptor blockers | vasopressin analogues |
| Alpha blockers | calcium |
| thiazide diuretics | allopurinol |
| loop diuretics | phosphate chelators |
| heparin | |
| Warfarin | |
| direct oral anticoagulant | |
| antiplatelet drugs | |
| 3-hydroxy-3-methylglutaryl-CoA reductase reductase inhibitors | |
| Digestive System | For Infections and Infestations |
| antacids | antibiotics |
| proton pump inhibitors (PPIs) | Anti-tuberculous drugs |
| H2-receptor antagonists | antivirals |
| Laxatives | |
| Central Nervous System | For the Immune System |
| hypnotics | vaccines |
| anesthetics | immunoglobulins |
| antidepressants (including tricyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs) anticonvulsants/antiepileptics | immunosuppressants |
| benzodiazepines | monoclonal antibodies |
| antihistamines | corticosteroids |
| Pain Killers | |
| Paracetamol | |
| Tramadol | |
| pioids |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana Machado, T.; Cerini, C.; Burtey, S. Emerging Roles of Aryl Hydrocarbon Receptors in the Altered Clearance of Drugs during Chronic Kidney Disease. Toxins 2019, 11, 209. https://doi.org/10.3390/toxins11040209
Santana Machado T, Cerini C, Burtey S. Emerging Roles of Aryl Hydrocarbon Receptors in the Altered Clearance of Drugs during Chronic Kidney Disease. Toxins. 2019; 11(4):209. https://doi.org/10.3390/toxins11040209
Chicago/Turabian StyleSantana Machado, Tacy, Claire Cerini, and Stéphane Burtey. 2019. "Emerging Roles of Aryl Hydrocarbon Receptors in the Altered Clearance of Drugs during Chronic Kidney Disease" Toxins 11, no. 4: 209. https://doi.org/10.3390/toxins11040209
APA StyleSantana Machado, T., Cerini, C., & Burtey, S. (2019). Emerging Roles of Aryl Hydrocarbon Receptors in the Altered Clearance of Drugs during Chronic Kidney Disease. Toxins, 11(4), 209. https://doi.org/10.3390/toxins11040209