An In-Silico Sequence-Structure-Function Analysis of the N-Terminal Lobe in CT Group Bacterial ADP-Ribosyltransferase Toxins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
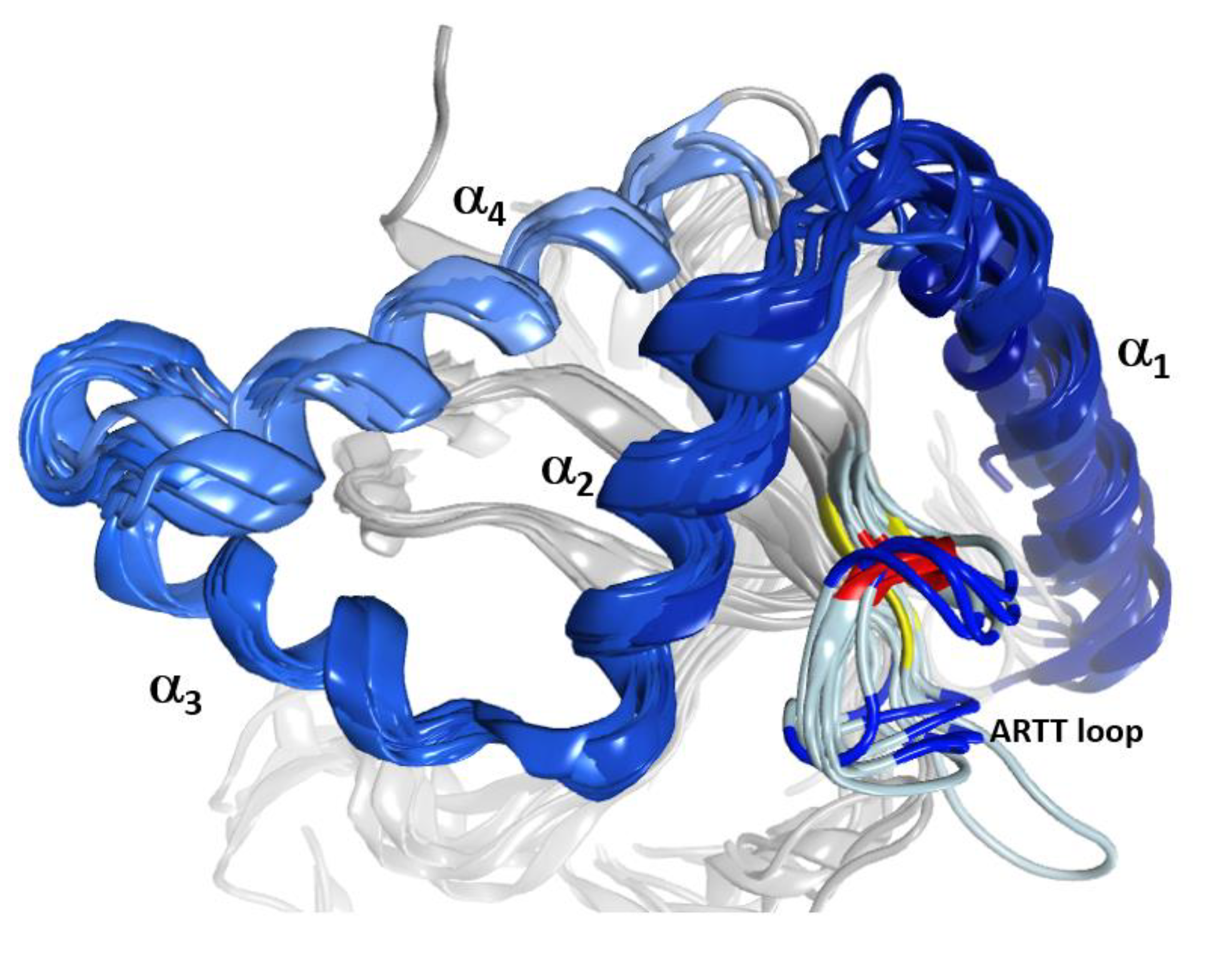
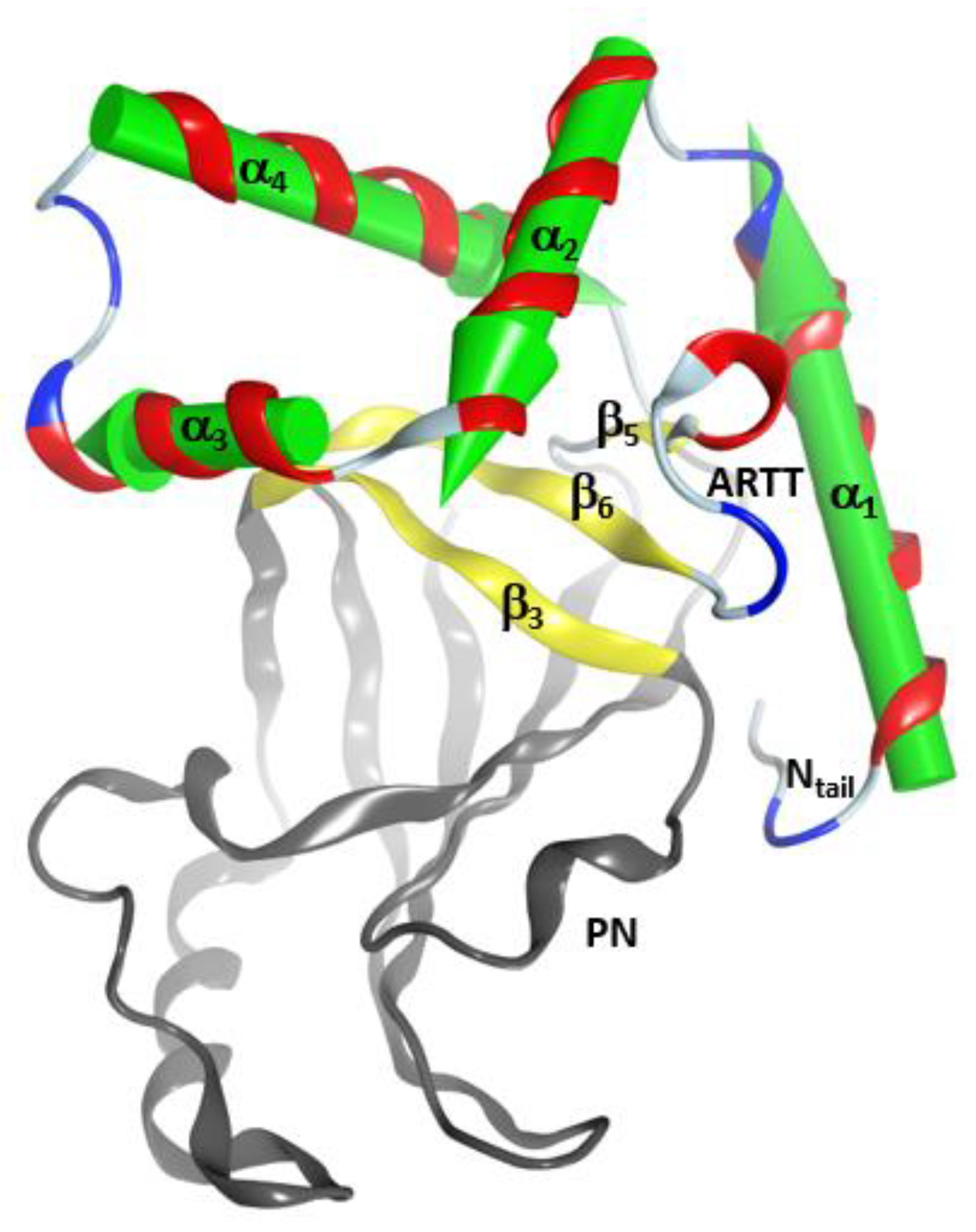
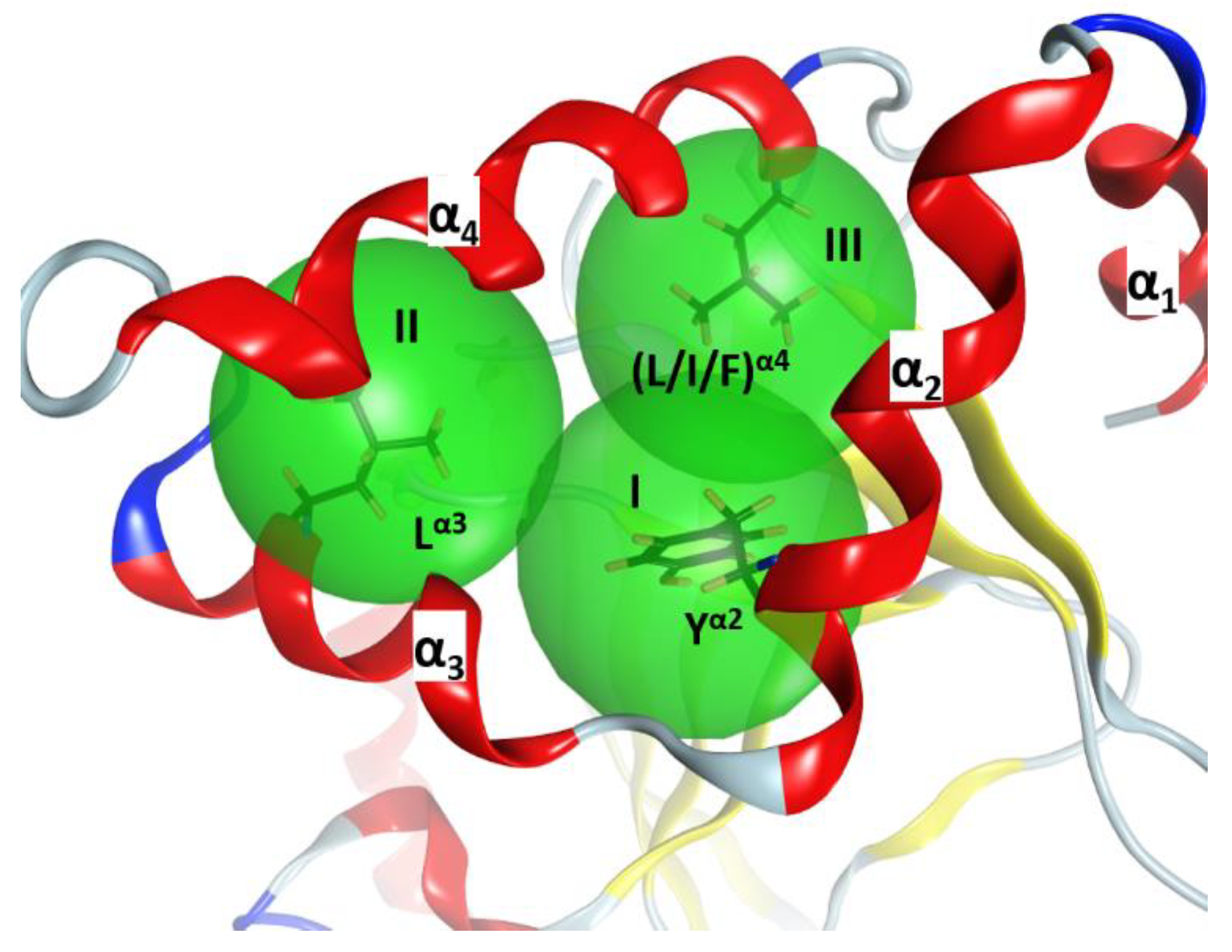
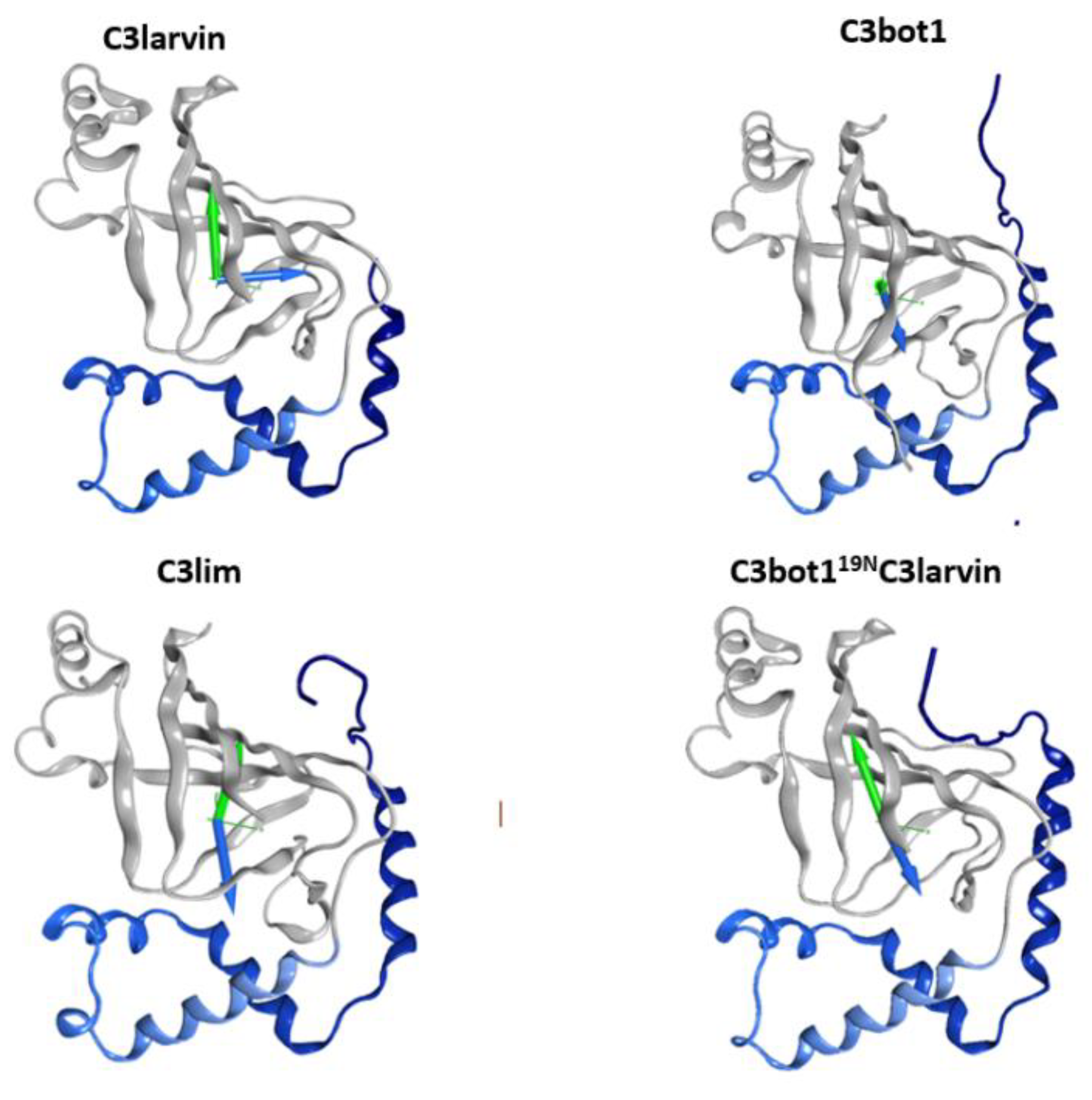
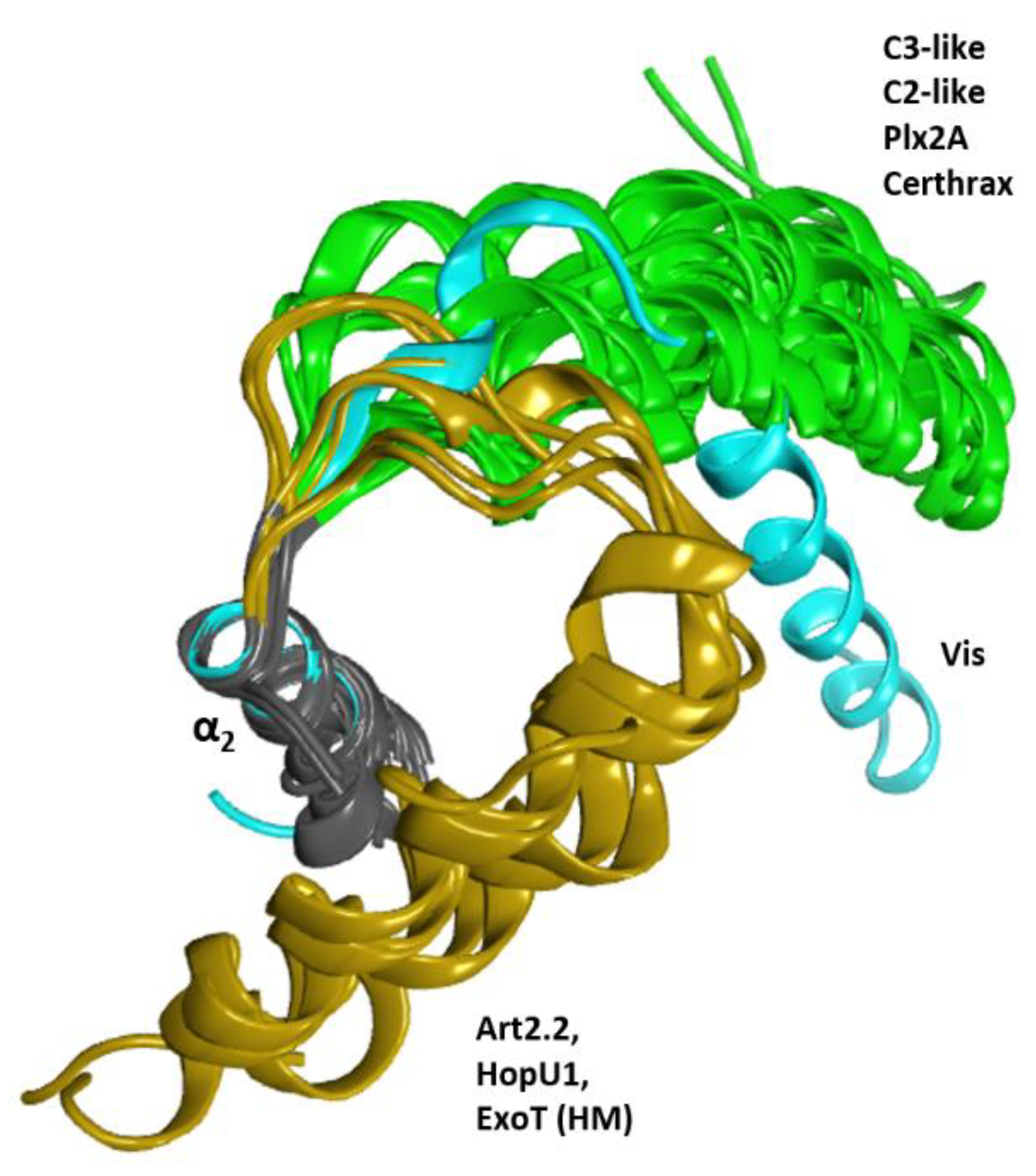
2.1. The Overall Structure of the N-Terminal α-Lobe
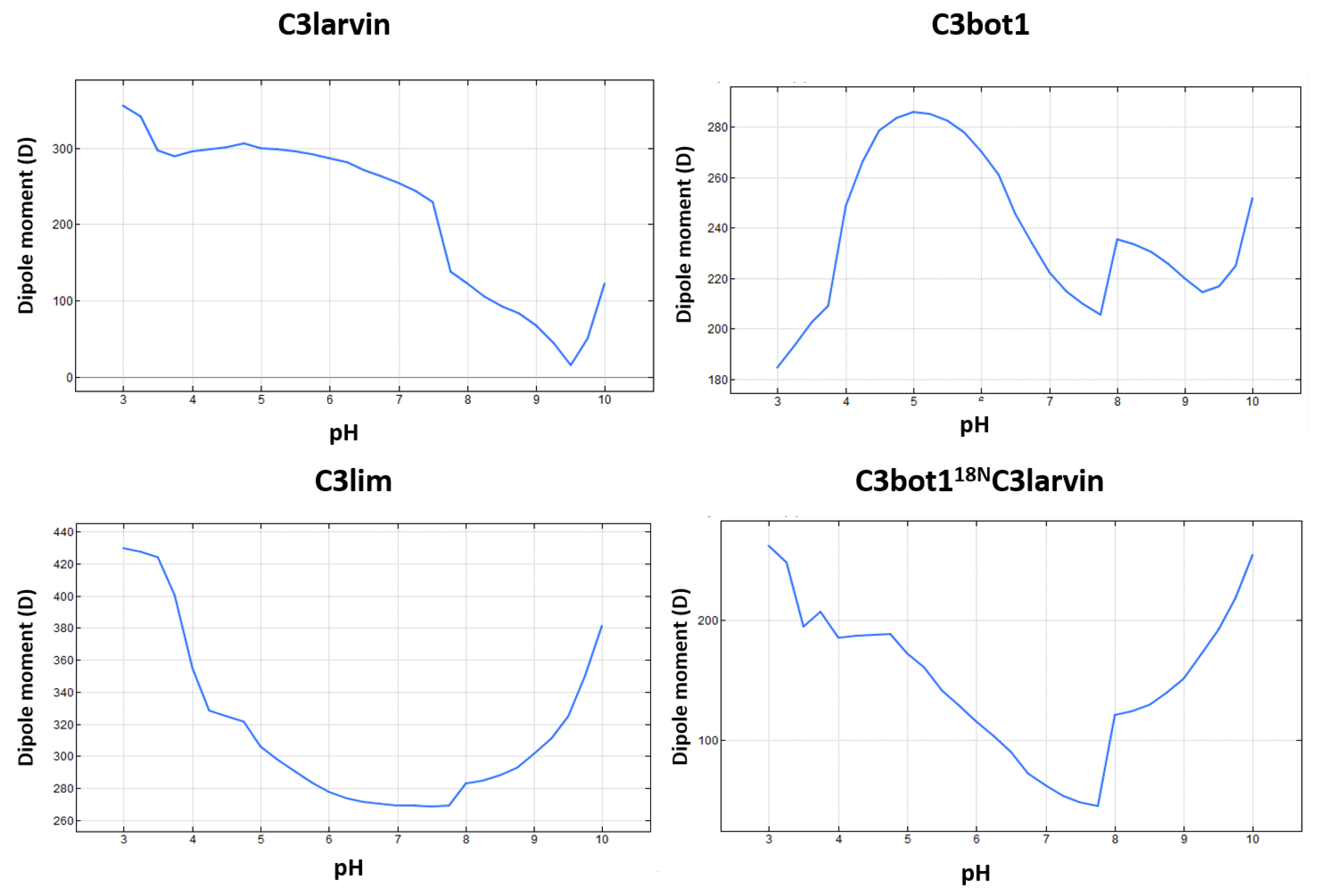
2.2. Stability of the α2−α4 Superstructure
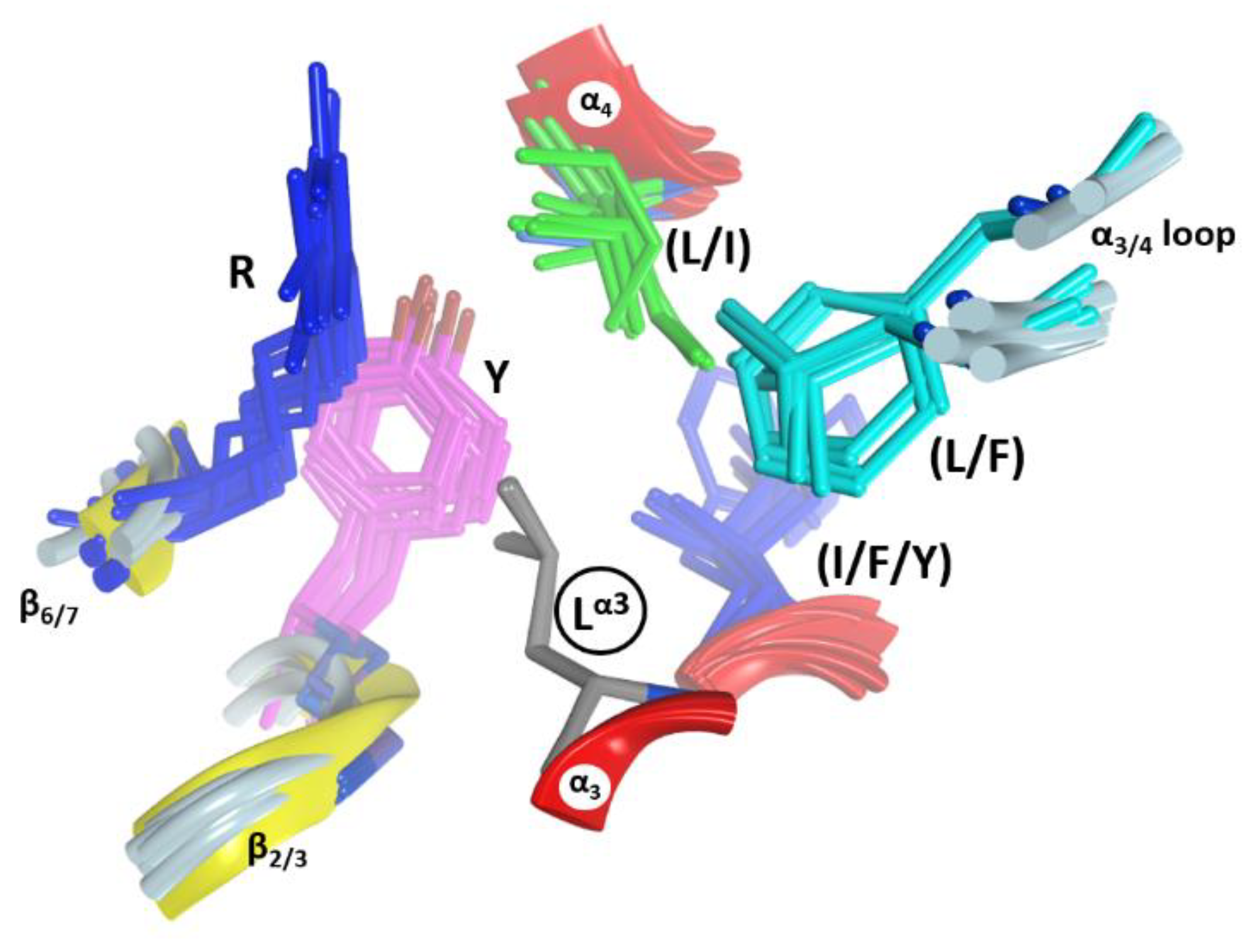
2.3. The α3-Helix and the α3-Motif
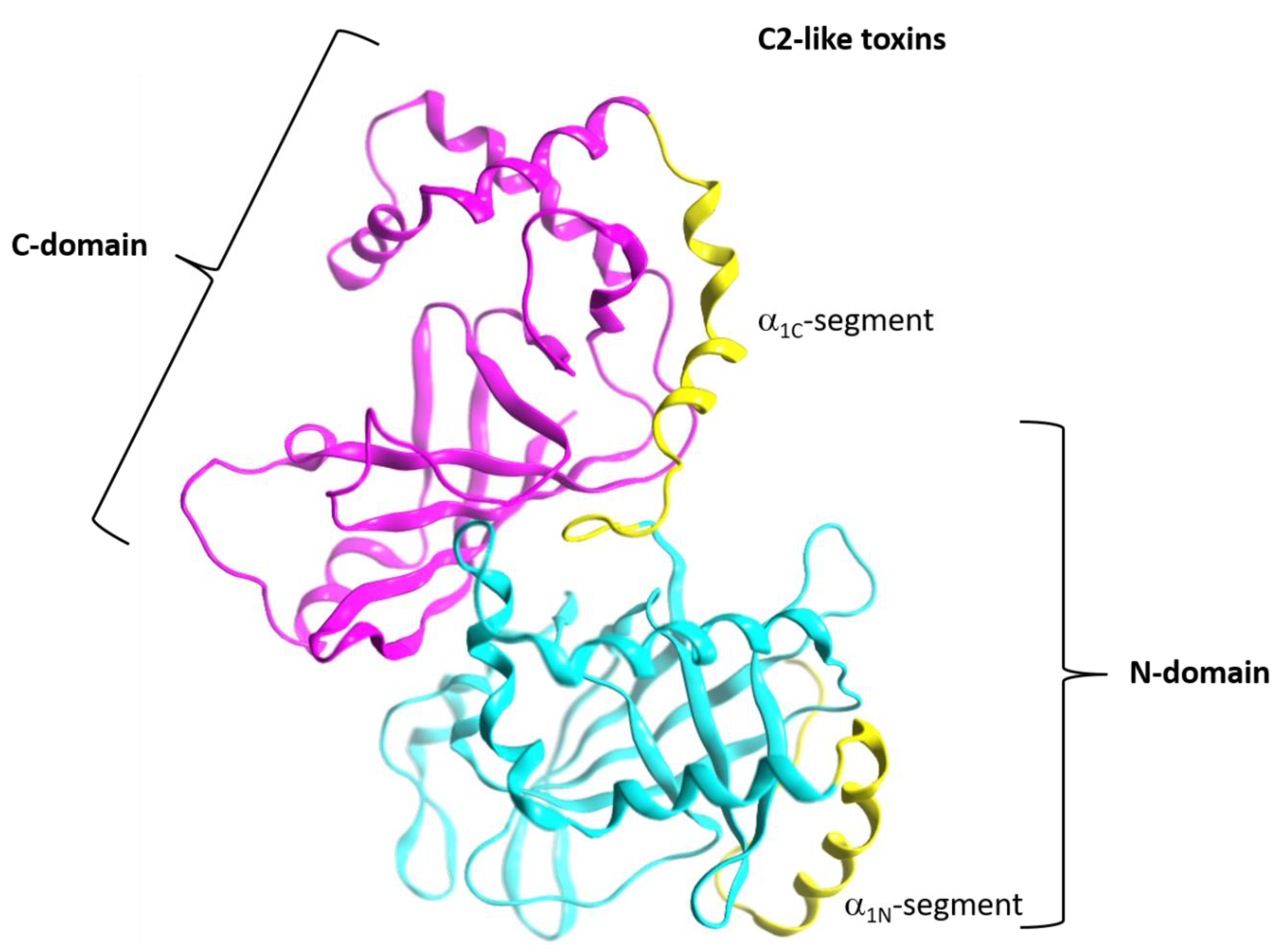
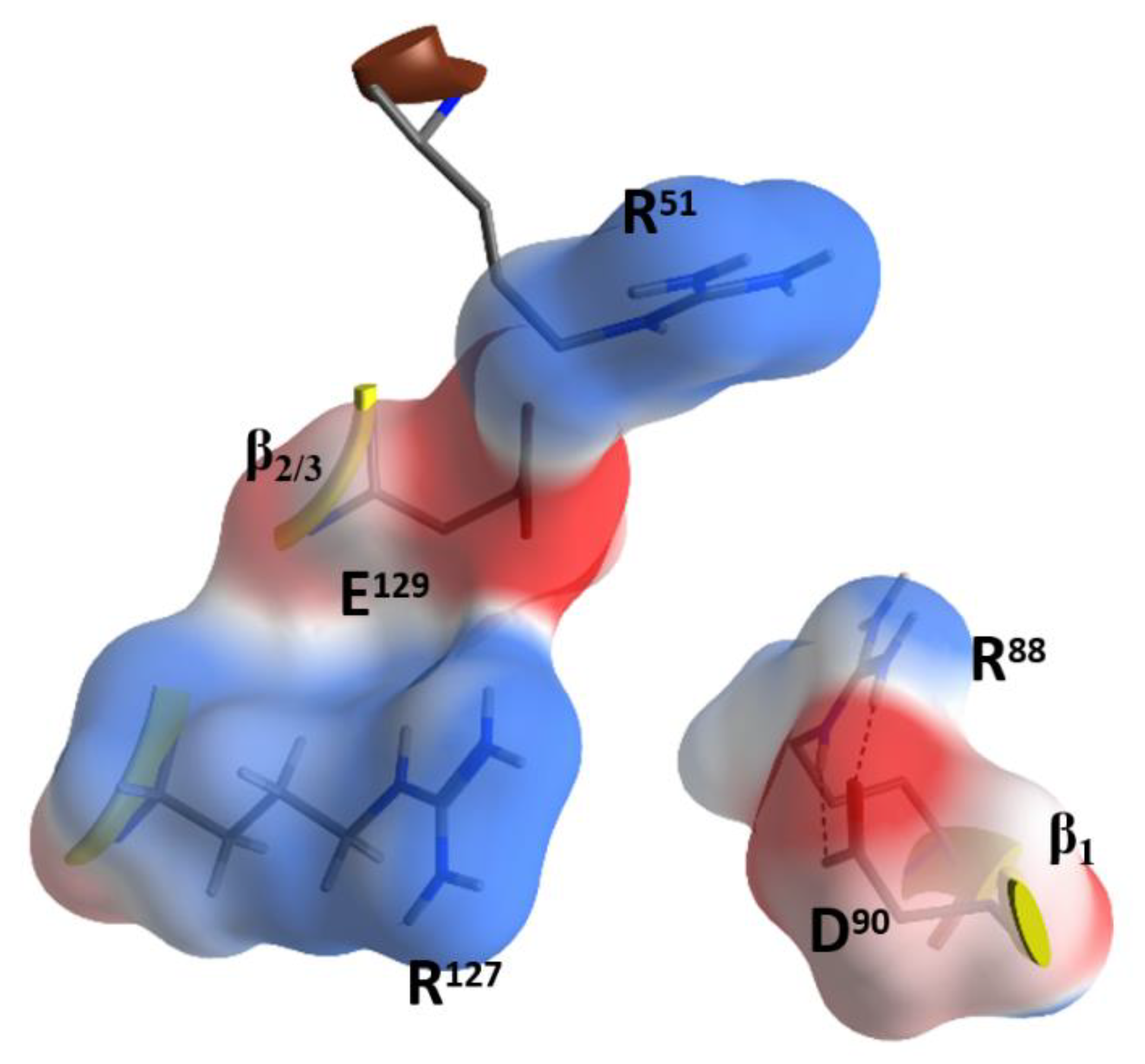
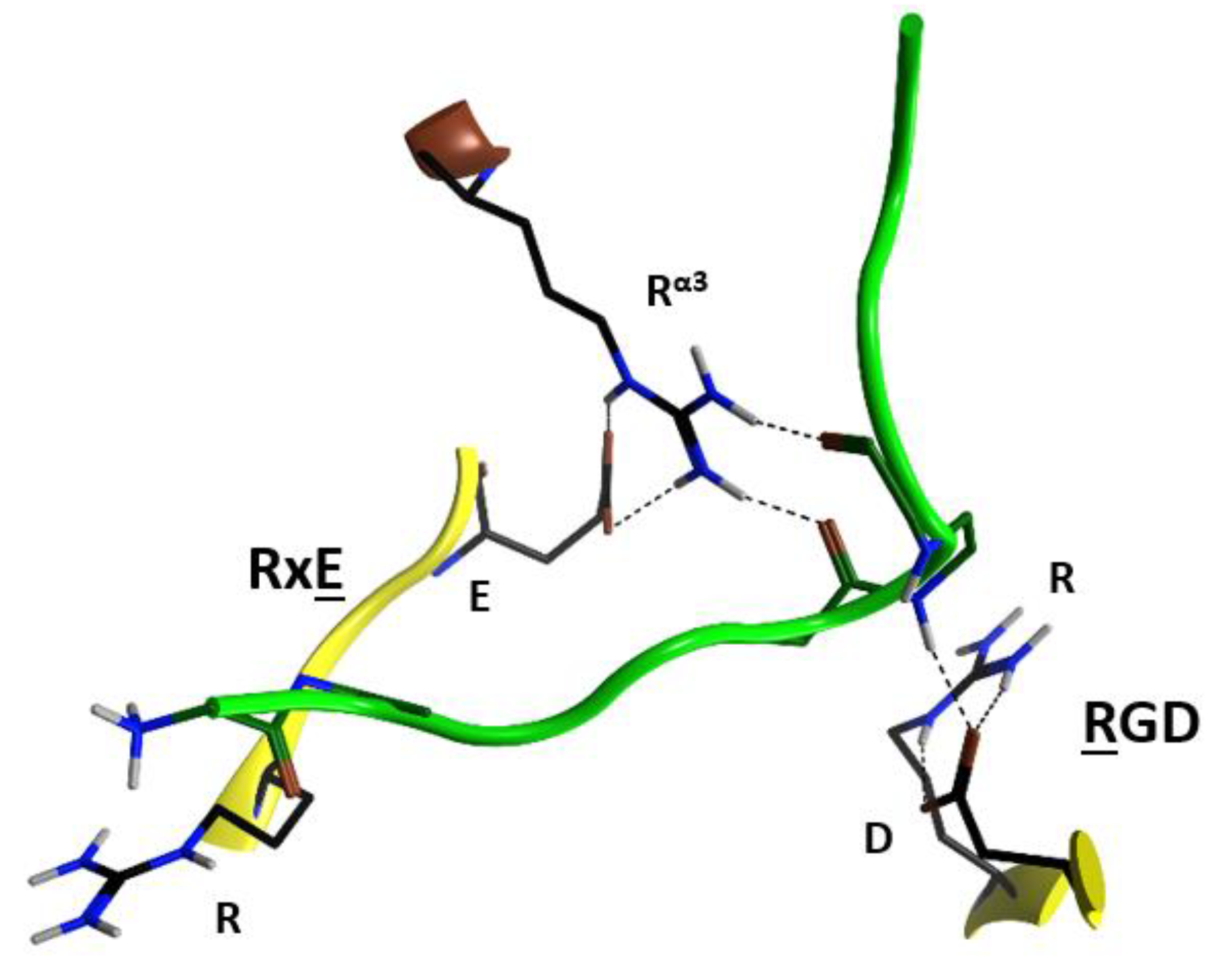
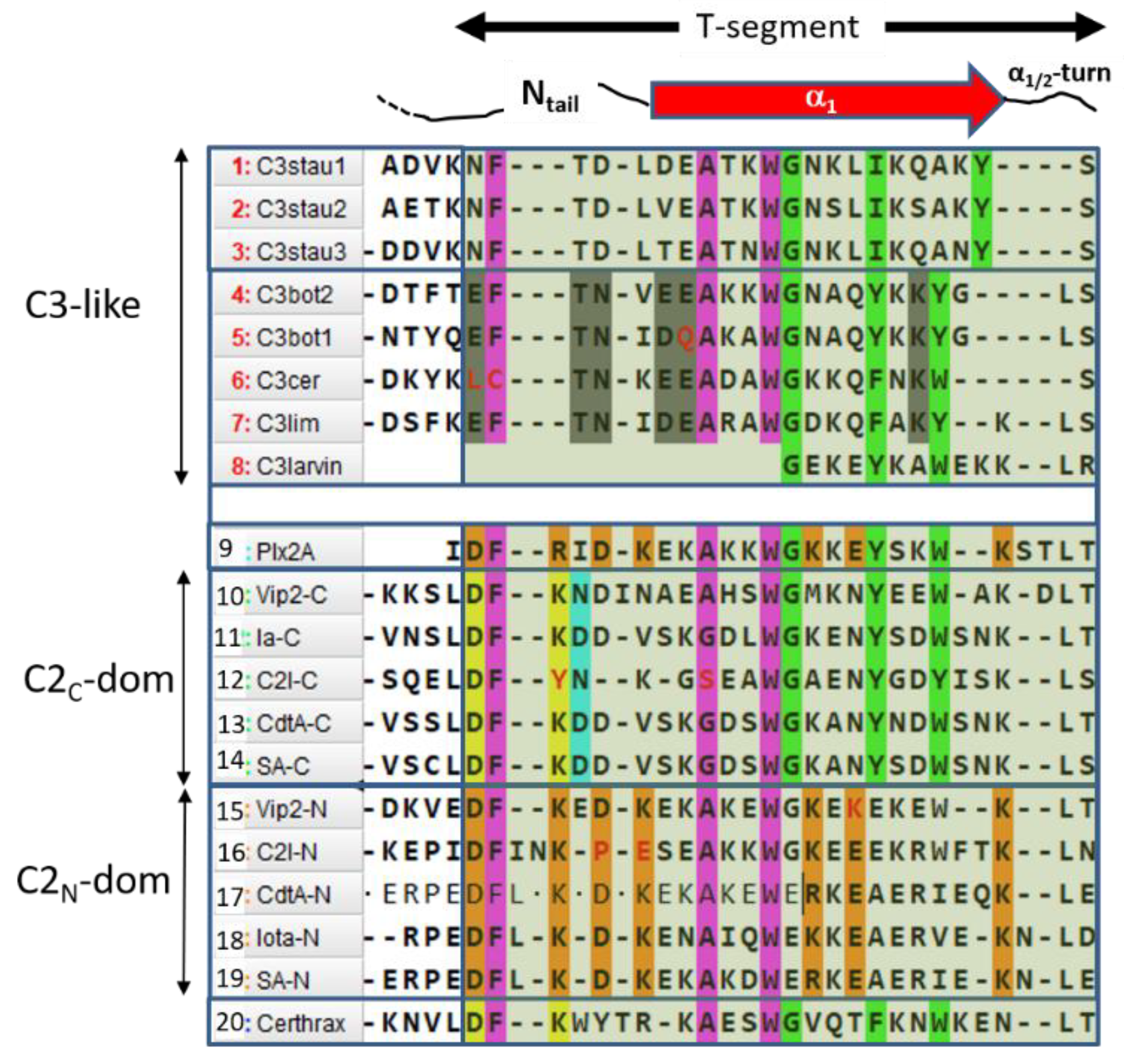
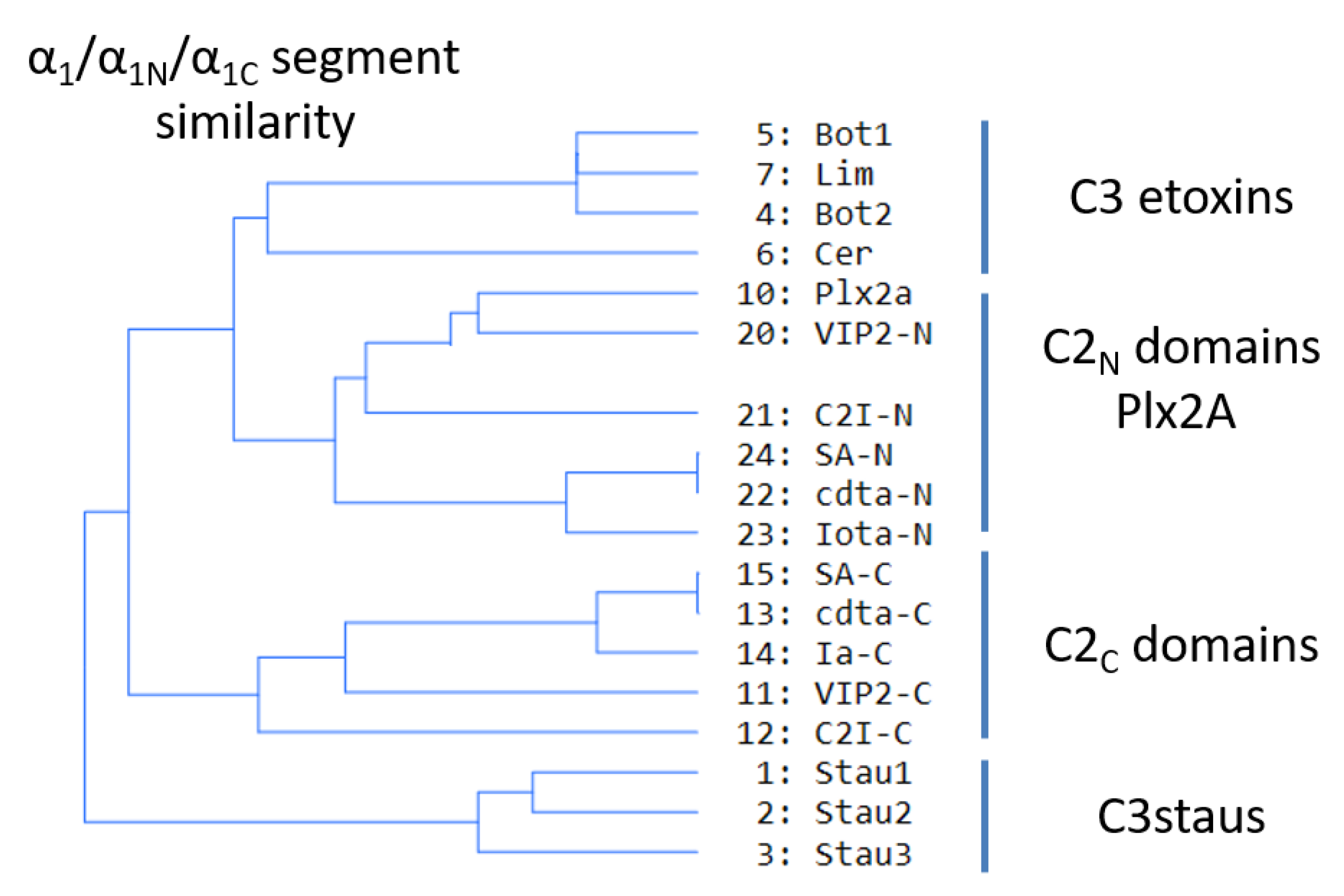
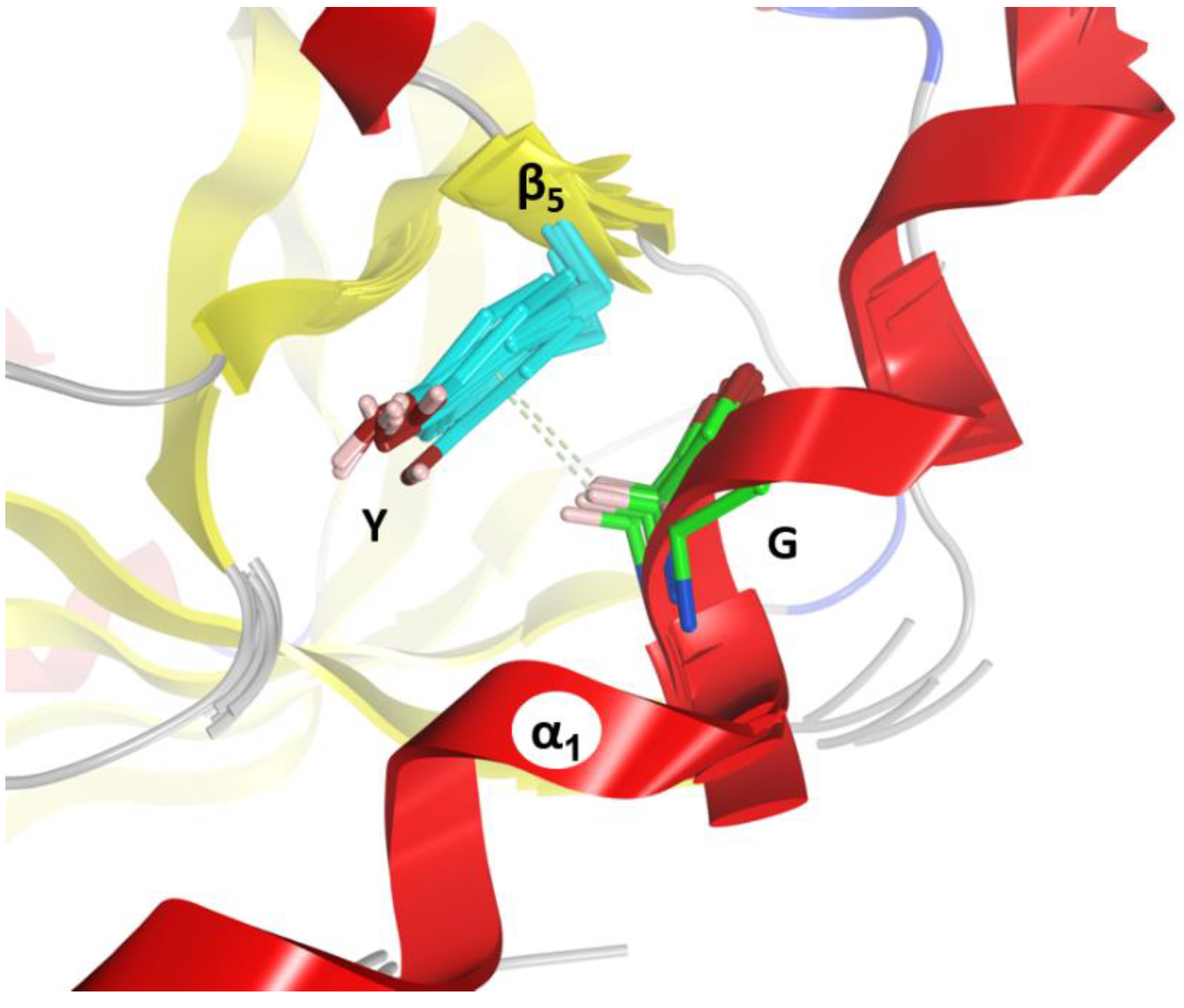
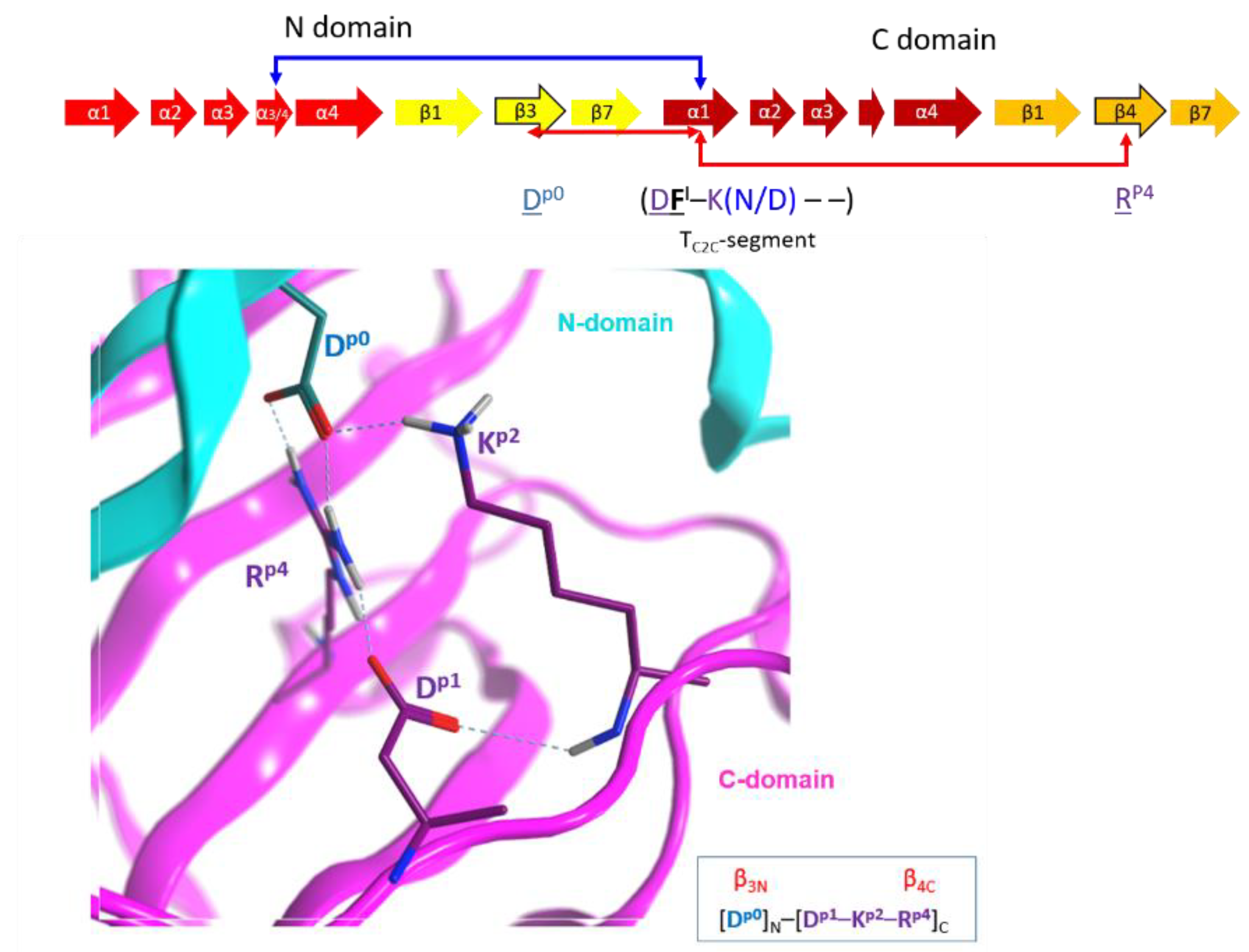
2.4. Role of the Ntail-α1 Segment in C3- and C2-Like Toxins
- (S1)
- FI–A–WIIG–(Y/F)III–(Y/W)IV, in Plx2A, and C3-etoxins,
- (S2)
- DFI–D–(A/G)–WIIG–YIII–WIV−K, in Plx2A, and C2C domains, and
- (S3)
- DFI–K–D–(K/R)–A–WII–(K/R)−E–K, in Plx2A, and C2N domains.
2.5. The Putative Role of the α3/4-Loop
2.6. The T-Segment in Other CT-Toxins
3. Conclusions
4. Materials and Methods
4.1. Ensemble of X-Ray Protein Structures
4.2. Force-Field Settings and Structure Preparation
4.3. Others
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simon, N.C.; Aktories, K.; Barbieri, J.T. Novel bacterial ADP-ribosylating toxins: Structure and function. Nat. Rev. Microbiol. 2014, 12, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Howorth, D.; Chothia, C.; Kulesha, E.; Murzin, A.G. Investigating protein structure and evolution with SCOP2. Curr. Protoc. Bioinform. 2015, 49, 1.26.1–1.26.21. [Google Scholar]
- Fieldhouse, R.J.; Merrill, A.R. Needle in the haystack: Structure-based toxin discovery. Trends Biochem. Sci. 2008, 33, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Fischer, S.; Moglich, A.; Fortsch, C. Clostridial C3 toxins target monocytes/macrophages and modulate their functions. Front. Immunol. 2015, 6, 339. [Google Scholar] [CrossRef]
- Ravulapalli, R.; Lugo, M.R.; Pfoh, R.; Visschedyk, D.; Poole, A.; Fieldhouse, R.J.; Pai, E.F.; Merrill, A.R. Characterization of vis toxin, a novel ADP-ribosyltransferase from Vibrio splendidus. Biochemistry 2015, 54, 5920–5936. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, J.T.; Sun, J. Pseudomonas aeruginosa ExoS and ExoT. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2004; Volume 152, pp. 79–92. [Google Scholar]
- Sakurai, J.; Nagahama, M.; Hisatsune, J.; Katunuma, N.; Tsuge, H. Clostridium perfringens iota-toxin, ADP-ribosyltransferase: Structure and mechanism of action. Adv. Enzyme Regul. 2003, 43, 361–377. [Google Scholar] [CrossRef]
- Sanchez, J.; Holmgren, J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell. Mol. Life Sci. 2008, 65, 1347–1360. [Google Scholar] [CrossRef]
- Visschedyk, D.; Rochon, A.; Tempel, W.; Dimov, S.; Park, H.W.; Merrill, A.R. Certhrax toxin, an anthrax-related ADP-ribosyltransferase from Bacillus cereus. J. Biol. Chem. 2012, 287, 41089–41102. [Google Scholar] [CrossRef]
- Simon, N.C.; Barbieri, J.T. Bacillus cereus Certhrax ADP-ribosylates vinculin to disrupt focal adhesion complexes and cell adhesion. J. Biol. Chem. 2014, 289, 10650–10659. [Google Scholar] [CrossRef]
- Simon, N.C.; Vergis, J.M.; Ebrahimi, A.V.; Ventura, C.L.; O’Brien, A.D.; Barbieri, J.T. Host cell cytotoxicity and cytoskeleton disruption by CerADPr, an ADP-ribosyltransferase of Bacillus cereus G9241. Biochemistry 2013, 52, 2309–2318. [Google Scholar] [CrossRef]
- Fieldhouse, R.J.; Turgeon, Z.; White, D.; Merrill, A.R. Cholera- and anthrax-like toxins are among several new ADP-ribosyltransferases. PLoS Comput. Biol. 2010, 6, e1001029. [Google Scholar] [CrossRef] [PubMed]
- Iglewski, B.H.; Sadoff, J.; Bjorn, M.J.; Maxwell, E.S. Pseudomonas aeruginosa exoenzyme S: An adenosine diphosphate ribosyltransferase distinct from toxin A. Proc. Natl. Acad. Sci. USA 1978, 75, 3211–3215. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Rokuda, M.; Park, K.S.; Cantarelli, V.V.; Matsuda, S.; Iida, T.; Honda, T. Identification and characterization of VopT, a novel ADP-ribosyltransferase effector protein secreted via the Vibrio parahaemolyticus type III secretion system 2. Cell. Microbiol. 2007, 9, 2598–2609. [Google Scholar] [CrossRef] [PubMed]
- Litvak, Y.; Selinger, Z. Aeromonas salmonicida toxin AexT has a Rho family GTPase-activating protein domain. J. Bacteriol. 2007, 189, 2558–2560. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Barmann, M.; Ohishi, I.; Tsuyama, S.; Jakobs, K.H.; Habermann, E. Botulinum C2 toxin ADP-ribosylates actin. Nature 1986, 322, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, J.; Schering, B.; Barmann, M.; Aktories, K. Clostridium perfringens iota toxin ADP-ribosylates skeletal muscle actin in Arg-177. FEBS Lett. 1987, 225, 48–52. [Google Scholar] [CrossRef]
- Popoff, M.R.; Milward, F.W.; Bancillon, B.; Boquet, P. Purification of the Clostridium spiroforme binary toxin and activity of the toxin on HEp-2 cells. Infect. Immun. 1989, 57, 2462–2469. [Google Scholar] [PubMed]
- Chakroun, M.; Banyuls, N.; Bel, Y.; Escriche, B.; Ferre, J. Bacterial vegetative insecticidal proteins (vip) from entomopathogenic bacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 329–350. [Google Scholar] [CrossRef]
- Popoff, M.R.; Rubin, E.J.; Gill, D.M.; Boquet, P. Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect. Immun. 1988, 56, 2299–2306. [Google Scholar]
- Yonogi, S.; Matsuda, S.; Kawai, T.; Yoda, T.; Harada, T.; Kumeda, Y.; Gotoh, K.; Hiyoshi, H.; Nakamura, S.; Kodama, T.; et al. BEC, a novel enterotoxin of Clostridium perfringens found in human clinical isolates from acute gastroenteritis outbreaks. Infect. Immun. 2014, 82, 2390–2399. [Google Scholar] [CrossRef]
- Tsuge, H.; Tsurumura, T.; Toda, A.; Murata, H.; Toniti, W.; Yoshida, T. Comparative studies of actin- and Rho-specific ADP-ribosylating toxins: Insight from structural biology. Curr. Top. Microbiol. Immunol. 2017, 399, 69–86. [Google Scholar] [PubMed]
- Aktories, K.; Rosener, S.; Blaschke, U.; Chhatwal, G.S. Botulinum ADP-ribosyltransferase C3. Purification of the enzyme and characterization of the ADP-ribosylation reaction in platelet membranes. Eur. J. Biochem. 1988, 172, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, Y.; Namba, T.; Kozaki, S.; Narumiya, S. Clostridium botulinum C3 ADP-ribosyltransferase gene. Cloning, sequencing, and expression of a functional protein in Escherichia coli. J. Biol. Chem. 1991, 266, 19312–19319. [Google Scholar] [PubMed]
- Just, I.; Mohr, C.; Schallehn, G.; Menard, L.; Didsbury, J.R.; Vandekerckhove, J.; Van Damme, J.; Aktories, K. Purification and characterization of an ADP-ribosyltransferase produced by Clostridium limosum. J. Biol. Chem. 1992, 267, 10274–10280. [Google Scholar] [PubMed]
- Wilde, C.; Vogelsgesang, M.; Aktories, K. Rho-specific Bacillus cereus ADP-ribosyltransferase C3cer cloning and characterization. Biochemistry 2003, 42, 9694–9702. [Google Scholar] [CrossRef] [PubMed]
- Krska, D.; Ravulapalli, R.; Fieldhouse, R.J.; Lugo, M.R.; Merrill, A.R. C3larvin toxin, an ADP-ribosyltransferase from Paenibacillus larvae. J. Biol. Chem. 2015, 290, 1639–1653. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, J.; Funfhaus, A.; Knispel, H.; Krska, D.; Ravulapalli, R.; Heney, K.A.; Lugo, M.R.; Merrill, A.R.; Genersch, E. Characterization of the toxin Plx2A, a RhoA-targeting ADP-ribosyltransferase produced by Paenibacillus larvae. Environ. Microbiol. 2017, 19, 5100–5116. [Google Scholar] [CrossRef]
- Wilde, C.; Just, I.; Aktories, K. Structure-function analysis of the Rho-ADP-ribosylating exoenzyme C3stau2 from Staphylococcus aureus. Biochemistry 2002, 41, 1539–1544. [Google Scholar] [CrossRef]
- Evans, H.R.; Sutton, J.M.; Holloway, D.E.; Ayriss, J.; Shone, C.C.; Acharya, K.R. The crystal structure of C3stau2 from Staphylococcus aureus and its complex with NAD. J. Biol. Chem. 2003, 278, 45924–45930. [Google Scholar] [CrossRef]
- Vogelsgesang, M.; Pautsch, A.; Aktories, K. C3 exoenzymes, novel insights into structure and action of Rho-ADP-ribosylating toxins. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 374, 347–360. [Google Scholar] [CrossRef]
- Gill, D.M.; Meren, R. Adp-ribosylation of membrane proteins catalyzed by cholera toxin: Basis of the activation of adenylate cyclase. Proc. Natl. Acad. Sci. USA 1978, 75, 3050–3054. [Google Scholar] [CrossRef] [PubMed]
- Farizo, K.M.; Fiddner, S.; Cheung, A.M.; Burns, D.L. Membrane localization of the S1 subunit of pertussis toxin in Bordetella pertussis and implications for pertussis toxin secretion. Infect. Immun. 2002, 70, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Taxt, A.; Aasland, R.; Sommerfelt, H.; Nataro, J.; Puntervoll, P. Heat-stable enterotoxin of enterotoxigenic Escherichia coli as a vaccine target. Infect. Immun. 2010, 78, 1824–1831. [Google Scholar] [CrossRef] [PubMed]
- Lyons, B.; Ravulapalli, R.; Lanoue, J.; Lugo, M.R.; Dutta, D.; Carlin, S.; Merrill, A.R. Scabin, a novel DNA-acting ADP-ribosyltransferase from Streptomyces scabies. J. Biol. Chem. 2016, 291, 11198–11215. [Google Scholar] [CrossRef]
- Coye, L.H.; Collins, C.M. Identification of SpyA, a novel ADP-ribosyltransferase of Streptococcus pyogenes. Mol. Microbiol. 2004, 54, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Icenogle, L.M.; Hengel, S.M.; Coye, L.H.; Streifel, A.; Collins, C.M.; Goodlett, D.R.; Moseley, S.L. Molecular and biological characterization of Streptococcal SpyA-mediated ADP-ribosylation of intermediate filament protein vimentin. J. Biol. Chem. 2012, 287, 21481–21491. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.J.; Chen, J.; Kant, S.; Rechkina, E.; Rush, J.S.; Forsberg, L.S.; Jaehrig, B.; Azadi, P.; Tchesnokova, V.; Sokurenko, E.V.; et al. SpyB, a small heme-binding protein, affects the composition of the cell wall in Streptococcus pyogenes. Front. Cell. Infect. Microbiol. 2016, 6, 126. [Google Scholar] [CrossRef]
- Shniffer, A.; Visschedyk, D.D.; Ravulapalli, R.; Suarez, G.; Turgeon, Z.J.; Petrie, A.A.; Chopra, A.K.; Merrill, A.R. Characterization of an actin-targeting ADP-ribosyltransferase from Aeromonas hydrophila. J. Biol. Chem. 2012, 287, 37030–37041. [Google Scholar] [CrossRef]
- Visschedyk, D.D.; Perieteanu, A.A.; Turgeon, Z.J.; Fieldhouse, R.J.; Dawson, J.F.; Merrill, A.R. Photox, a novel actin-targeting mono-ADP-ribosyltransferase from Photorhabdus luminescens. J. Biol. Chem. 2010, 285, 13525–13534. [Google Scholar] [CrossRef]
- Sha, J.; Wang, S.F.; Suarez, G.; Sierra, J.C.; Fadl, A.A.; Erova, T.E.; Foltz, S.M.; Khajanchi, B.K.; Silver, A.; Graf, J.; et al. Further characterization of a type III secretion system (T3SS) and of a new effector protein from a clinical isolate of Aeromonas hydrophila—Part I. Microb. Pathog. 2007, 43, 127–146. [Google Scholar] [CrossRef]
- Greene, L.H.; Lewis, T.E.; Addou, S.; Cuff, A.; Dallman, T.; Dibley, M.; Redfern, O.; Pearl, F.; Nambudiry, R.; Reid, A.; et al. The CATH domain structure database: New protocols and classification levels give a more comprehensive resource for exploring evolution. Nucleic Acids Res. 2007, 35, D291–D297. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Maresso, A.W.; Kim, J.J.; Barbieri, J.T. How bacterial ADP-ribosylating toxins recognize substrates. Nat. Struct. Mol. Biol. 2004, 11, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.R.; Lin, Y.; Joe, A.; Guo, M.; Korneli, C.; Yang, H.; Wang, P.; Yu, M.; Cerny, R.L.; Staiger, D.; et al. Structure function analysis of an ADP-ribosyltransferase type III effector and its RNA-binding target in plant immunity. J. Biol. Chem. 2011, 286, 43272–43281. [Google Scholar] [CrossRef]
- Lang, A.E.; Schmidt, G.; Schlosser, A.; Hey, T.D.; Larrinua, I.M.; Sheets, J.J.; Mannherz, H.G.; Aktories, K. Photorhabdus luminescens toxins ADP-ribosylate actin and RhoA to force actin clustering. Science 2010, 327, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, P.; Yang, H.; Xu, Y. Crystallization and preliminary crystallographic analysis of the ADP-ribosyltransferase HopU1. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Von Elsner, L.; Hagemann, S.; Just, I.; Rohrbeck, A. C3 exoenzyme impairs cell proliferation and apoptosis by altering the activity of transcription factors. Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 1021–1031. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [PubMed]
- Winton, M.J.; Dubreuil, C.I.; Lasko, D.; Leclerc, N.; McKerracher, L. Characterization of new cell permeable C3-like proteins that inactivate Rho and stimulate neurite outgrowth on inhibitory substrates. J. Biol. Chem. 2002, 277, 32820–32829. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Kuban, J.; Heine, K.; Rupps, G.; Kaiser, E.; Felder, E.; Benz, R.; Barth, H. Selective and specific internalization of clostridial C3 ADP-ribosyltransferases into macrophages and monocytes. Cell. Microbiol. 2010, 12, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Rohrbeck, A.; Schroder, A.; Hagemann, S.; Pich, A.; Holtje, M.; Ahnert-Hilger, G.; Just, I. Vimentin mediates uptake of C3 exoenzyme. PLoS ONE 2014, 9, e101071. [Google Scholar] [CrossRef] [PubMed]
- Rohrbeck, A.; Just, I. Cell entry of C3 exoenzyme from Clostridium botulinum. Curr. Top. Microbiol. Immunol. 2017, 406, 97–118. [Google Scholar] [PubMed]
- Rohrbeck, A.; Holtje, M.; Adolf, A.; Oms, E.; Hagemann, S.; Ahnert-Hilger, G.; Just, I. The Rho ADP-ribosylating C3 exoenzyme binds cells via an Arg-Gly-Asp motif. J. Biol. Chem. 2017, 292, 17668–17680. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, I.; Yanagimoto, A. Visualizations of binding and internalization of two nonlinked protein components of botulinum C2 toxin in tissue culture cells. Infect. Immun. 1992, 60, 4648–4655. [Google Scholar] [PubMed]
- Barth, H.; Preiss, J.C.; Hofmann, F.; Aktories, K. Characterization of the catalytic site of the ADP-ribosyltransferase Clostridium botulinum C2 toxin by site-directed mutagenesis. J. Biol. Chem. 1998, 273, 29506–29511. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.G.; Blocker, D.; Hale, M.L.; Guetthoff, M.A.; Barth, H. Clostridium botulinum C2 toxin: Binding studies with fluorescence-activated cytometry. Toxicon 2002, 40, 1135–1140. [Google Scholar] [CrossRef]
- Barth, H.; Roebling, R.; Fritz, M.; Aktories, K. The binary Clostridium botulinum C2 toxin as a protein delivery system: Identification of the minimal protein region necessary for interaction of toxin components. J. Biol. Chem. 2002, 277, 5074–5081. [Google Scholar] [CrossRef] [PubMed]
- Funfhaus, A.; Poppinga, L.; Genersch, E. Identification and characterization of two novel toxins expressed by the lethal honey bee pathogen Paenibacillus larvae, the causative agent of American foulbrood. Environ. Microbiol. 2013, 15, 2951–2965. [Google Scholar] [PubMed]
- Vogelsgesang, M.; Aktories, K. Exchange of glutamine-217 to glutamate of Clostridium limosum exoenzyme C3 turns the asparagine-specific ADP-ribosyltransferase into an arginine-modifying enzyme. Biochemistry 2006, 45, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Pautsch, A.; Vogelsgesang, M.; Trankle, J.; Herrmann, C.; Aktories, K. Crystal structure of the C3bot-RalA complex reveals a novel type of action of a bacterial exoenzyme. EMBO J. 2005, 24, 3670–3680. [Google Scholar] [CrossRef] [PubMed]
- Ammam, F.; Marvaud, J.C.; Lambert, T. Distribution of the vanG-like gene cluster in Clostridium difficile clinical isolates. Can. J. Microbiol. 2012, 58, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Craig, J.A.; Putnam, C.D.; Carozzi, N.B.; Tainer, J.A. Evolution and mechanism from structures of an ADP-ribosylating toxin and NAD complex. Nat. Struct. Biol. 1999, 6, 932–936. [Google Scholar] [PubMed]


















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lugo, M.R.; Merrill, A.R. An In-Silico Sequence-Structure-Function Analysis of the N-Terminal Lobe in CT Group Bacterial ADP-Ribosyltransferase Toxins. Toxins 2019, 11, 365. https://doi.org/10.3390/toxins11060365
Lugo MR, Merrill AR. An In-Silico Sequence-Structure-Function Analysis of the N-Terminal Lobe in CT Group Bacterial ADP-Ribosyltransferase Toxins. Toxins. 2019; 11(6):365. https://doi.org/10.3390/toxins11060365
Chicago/Turabian StyleLugo, Miguel R., and A. Rod Merrill. 2019. "An In-Silico Sequence-Structure-Function Analysis of the N-Terminal Lobe in CT Group Bacterial ADP-Ribosyltransferase Toxins" Toxins 11, no. 6: 365. https://doi.org/10.3390/toxins11060365
APA StyleLugo, M. R., & Merrill, A. R. (2019). An In-Silico Sequence-Structure-Function Analysis of the N-Terminal Lobe in CT Group Bacterial ADP-Ribosyltransferase Toxins. Toxins, 11(6), 365. https://doi.org/10.3390/toxins11060365