1. Introduction
Vip3A proteins are produced during the vegetative phase of growth of
Bacillus thuringiensis and are of practical interest because of their insecticidal activity against Lepidoptera [
1]. Because Vip3A proteins share no sequence and structural homology with
B. thuringiensis Cry proteins, they are considered an excellent complement to Cry proteins in crop protection and resistance management. Some commercial Bt-crops (crops protected from insect attacks by expressing insecticidal proteins from
B. thuringiensis) combine Cry and Vip3 proteins, and this strategy of pyramiding proteins with different modes of action is expected to continue in the future [
2].
Despite the increasing interest in Vip3 proteins, their mode of action is not completely understood, and their 3D structure still remains unknown. Recently, a number of studies have provided valuable information toward the structure of these proteins. Multiple-sequence alignments of Vip3 proteins have shown that they contain between 786 and 803 amino acids (corresponding to a molecular weight of around 89 kDa), with a highly conserved N-terminal part (up to residue 334) and a highly variable C-terminal region [
1]. Proteolytical activation in the midgut of insects eliminates a small part of the N-terminus, which, in the case of Vip3A, takes place at residue R11/12 [
3,
4] and, in the case of Vip3Bc1, at R20 [
3], followed by the cleavage of the protein at the primary cleavage site, which in Vip3Aa and Vip3Af is K198/D199 [
4,
5]. Then, two peptides, of about 19 and 65 kDa, are generated, and these remain strongly bound to each other [
3,
6,
7]. More recently, it has been shown that Vip3 proteins are found in solution as homo-tetramers, both as protoxins and after activation by proteases [
3,
7,
8,
9].
To date, a high resolution 3D structure of a Vip3 tetrameric protein is lacking, though low resolution images have been obtained [
8,
10]. In an attempt to propose a 3D structure for Vip3 proteins, Vip3Af1 and Vip3Aa16 have been subjected to in silico modelling, and several domains have been proposed. For Vip3Af, five structural domains were proposed [
4], with domain 1 spanning from the N-terminus to residue 188, domain 2 from residue 189 to 272, domain 3 from 273 to 542, domain 4 from 543 to 715, and domain 5 from 716 to the end. For Vip3Aa16, three domains were proposed, though domain 1 was further subdivided into three domains [
11]: Subdomain 1.1 spanned from the N-terminus to residue 313, subdomain 1.2.1 from 314 to 441, subdomain 1.2.2 from 442 to 532, domain 2 from 533 to 667, and domain 3 from 668 to the end. Given the high sequence similarity between the two proteins (92.7%), the discrepancy between them regarding the regions spanned by the domains just reflects inaccuracies of the modelling programs used, probably due to the low availability of reference sequences with known 3D structures.
With the aim of shedding light on the putative functional and structural domains of Vip3 proteins, we have made use of selected Vip3Af alanine mutants (Ala-mutants) from a previous work [
4] which drastically affect insecticidal activity. Most of these mutants are distributed in three clusters along the length of the protein and show altered proteolytic patterns upon trypsin digestion [
4]. Banyuls et al. labeled these altered patterns as “a” to “f” [
4]. In the present work, we have made use of these critical Ala-mutants with the rationale that the altered patterns, generated by conformational changes due to the residue substitution, may unravel structural and functional domains. The results, based on protease digestion patterns, oligomer formation, and theoretical tryptic sites, have allowed us to propose a map of the Vip3Af protein with five domains. The information thus generated will contribute to the better understanding of the structure of Vip3 proteins and may be useful in the search of the 3D structure of this family of proteins.
3. Discussion
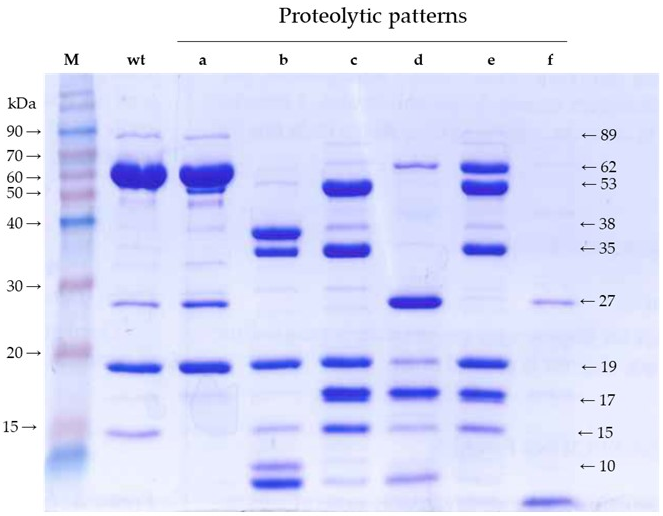
Banyuls et al. [
4] defined six proteolytic patterns of mutants with strongly impaired insecticidal activity. With minor modifications in the methodology, we have confirmed and refined such patterns with the aim of revealing the major fragments generated by trypsin and then identifying their position in the primary structure of the protein. The only difference observed with the previous proteolytic patterns is that, using the irreversible trypsin inhibitor to stop the reaction, we obtained a strong band of 35 kDa in patterns “b” and “c”, which was not observed before. We also detected bands smaller than 19 kDa by stopping the electrophoresis before they ran out of gel. Altogether, we ended up with fragments of 53, 38, 35, 27, 19, 17, 15, and <10 kDa, most of them shared by various patterns. We hypothesized that the limits of these fragments may correlate with the structural domains of the wild type protein.
In a previous study, Banyuls et al. [
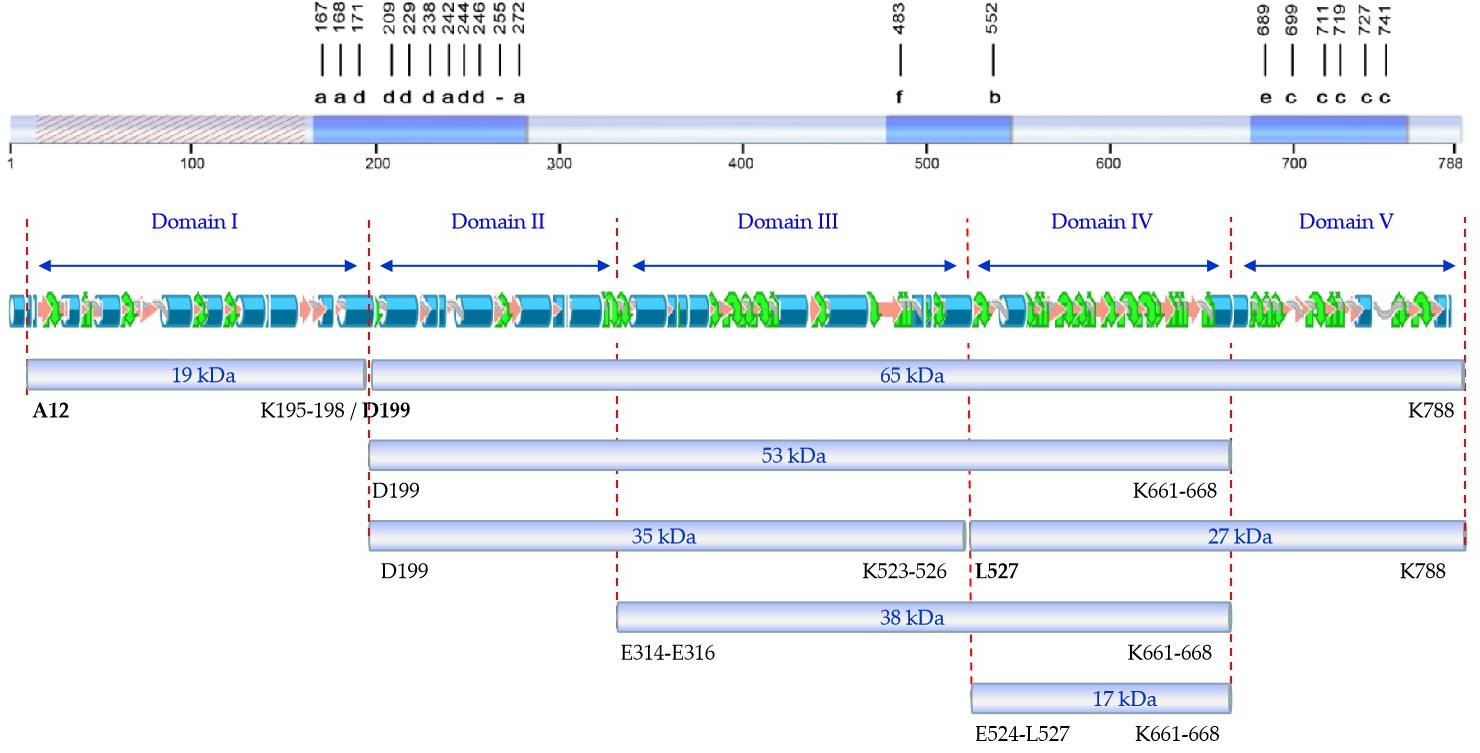
4] identified the tryptic fragments of 62 (here referred to as 65), 55 (here referred to as 53), 27, and 20 (here referred to as 19) kDa. Our peptide fingerprint results of fragments of 17, 27, and 38 kDa, taking into account the tryptic sites in the sequence of Vip3Af, allowed us to define their position in the sequence of the protein. Putting all this information together, we propose a map of the tryptic fragments such as the one shown in
Figure 4, which defines five domains. In this map, domain I spans the region covered by the 19 kDa fragment (from residues 12 to 198); domain II spans the region from the primary cleavage site to the start of the 38 kDa fragment (from residues 200 to 313/315); domain III spans from the start of the 38 kDa fragment up to the start of fragments of 17 and 27 kDa (from residues E314-E316 to 523/526); domain IV spans the 17 kDa fragment (from residue 524/527 to residue 661/668) and basically consists of the carbohydrate-binding motif common to all Vip3 proteins with the exception of Vip3Ba [
1]; and domain V spans from the end of the 17 kDa fragment (and also the end of the 38 and 53 kDa fragments) to the end of the protein (from residue 662/669 to 788). Compared with the proposed domains by in silico modelling, the domain I proposed by us is in good agreement with domain 1 proposed by Banyuls et al. [
4] for Vip3Af (from 1 to 188), though there is no further correlation between both models for the rest of domains. However, the boundaries between domain II and III, III and IV, and IV and V in our proposed map have their correspondence with the domains proposed by Sellami et al. [
11] for Vip3Aa (at residues 313, 532, and 667, respectively). The agreement between the domain limits proposed by us with some of those defined by in silico modelling supports the predictive value of the tryptic fragments approach to unravel structural domains of the Vip3A proteins.
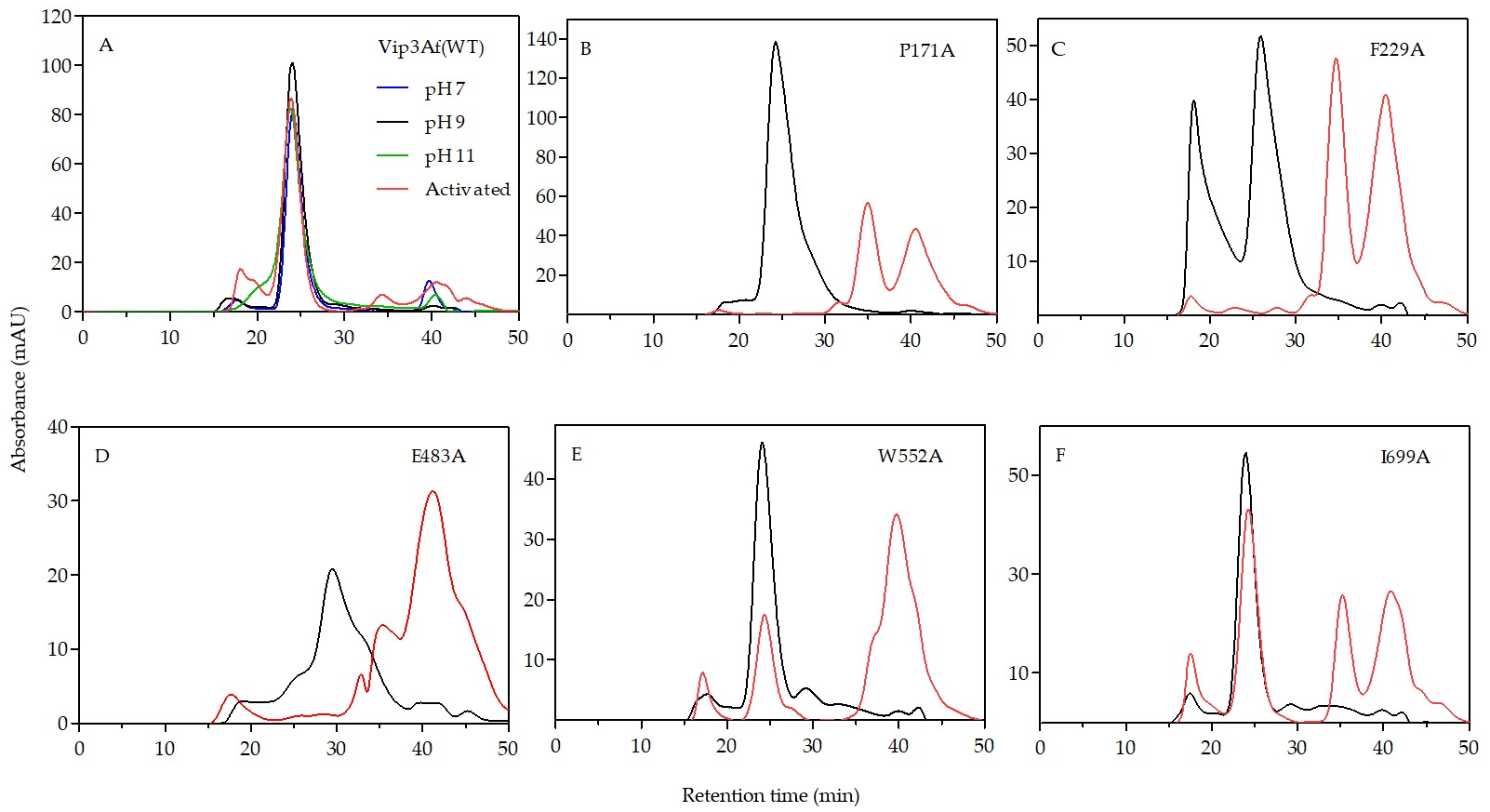
The results from gel filtration chromatography of the Ala-mutants shed light on the structural role of the proposed domains. Mutants rendering patterns “b”, “c”, and “e” are found forming a tetramer both as protoxins and also after trypsin treatment. Since trypsin digests the 27 kDa fragment (the only one containing domain V), we can conclude that domain V is not necessary to maintain the oligomeric structure. All these mutants, after trypsin treatment, have in common fragments of 19 kDa (domain I) and 35 kDa (domains II and III), plus another larger fragment (either of 38 or 53 kDa) which includes domain IV. Despite the fact that the 17 kDa fragment (which corresponds to domain IV) elutes separately from the tetramer in the chromatography of mutants with patterns “c” and “e”, the tetramer contains domain IV in the structure as part of the 53 kDa fragment. Therefore, according to the results, domains I–III are required to form the tetrameric structure, the need for domain IV is not clear, and domain V is not necessary.
An interesting observation from patterns “b” and “c” is that, in addition to the 19 kDa band, the sum of the remaining main bands gives an apparent molecular weight exceeding that of 65 kDa. In the case of pattern “b”, these bands correspond to fragments of 35 and 38 kDa. In the case of pattern “c”, the strongest bands are those corresponding to fragments of 15, 17, 19, 35, and 53 kDa. Therefore, there must be an alternative splicing of the 65 kDa fragment in mutants rendering these two patterns.
The requirement of domain I to form the tetramer, along with domain exchange studies between the 19 kDa fragment and the rest of the protein with Vip3Ab and Vip3Bb [
3], support the functional role of this domain and rules out the early beliefs that the 19 kDa fragment was non-essential in the insecticidal activity of Vip3 proteins and that only the 65 kDa fragment was the active core. It has been reported that complete deletion of the first 198 N-terminal amino acids in Vip3Aa completely abolishes its toxicity and produces a 62 kDa protein highly sensitive to trypsin degradation [
12]. However, some studies have shown that domain I can withstand short N-terminal deletions without affecting the insecticidal activity [
13,
14]. In contradiction to the above results, Gayen et al. [
15] reported an active Vip3Aa protein without domain I.
From the distribution of Ala-mutants with decreased insecticidal activity in the primary structure of Vip3Af (
Figure 4), we can observe that they gather into two clusters, except for mutant E483A (the only representative of pattern “f”) and mutant W552A (the only representative of pattern “b”). The first cluster contains all mutants with either pattern “a” or “d”. Mutations altering the structure and giving pattern “d” are concentrated at the end of domain I and the first part of domain II. This region of the protein, around the primary cleavage site, must have an important role in maintaining the 19 and 65 kDa fragments together, and this might be essential to preserve the overall structure of the tetrameric protein. The second cluster is in domain V and contains all the mutants with either pattern “c” or “e”. These mutants destabilize domain V, which is further digested by trypsin with the result of fragment 27 kDa being converted to the 17 kDa fragment.
5. Materials and Methods
5.1. Protein Source, Expression and Purification
The source of the 788 amino acid protein Vip3Af1(WT) (NCBI accession No. CAI43275) and that of its mutant proteins has been described in Banyuls et al. [
4]. The mutant proteins, all with decreased insecticidal activity, differed from Vip3Af(WT) and from each other, by a single amino acid residue which had been changed to an alanine residue. For this work, we selected the mutants which decreased the toxicity: T167A, E168A, P171A, L209A, F229A, M238A, E483A, W552A, G689A, I699A, Y719, and G727. The expression and purification of Vip3Af(WT) and the mutant proteins was carried out as described before [
4], using 1 mL HisTrap FF columns (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Vip3Af proteins were eluted with a phosphate buffer (50 mM phosphate, 300 mM NaCl, pH 7.4) containing 150 mM imidazole, and 1 mL fractions were collected in tubes containing 50 µL of 0.1 M ethylenediaminetetraacetic acid (EDTA). Fractions with a high protein concentration (determined photometrically at 280 nm) were pooled and dialyzed overnight at 4 °C against a TNE buffer (20 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, pH 8.6). The purity of the preparation (10 µL) was checked by SDS-PAGE, and the protein concentration was determined by the Bradford’s method. After dialysis, the proteins were stored at −20 °C until used.
5.2. Trypsin Treatment and SDS-PAGE Analysis of the Tryptic Fragments
The purified Vip3Af protoxins were subjected to proteolytic activation with commercial trypsin (trypsin from bovine pancreas, SIGMA T8003, Sigma-Aldrich, St. Louis, MO, USA). A mixture of protein:trypsin (5:100, w/w), in a TNE buffer was incubated at 30 °C for 24 h. Aliquots (10 µL ) of the trypsinized proteins were subjected to 12% SDS-PAGE. Prior to electrophoresis, the samples were made 1 mM with an 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) protease inhibitor (ThermoFisher, Waltham, MA, USA), left standing for 10 min at room temperature, and then heated at 100 °C for 5 min with a loading buffer (0.2 M Tris-HCl pH 6.8, 1 M sucrose, 5 mM EDTA, 0.1% bromophenol blue, 2.5% SDS, and 5% β-mercaptoethanol) (2:1, sample:loading buffer). The trypsin-treated samples to be used for chromatography and bioassays were stored at −20 °C for less than one week.
5.3. Insect Rearing and Bioassays
Insect rearing and bioassays were carried out in a rearing chamber maintained at 25 ± 2 °C, 70 ± 5% relative humidity, and 16:8 h light:dark on a semi-synthetic diet based on corn flour and wheat germ that contained yeast, ascorbic acid, and nipagin. Surface contamination assays were performed with 50 μL of protein sample on 2 cm2 diameter well plates. The concentration of Vip3Af protein was 1 µg/cm2, a concentration at which the Vip3Af(WT) kills 100% of the larvae. A Tris buffer (20 mM Tris-HCl, 150 mM NaCl, pH 8.6) was used as a blank control. Once the surface was dry, a neonate S. frugiperda larvae was gently placed into the well and then sealed. The number of dead and 1-instar larvae were recorded after 7 days. A larva was considered dead if it did not respond to mechanical stimulation. The mean mortality and functional mortality (dead larvae plus larvae that had not developed beyond the first instar) were determined from two replicates of 32 insects each.
5.4. Gel Filtration Chromatography
Gel filtration chromatography was performed with an ÄKTA explorer 100 chromatography system in a Superdex 200 10/300 GL column (GE Healthcare Life Sciences, Uppsala, Sweden) at a flow rate of 0.5 mL/min of a Tris buffer (50 mM Tris-HCl, 150 mM NaCl, pH 9.0), unless otherwise indicated. To estimate the molecular weight of the peaks, the column was calibrated with the following mix of standards: 4 mg/mL ovalbumin (44 kDa), 3 mg/mL conalbumin (75 kDa), 4 mg/mL aldolase (158 kDa), 0.3 mg/mL ferritin (440 kDa), 5 mg/mL thyroglobulin (6690 kDa), and Blue Dextran 200 (exclusion limit), dissolved in water.
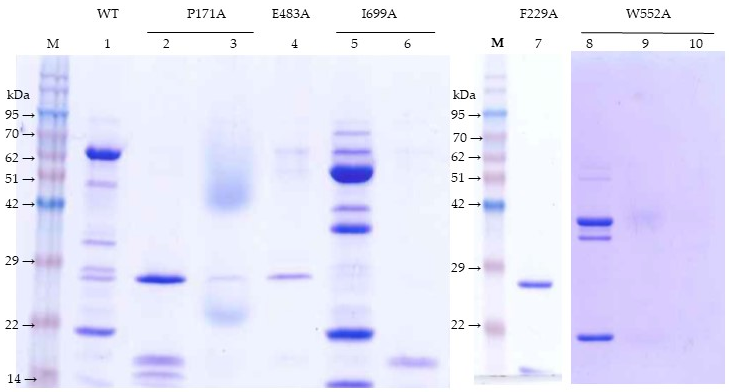
5.5. Identification of Tryptic Fragments
Major bands (27 and 17 kDa) from the trypsin-treated F229A mutant were identified after separation in a 2D-gel. The 17 kDa band from the trypsin-treated I699A mutant was first separated by chromatography in the Superdex 200 column and then by SDS-PAGE. The 38 kDa band from the trypsin-treated W552A mutant was first isolated by Superdex 200 chromatography and then by SDS-PAGE.
For the peptide identification, protein bands were directly cut out from the gel and digested with trypsin. The peptide mass and sequence were determined by liquid chromatography and tandem mass spectrometry (LC-MS/MS) in a nanoESI qQTOF (5600 TripleTOF, ABSCIEX, Framingham, MA, USA). The mass transitions were scanned first from 350–1250 m/z and then followed by a second scan from 100–1500 m/z. The peptides sequence identified were compared to the Vip3Af1(WT) protein sequence to match the region corresponding to each SDS-PAGE proteolytic band. Expected molecular weights were calculated using the online SIB Compute pI/Mw tool (
https://web.expasy.org/compute_pi).








{kind=link}
{kind=link}
{kind=link}
{kind=link}