Structural Biology and Molecular Modeling to Analyze the Entry of Bacterial Toxins and Virulence Factors into Host Cells



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Bacterial Toxins
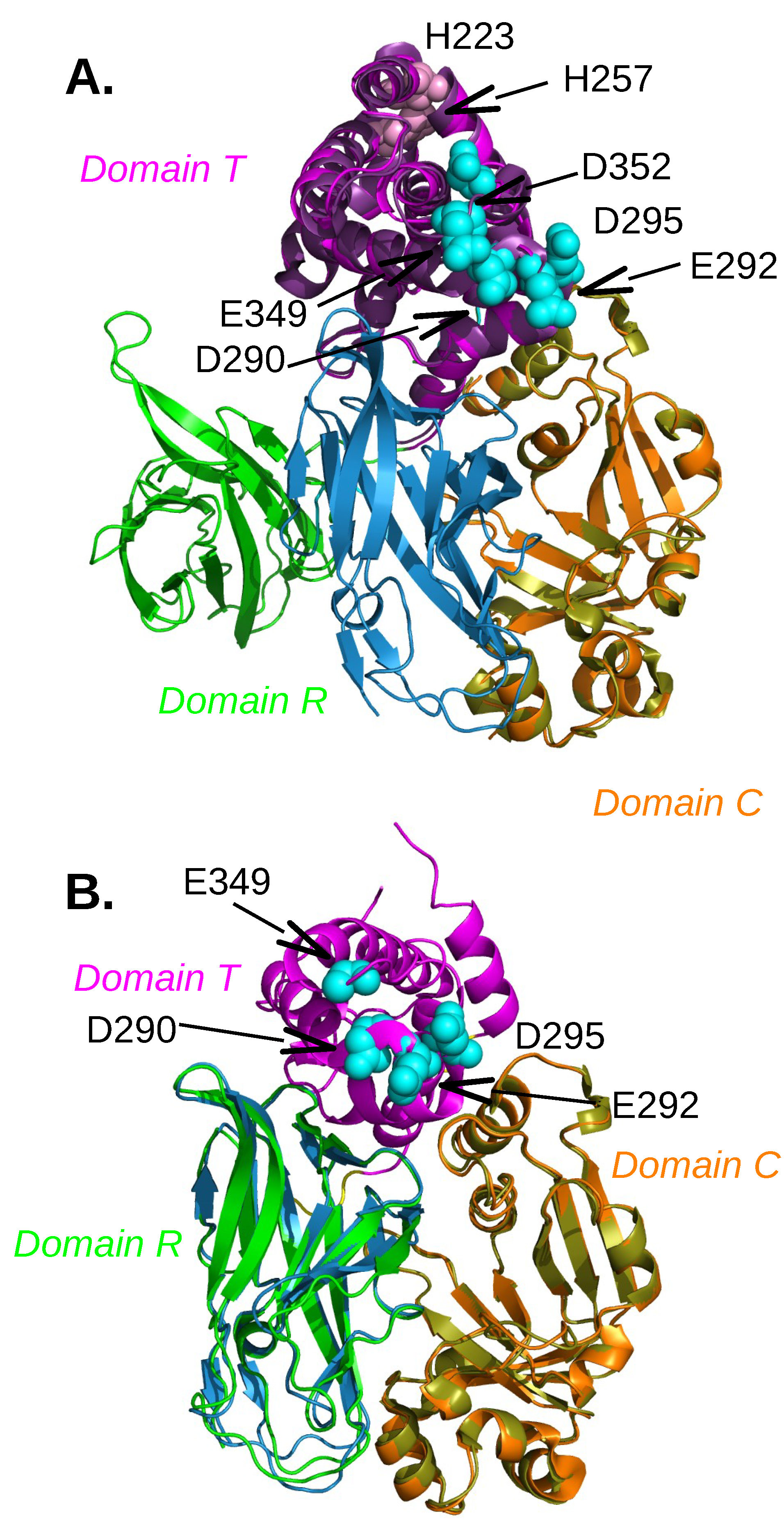
2.1. Diphtheria Toxin
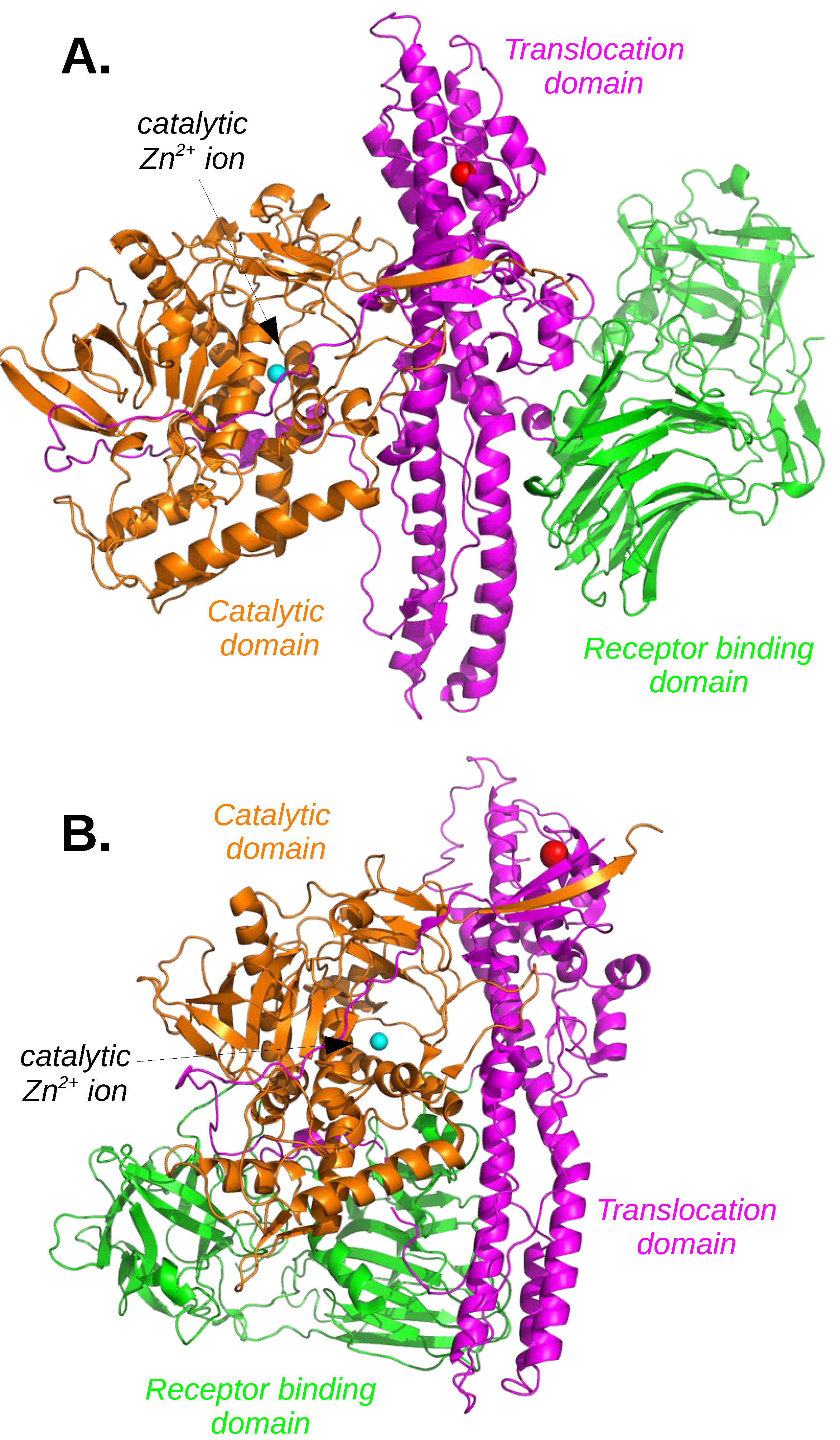
2.2. Botulinium and Tetanus Toxins
3. Adenylyl Cyclase Virulence Factors
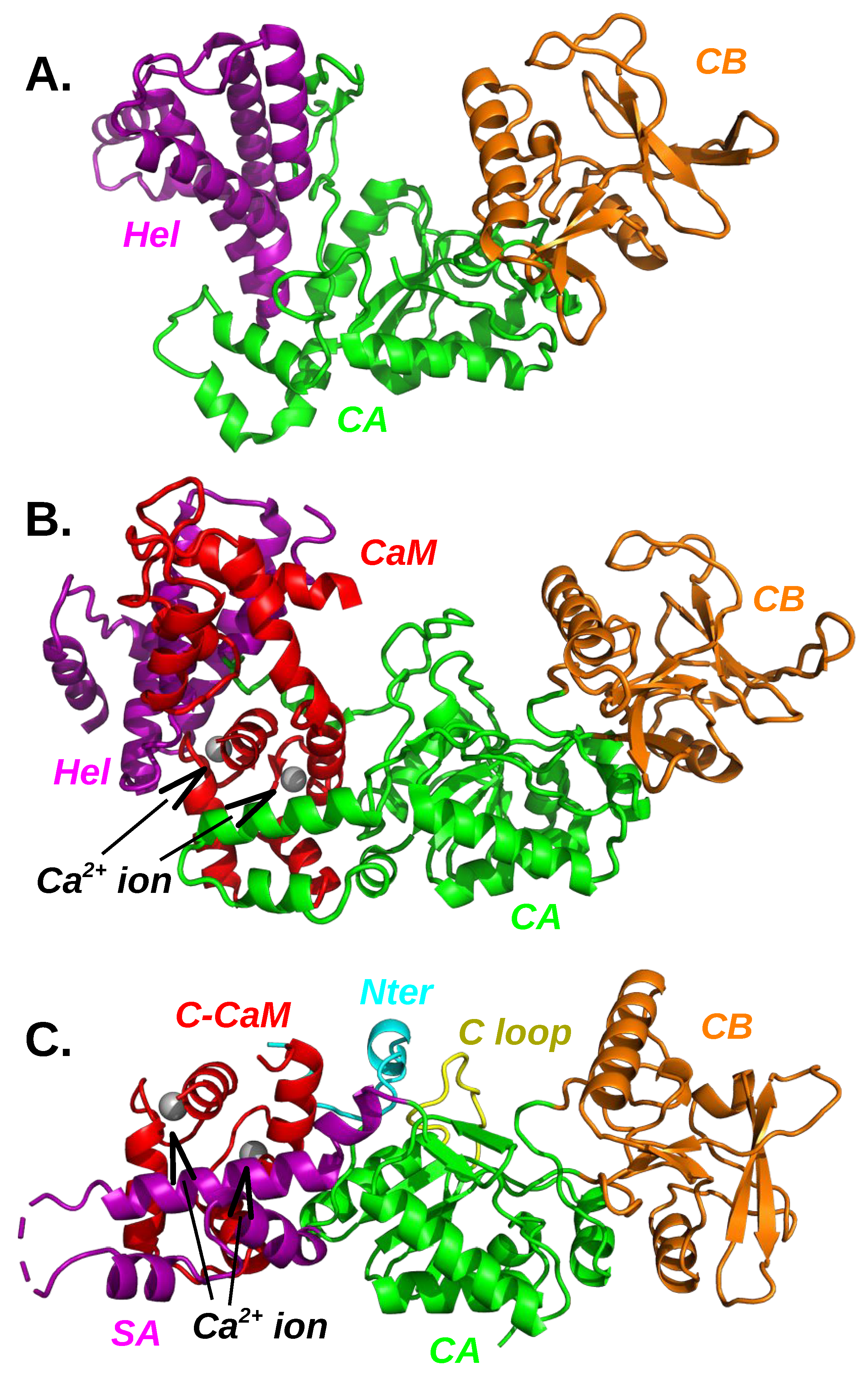
3.1. Plasticity of the Adenylyl Cyclase Interaction with Calmodulin
3.2. Searching Inhibitors for Adenylyl Cyclases
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kendrew, J.; Bodo, G.; Dintzis, H.; Parrish, R.; Wyckoff, H.; Phillips, D. A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef]
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztanyi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered: Why these proteins are intrinsically disordered. Intrinsically Disord Proteins 2013, 1, e24157. [Google Scholar] [CrossRef]
- Bernardi, R.; Melo, M.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta 2015, 1850, 872–877. [Google Scholar] [CrossRef]
- Fujisaki, H.; Moritsugu, K.; Matsunaga, Y.; Morishita, T.; Maragliano, L. Extended Phase-Space Methods for Enhanced Sampling in Molecular Simulations: A Review. Front. Bioeng. Biotechnol. 2015, 3, 125. [Google Scholar] [CrossRef]
- Miao, Y.; McCammon, J. Unconstrained Enhanced Sampling for Free Energy Calculations of Biomolecules: A Review. Mol. Simul. 2016, 42, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Jiang, F.; Wu, Y. Significantly Improved Protein Folding Thermodynamics Using a Dispersion-Corrected Water Model and a New Residue-Specific Force Field. J. Phys. Chem. Lett. 2017, 8, 3199–3205. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Nussinov, R. Release factors eRF1 and RF2: A universal mechanism controls the large conformational changes. J. Biol. Chem. 2004, 279, 53875–53885. [Google Scholar] [CrossRef] [PubMed]
- Zachariae, U.; Grubmuller, H. A highly strained nuclear conformation of the exportin Cse1p revealed by molecular dynamics simulations. Structure 2006, 14, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Hsin, J.; Sotomayor, M.; Comellas, G.; Schulten, K. Discovery through the computational microscope. Structure 2009, 17, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.J.; Eisenberg, D. Refined structure of monomeric diphtheria toxin at 2.3 A resolution. Protein Sci. 1994, 3, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.J.; Choe, S.; Eisenberg, D. Domain swapping: Entangling alliances between proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 3127–3131. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.E.; Eisenberg, D. Crystal structure of diphtheria toxin bound to nicotinamide adenine dinucleotide. Biochemistry 1996, 35, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.E.; Eisenberg, D. Crystal structure of nucleotide-free diphtheria toxin. Biochemistry 1997, 36, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.F.; Barbieri, J.T.; Collier, R.J. Dimeric form of diphtheria toxin: Purification and characterization. Biochemistry 1986, 25, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
- Leka, O.; Vallese, F.; Pirazzini, M.; Berto, P.; Montecucco, C.; Zanotti, G. Diphtheria toxin conformational switching at acidic pH. FEBS J. 2014, 281, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Steere, B. Characterization of High-Order Oligomerization and Energetics in Diphteria Toxin; University of California: Los Angeles, CA, USA, 2001. [Google Scholar]
- Rosconi, M.P.; Zhao, G.; London, E. Analyzing topography of membrane-inserted diphtheria toxin T domain using BODIPY-streptavidin: At low pH, helices 8 and 9 form a transmembrane hairpin but helices 5-7 form stable nonclassical inserted segments on the cis side of the bilayer. Biochemistry 2004, 43, 9127–9139. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; London, E. The membrane topography of the diphtheria toxin T domain linked to the a chain reveals a transient transmembrane hairpin and potential translocation mechanisms. Biochemistry 2009, 48, 10446–10456. [Google Scholar] [CrossRef]
- Chassaing, A.; Pichard, S.; Araye-Guet, A.; Barbier, J.; Forge, V.; Gillet, D. Solution and membrane-bound chaperone activity of the diphtheria toxin translocation domain towards the catalytic domain. FEBS J. 2011, 278, 4516–4525. [Google Scholar] [CrossRef]
- Ghalak, C.; Rodnin, M.; Vargas-Uribe, M.; McCluskey, A.; Flores-Canales, J.; Kurnikova, M.; Ladokhin, A. Role of Acidic Residues in Helices TH8–TH9 in Membrane Interactions of the Diphtheria Toxin T Domain. Toxins 2015, 7, 1303–1323. [Google Scholar]
- Kyrychenko, A.; Lim, N.; Vasquez-Montes, V.; Rodnin, M.; Freites, J.; Nguyen, L.; Tobias, D.; Mobley, D.; Ladokhin, A. Refining Protein Penetration into the Lipid Bilayer Using Fluorescence Quenching and Molecular Dynamics Simulations: The Case of Diphtheria Toxin Translocation Domain. J. Membrane Biol. 2018, 251, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Chenal, A.; Prongidi-Fix, L.; Perier, A.; Aisenbrey, C.; Vernier, G.; Lambotte, S.; Haertlein, M.; Dauvergne, M.T.; Fragneto, G.; Bechinger, B.; et al. Deciphering membrane insertion of the diphtheria toxin T domain by specular neutron reflectometry and solid-state NMR spectroscopy. J. Mol. Biol. 2009, 391, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Kienker, P.; Wu, Z.; Finkelstein, A. Mapping the membrane topography of the TH6–TH7 segment of the diphtheria toxin T-domain channel. J. Gen. Physiol. 2015, 145, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Man, P.; Montagner, C.; Vitrac, H.; Kavan, D.; Pichard, S.; Gillet, D.; Forest, E.; Forge, V. Accessibility changes within diphtheria toxin T domain when in the functional molten globule state, as determined using hydrogen/deuterium exchange measurements. FEBS J. 2010, 277, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rodnin, M.; Ladokhin, A.; Gross, M. Hydrogen-Deuterium Exchange and Mass Spectrometry Reveal the pH-Dependent Conformational Changes of Diphtheria Toxin T Domain. Biochemistry 2014, 53, 6849–6856. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Zhan, H.; Cui, C.; Altenbach, C.; Hubbell, W.; Collier, R. Conformation of the Diphtheria Toxin T Domain in Membranes: A Site-Directed Spin-Labeling Study of the TH8 Helix and TL5 Loop. Biochemistry 1999, 38, 10336–10343. [Google Scholar] [CrossRef] [PubMed]
- Rodnin, M.V.; Kyrychenko, A.; Kienker, P.; Sharma, O.; Posokhov, Y.O.; Collier, R.J.; Finkelstein, A.; Ladokhin, A.S. Conformational switching of the diphtheria toxin T domain. J. Mol. Biol. 2010, 402, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rodnin, M.V.; Li, J.; Gross, M.L.; Ladokhin, A.S. The pH-Dependent Trigger in Diphtheria Toxin T Domain Comes with a Safety Latch. Biophys. J. 2016, 111, 1946–1953. [Google Scholar] [CrossRef] [Green Version]
- D’Silva, P.R.; Lala, A.K. Organization of diphtheria toxin in membranes. A hydrophobic photolabeling study. J. Biol. Chem. 2000, 275, 11771–11777. [Google Scholar] [CrossRef]
- Falnes, P.O.; Choe, S.; Madshus, I.H.; Wilson, B.A.; Olsnes, S. Inhibition of membrane translocation of diphtheria toxin A-fragment by internal disulfide bridges. J. Biol. Chem. 1994, 269, 8402–8407. [Google Scholar] [PubMed]
- Sharpe, J.C.; London, E. Diphtheria Toxin Forms Pores of Different Sizes Depending on Its Concentration in Membranes: Probable Relationship to Oligomerization. J. Membr. Biol. 1999, 171, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Ladokhin, A. pH-Triggered Conformational Switching along the Membrane Insertion Pathway of the Diphtheria Toxin T-Domain. Toxins 2013, 5, 1362–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, M.; Finkelstein, A. The number of subunits comprising the channel formed by the T domain of diphtheria toxin. J. Gen. Physiol. 2001, 118, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Malenbaum, S.E.; Kachel, K.; Zhan, H.; Collier, R.J.; London, E. Identification of shallow and deep membrane-penetrating forms of diphtheria toxin T domain that are regulated by protein concentration and bilayer width. J. Biol. Chem. 1997, 272, 25091–25098. [Google Scholar] [CrossRef] [PubMed]
- Rosconi, M.P.; London, E. Topography of helices 5-7 in membrane-inserted diphtheria toxin T domain: Identification and insertion boundaries of two hydrophobic sequences that do not form a stable transmembrane hairpin. J. Biol. Chem. 2002, 277, 16517–16527. [Google Scholar] [CrossRef] [PubMed]
- Flores-Canales, J.; Vargas-Uribe, M.; Ladokhin, A.; Kurnikova, M. Membrane Association of the Diphtheria Toxin Translocation Domain Studied by Coarse-Grained Simulations and Experiment. J. Membrane Biol. 2015, 248, 529–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Canales, J.; Kurnikova, M. Targeting Electrostatic Interactions in Accelerated Molecular Dynamics with Application to Protein Partial Unfolding. J. Chem. Theory Comput. 2015, 11, 2550–2559. [Google Scholar] [CrossRef] [Green Version]
- Flores-Canales, J.; Kurnikova, M. Microsecond Simulations of the Diphtheria Toxin Translocation Domain in Association with Anionic Lipid Bilayers. J. Phys. Chem. B 2015, 119, 12074–12085. [Google Scholar] [CrossRef] [Green Version]
- Kurnikov, I.; Kyrychenko, A.; Flores-Canales, J.; Rodnin, M.; Simakov, N.; Vargas-Uribe, M.; Posokhov, Y.; Kurnikova, M.; Ladokhin, A. pH-Triggered Conformational Switching of the Diphtheria Toxin T-Domain: The Roles of N-Terminal Histidines. J. Mol. Biol. 2013, 425, 2752–2764. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.; Masuyer, G.; Stenmark, P. Botulinum and Tetanus Neurotoxins. Annu. Rev. Biochem. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Miethe, S.; Mazuet, C.; Liu, Y.; Tierney, R.; Rasetti-Escargueil, C.; Avril, A.; Frenzel, A.; Thullier, P.; Pelat, T.; Urbain, R.; et al. Development of Germline-Humanized Antibodies Neutralizing Botulinum Neurotoxin A and B. PLoS ONE 2016, 11, e0161446. [Google Scholar] [CrossRef] [PubMed]
- Derman, Y.; Selby, K.; Miethe, S.; Frenzel, A.; Liu, Y.; Rasetti-Escargueil, C.; Avril, A.; Pelat, T.; Urbain, R.; Fontayne, A.; et al. Neutralization of Botulinum Neurotoxin Type E by a Humanized Antibody. Toxins (Basel) 2016, 8, 257. [Google Scholar] [CrossRef] [PubMed]
- Rasetti-Escargueil, C.; Avril, A.; Miethe, S.; Mazuet, C.; Derman, Y.; Selby, K.; Thullier, P.; Pelat, T.; Urbain, R.; Fontayne, A.; et al. The European AntibotABE Framework Program and Its Update: Development of Innovative Botulinum Antibodies. Toxins (Basel) 2017, 9, 309. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, J.E.; Schmidt, J.J.; Fenn, T.; Burnett, J.C.; Arac, D.; Gussio, R.; Stafford, R.G.; Badie, S.S.; Bavari, S.; Brunger, A.T. A potent peptidomimetic inhibitor of botulinum neurotoxin serotype A has a very different conformation than SNAP-25 substrate. Structure 2008, 16, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Legler, P.M.; Southall, N.; Maloney, D.J.; Simeonov, A.; Jadhav, A. Structural insight into exosite binding and discovery of novel exosite inhibitors of botulinum neurotoxin serotype A through in silico screening. J. Comput. Aided Mol. Des. 2014, 28, 765–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.H.; Berger, W.T.; Nesbitt, N.M.; Kumar, K.; Balius, T.E.; Rizzo, R.C.; Tonge, P.J.; Ojima, I.; Swaminathan, S. Computer-aided identification, synthesis, and biological evaluation of novel inhibitors for botulinum neurotoxin serotype A. Bioorg. Med. Chem. 2015, 23, 5489–5495. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Wang, J.; Gao, S.; Ji, B.; Waichi Chan, E.; Chen, S. Substrate-based inhibitors exhibiting excellent protective and therapeutic effects against Botulinum Neurotoxin A intoxication. Sci. Rep. 2015, 5, 16981. [Google Scholar] [CrossRef] [Green Version]
- Kumaran, D.; Adler, M.; Levit, M.; Krebs, M.; Sweeney, R.; Swaminathan, S. Interactions of a potent cyclic peptide inhibitor with the light chain of botulinum neurotoxin A: Insights from X-ray crystallography. Bioorg. Med. Chem. 2015, 23, 7264–7273. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.; Agarwal, R.; Swaminathan, S. Small molecule non-peptide inhibitors of botulinum neurotoxin serotype E: Structure-activity relationship and a pharmacophore model. Bioorg. Med. Chem. 2016, 24, 3978–3985. [Google Scholar] [CrossRef]
- Minnow, Y.V.; Goldberg, R.; Tummalapalli, S.R.; Rotella, D.P.; Goodey, N.M. Mechanism of inhibition of botulinum neurotoxin type A light chain by two quinolinol compounds. Arch. Biochem. Biophys. 2017, 618, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, A.R.; Adler, M.; Silvaggi, N.R.; Allen, K.N.; Smith, G.M.; Fredenburg, R.A.; Stein, R.L.; Park, J.B.; Feng, X.; Shoemaker, C.B.; et al. Small molecule metalloprotease inhibitor with in vitro, ex vivo and in vivo efficacy against botulinum neurotoxin serotype A. Toxicon 2017, 137, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Lacy, D.B.; Tepp, W.; Cohen, A.C.; DasGupta, B.R.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 1998, 5, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Eswaramoorthy, S. Structural analysis of the catalytic and binding sites of Clostridium botulinum neurotoxin B. Nat. Struct. Biol. 2000, 7, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, D.; Eswaramoorthy, S.; Furey, W.; Navaza, J.; Sax, M.; Swaminathan, S. Domain organization in Clostridium botulinum neurotoxin type E is unique: Its implication in faster translocation. J. Mol. Biol. 2009, 386, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Masuyer, G.; Conrad, J.; Stenmark, P. The structure of the tetanus toxin reveals pH-mediated domain dynamics. EMBO Rep. 2017, 18, 1306–1317. [Google Scholar] [CrossRef]
- Gu, S.; Rumpel, S.; Zhou, J.; Strotmeier, J.; Bigalke, H.; Perry, K.; Shoemaker, C.B.; Rummel, A.; Jin, R. Botulinum neurotoxin is shielded by NTNHA in an interlocked complex. Science 2012, 335, 977–981. [Google Scholar] [CrossRef]
- Eswaramoorthy, S.; Sun, J.; Li, H.; Singh, B.R.; Swaminathan, S. Molecular Assembly of Clostridium botulinum progenitor M complex of type E. Sci Rep 2015, 5, 17795. [Google Scholar] [CrossRef]
- Eswaramoorthy, S.; Kumaran, D.; Keller, J.; Swaminathan, S. Role of metals in the biological activity of Clostridium botulinum neurotoxins. Biochemistry 2004, 43, 2209–2216. [Google Scholar] [CrossRef]
- Chellappan, G.; Kumar, R.; Santos, E.; Goyal, D.; Cai, S.; Singh, B.R. Structural and functional analysis of botulinum neurotoxin subunits for pH-dependent membrane channel formation and translocation. Biochim. Biophys. Acta 2015, 1854, 1510–1516. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Deng, Y. Long-time molecular dynamics simulations of botulinum biotoxin type-A at different pH values and temperatures. J. Mol. Model. 2007, 13, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Guo, Q. The adenylyl cyclase activity of anthrax edema factor. Mol. Asoects Med. 2009, 30, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyy, A.; Mechold, U.; Renault, L.; Ladant, D. ExoY, an actin-activated nucleotidyl cyclase toxin from P. aeruginosa: A minireview. Toxicon 2018, 149, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drum, C.; Yan, S.; Bard, J.; Shen, Y.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 2002, 415, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Shen, Y.; Lee, Y.; Gibbs, C.; Mrksich, M.; Tang, W. Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 2005, 24, 3190–3201. [Google Scholar] [CrossRef] [PubMed]
- Khanppnavar, B.; Datta, S. Crystal structure and substrate specificity of ExoY, a unique T3SS mediated secreted nucleotidyl cyclase toxin from Pseudomonas aeruginosa. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2090–2103. [Google Scholar] [CrossRef]
- Malliavin, T.E. Molecular Modeling of the Catalytic Domain of CyaA Deepened the Knowledge of Its Functional Dynamics. Toxins (Basel) 2017, 9, 199. [Google Scholar] [CrossRef]
- Laine, E.; Martinez, L.; Ladant, D.; Malliavin, T.; Blondel, A. Molecular motions as a drug target: Mechanistic simulations of anthrax toxin edema factor function led to the discovery of novel allosteric inhibitors. Toxins (Basel) 2012, 4, 580–604. [Google Scholar] [CrossRef]
- Ladant, D.; Ullmann, A. Bordetella pertussis adenylate cyclase: A toxin with multiple talents. Trends Microbiol. 1999, 7, 172–176. [Google Scholar] [CrossRef]
- Karimova, G.; Pidoux, J.; Ullmann, A.; Ladant, D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 5752–5756. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, N.; Lacy, C.R.; Cruz, J.E.; Nahass, M.; Karp, J.; Barone, J.A.; Hermes-DeSantis, E.R. Disaster Preparedness: Biological Threats and Treatment Options. Pharmacotherapy 2018, 38, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jureller, J.; Warren, J.; Solomaha, E.; Florián, J.; Tang, W. Protein-protein docking and analysis reveal that two homologous bacterial adenylyl cyclase toxins interact with calmodulin differently. J. Biol. Chem. 2008, 283, 23836–23845. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Guo, Q.; Zhukovskaya, N.L.; Drum, C.L.; Bohm, A.; Tang, W.J. Structure of anthrax edema factor-calmodulin-adenosine 5’-(alpha,beta-methylene)-triphosphate complex reveals an alternative mode of ATP binding to the catalytic site. Biochem. Biophys. Res. Commun. 2004, 317, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Shen, Y.; Zhukovskaya, N.L.; Florian, J.; Tang, W.J. Structural and kinetic analyses of the interaction of anthrax adenylyl cyclase toxin with reaction products cAMP and pyrophosphate. J. Biol. Chem. 2004, 279, 29427–29435. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhukovskaya, N.L.; Guo, Q.; Florian, J.; Tang, W.J. Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J. 2005, 24, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Laine, E.; Malliavin, T.; Nilges, M.; Blondel, A. ATP conformations and ion binding modes in the active site of anthrax edema factor: A computational analysis. Proteins 2009, 77, 971–983. [Google Scholar] [CrossRef]
- Martínez, L.; Malliavin, T.; Blondel, A. Mechanism of reactant and product dissociation from the Anthrax Edema Factor: A Locally Enhanced Sampling and Steered Molecular Dynamics Study. Proteins 2011, in press. [Google Scholar]
- Shen, Y.; Lee, Y.; Soelaiman, S.; Bergson, P.; Lu, D.; Chen, A.; Beckingham, K.; Grabarek, Z.; Mrksich, M.; Tang, W. Physiological calcium concentrations regulate calmodulin binding and catalysis of adenylyl cyclase exotoxins. EMBO J. 2002, 21, 6721–6732. [Google Scholar] [CrossRef] [Green Version]
- Glaser, P.; Elmaoglou-Lazaridou, A.; Krin, E.; Ladant, D.; Bárzu, O.; Danchin, A. Identification of residues essential for catalysis and binding of calmodulin in Bordetella pertussis adenylate cyclase by site-directed mutagenesis. EMBO J. 1989, 8, 967–972. [Google Scholar] [CrossRef]
- Laine, E.; Martínez, L.; Blondel, A.; Malliavin, T. Activation of the edema factor of Bacillus anthracis by calmodulin: Evidence of an interplay between the EF-calmodulin interaction and calcium binding. Biophys. J. 2010, 99, 2264–2272. [Google Scholar] [CrossRef]
- Selwa, E.; Laine, E.; Malliavin, T. Differential role of Calmodulin and Calcium ions in the stabilization of the catalytic domain of adenyl cyclase CyaA from Bordetella pertussis. Proteins 2012, 80, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Jas, G.; Kuczera, K. Structure, dynamics and interaction with kinase targets: Computer simulations of calmodulin. Biochim. Biophys. Acta 2004, 1697, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Vougier, S.; Mary, J.; Dautin, N.; Vinh, J.; Friguet, B.; Ladant, D. Essential role of methionine residues in calmodulin binding to Bordetella pertussis adenylate cyclase, as probed by selective oxidation and repair by the peptide methionine sulfoxide reductases. J. Biol. Chem. 2004, 279, 30210–30218. [Google Scholar] [CrossRef] [PubMed]
- Prêcheur, B.; Siffert, O.; Bârzu, O.; Craescu, C. NMR and circular dichroic studies on the solution conformation of a synthetic peptide derived from the calmodulin-binding domain of Bordetella pertussis adenylate cyclase. Eur. J. Biochem. 1991, 196, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Craescu, C.; Bouhss, A.; Mispelter, J.; Diesis, E.; Popescu, A.; Chiriac, M.; Bârzu, O. Calmodulin binding of a peptide derived from the regulatory domain of Bordetella pertussis adenylate cyclase. J. Biol. Chem. 1995, 270, 7088–7096. [Google Scholar] [CrossRef] [PubMed]
- Selwa, E.; Huynh, T.; Ciccotti, G.; Maragliano, L.; Malliavin, T. Temperature-accelerated molecular dynamics gives insights into globular conformations sampled in the free state of the AC catalytic domain. Proteins 2014, 82, 2483–2496. [Google Scholar] [CrossRef] [Green Version]
- Springer, T.; Emerson, C.; Johns, C.; Finley, N. Interaction with adenylate cyclase toxin from Bordetella pertussis affects the metal binding properties of calmodulin. FEBS Open Bio 2017, 7, 25–34. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar]
- O’Brien, D.P.; Durand, D.; Voegele, A.; Hourdel, V.; Davi, M.; Chamot-Rooke, J.; Vachette, P.; Brier, S.; Ladant, D.; Chenal, A. Calmodulin fishing with a structurally disordered bait triggers CyaA catalysis. PLoS Biol. 2017, 15, e2004486. [Google Scholar] [CrossRef]
- Ulmer, T.; Soelaiman, S.; Li, S.; Klee, C.; Tang, W.; Bax, A. Calcium dependence of the interaction between calmodulin and anthrax edema factor. J. Biol. Chem. 2003, 278, 29261–29266. [Google Scholar] [CrossRef]
- Laine, E.; Yoneda, J.; Blondel, A.; Malliavin, T. The conformational plasticity of calmodulin upon calcium complexation gives a model of its interaction with the oedema factor of Bacillus anthracis. Proteins 2008, 71, 1813–1829. [Google Scholar] [CrossRef] [PubMed]
- Laine, E.; Blondel, A.; Malliavin, T. Dynamics and energetics: A consensus analysis of the impact of calcium on EF-CaM protein complex. Biophys. J. 2009, 96, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Tanaka, T.; Ikura, M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 1995, 2, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Finn, B.; Evenas, J.; Drakenberg, T.; Waltho, J.; Thulin, E.; Forsen, S. Calcium-induced structural changes and domain autonomy in calmodulin. Nat. Struct. Biol. 1995, 2, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Maragliano, L.; Vanden-Eijnden, E. A temperature accelerated method for sampling free energy and determining reaction pathways in rare events simulations. Chem. Phys. Lett. 2006, 426, 168–175. [Google Scholar] [CrossRef]
- Maragliano, L.; Cottone, G.; Ciccotti, G.; Vanden-Eijnden, E. Mapping the network of pathways of CO diffusion in myoglobin. J. Am. Chem. Soc. 2010, 132, 1010–1017. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Bouvier, G.; Nilges, M.; Maragliano, L.; Malliavin, T. Temperature Accelerated Molecular Dynamics with Soft-Ratcheting Criterion Orients Enhanced Sampling by Low-Resolution Information. J. Chem. Theory Comput. 2015, 11, 3446–3454. [Google Scholar] [CrossRef]
- Springer, T.; Goebel, E.; Hariraju, D.; Finley, N. Mutation in the β-hairpin of the Bordetella pertussis adenylate cyclase toxin modulates N-lobe conformation in calmodulin. Biochem. Biophys. Res. Commun. 2014, 453, 43–48. [Google Scholar] [CrossRef]
- Seifert, R.; Dove, S. Towards selective inhibitors of adenylyl cyclase toxin from Bordetella pertussis. Trends Microbiol. 2012, 20, 343–351. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, K.; Ma, X.; Shi, W.; Yuan, L.; Yang, Y. Susceptibility to Erythromycin and Virulence-Related Genotype Changes in China (1970–2014). PLoS ONE 2015, 10, e0138941. [Google Scholar] [CrossRef]
- Soelaiman, S.; Wei, B.Q.; Bergson, P.; Lee, Y.S.; Shen, Y.; Mrksich, M.; Shoichet, B.K.; Tang, W.J. Structure-based inhibitor discovery against adenylyl cyclase toxins from pathogenic bacteria that cause anthrax and whooping cough. J. Biol. Chem. 2003, 278, 25990–25997. [Google Scholar] [CrossRef]
- Gottle, M.; Dove, S.; Steindel, P.; Shen, Y.; Tang, W.J.; Geduhn, J.; Konig, B.; Seifert, R. Molecular analysis of the interaction of Bordetella pertussis adenylyl cyclase with fluorescent nucleotides. Mol. Pharmacol. 2007, 72, 526–535. [Google Scholar] [CrossRef]
- Taha, H.M.; Schmidt, J.; Gottle, M.; Suryanarayana, S.; Shen, Y.; Tang, W.J.; Gille, A.; Geduhn, J.; Konig, B.; Dove, S.; et al. Molecular analysis of the interaction of anthrax adenylyl cyclase toxin, edema factor, with 2′(3′)-O-(N-(methyl)anthraniloyl)-substituted purine and pyrimidine nucleotides. Mol. Pharmacol. 2009, 75, 693–703. [Google Scholar] [CrossRef]
- Geduhn, J.; Dove, S.; Shen, Y.; Tang, W.J.; Konig, B.; Seifert, R. Bis-halogen-anthraniloyl-substituted nucleoside 5′-triphosphates as potent and selective inhibitors of Bordetella pertussis adenylyl cyclase toxin. J. Pharmacol. Exp. Ther. 2011, 336, 104–115. [Google Scholar] [CrossRef]
- Taha, H.; Dove, S.; Geduhn, J.; Konig, B.; Shen, Y.; Tang, W.J.; Seifert, R. Inhibition of the adenylyl cyclase toxin, edema factor, from Bacillus anthracis by a series of 18 mono- and bis-(M)ANT-substituted nucleoside 5′-triphosphates. Naunyn Schmiedebergs Arch. Pharmacol. 2012, 385, 57–68. [Google Scholar] [CrossRef]
- Gille, A.; Lushington, G.H.; Mou, T.C.; Doughty, M.B.; Johnson, R.A.; Seifert, R. Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides. J. Biol. Chem. 2004, 279, 19955–19969. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Bergson, P.; He, W.S.; Mrksich, M.; Tang, W.J. Discovery of a small molecule that inhibits the interaction of anthrax edema factor with its cellular activator, calmodulin. Chem. Biol. 2004, 11, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhukovskaya, N.L.; Zimmer, M.I.; Soelaiman, S.; Bergson, P.; Wang, C.R.; Gibbs, C.S.; Tang, W.J. Selective inhibition of anthrax edema factor by adefovir, a drug for chronic hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 3242–3247. [Google Scholar] [CrossRef] [Green Version]
- Suryanarayana, S.; Wang, J.L.; Richter, M.; Shen, Y.; Tang, W.J.; Lushington, G.H.; Seifert, R. Distinct interactions of 2′- and 3′-O-(N-methyl)anthraniloyl-isomers of ATP and GTP with the adenylyl cyclase toxin of Bacillus anthracis, edema factor. Biochem. Pharmacol. 2009, 78, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Schein, C.H.; Chen, D.; Ma, L.; Kanalas, J.J.; Gao, J.; Jimenez, M.E.; Sower, L.E.; Walter, M.A.; Gilbertson, S.R.; Peterson, J.W. Pharmacophore selection and redesign of non-nucleotide inhibitors of anthrax edema factor. Toxins (Basel) 2012, 4, 1288–1300. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ma, L.; Kanalas, J.J.; Gao, J.; Pawlik, J.; Jimenez, M.E.; Walter, M.A.; Peterson, J.W.; Gilbertson, S.R.; Schein, C.H. Structure-based redesign of an edema toxin inhibitor. Bioorg. Med. Chem. 2012, 20, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, R.; Dove, S. Inhibitors of Bacillus anthracis edema factor. Pharmacol. Ther. 2013, 140, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Jiao, G.S.; Kim, S.; Moayeri, M.; Thai, A.; Cregar-Hernandez, L.; McKasson, L.; O’Malley, S.; Leppla, S.H.; Johnson, A.T. Small molecule inhibitors of anthrax edema factor. Bioorg. Med. Chem. Lett. 2018, 28, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Lubker, C.; Seifert, R. Effects of 39 Compounds on Calmodulin-Regulated Adenylyl Cyclases AC1 and Bacillus anthracis Edema Factor. PLoS ONE 2015, 10, e0124017. [Google Scholar] [CrossRef] [PubMed]
- Česnek, M.; Skácel, J.; Jansa, P.; Dračinský, M.; Šmîdková, M.; Mertliková-Kaiserová, H.; Soto-Velasquez, M.P.; Watts, V.J.; Janeba, Z. Nucleobase Modified Adefovir (PMEA) Analogues as Potent and Selective Inhibitors of Adenylate Cyclases from Bordetella pertussis and Bacillus anthracis. ChemMedChem 2018, 13, 1779–1796. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Moayeri, M.; Zhao, H.; Crown, D.; Leppla, S.H.; Purcell, R.H. Potent neutralization of anthrax edema toxin by a humanized monoclonal antibody that competes with calmodulin for edema factor binding. Proc. Natl. Acad. Sci. USA 2009, 106, 13487–13492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, E.; Goncalves, C.; Karst, J.; Lesnard, A.; Rault, S.; Tang, W.; Malliavin, T.; Ladant, D.; Blondel, A. Use of allostery to identify inhibitors of calmodulin- induced activation of Bacillus anthracis Edema Factor. Proc. Natl. Acad. Sci. USA 2010, 107, 11277–11282. [Google Scholar] [CrossRef] [PubMed]
- Belyy, A.; Raoux-Barbot, D.; Saveanu, C.; Namane, A.; Ogryzko, V.; Worpenberg, L.; David, V.; Henriot, V.; Fellous, S.; Merrifield, C.; et al. Actin activates Pseudomonas aeruginosa ExoY nucleotidyl cyclase toxin and ExoY-like effector domains from MARTX toxins. Nat. Commun. 2016, 7, 135–182. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitard, I.; Malliavin, T.E. Structural Biology and Molecular Modeling to Analyze the Entry of Bacterial Toxins and Virulence Factors into Host Cells. Toxins 2019, 11, 369. https://doi.org/10.3390/toxins11060369
Pitard I, Malliavin TE. Structural Biology and Molecular Modeling to Analyze the Entry of Bacterial Toxins and Virulence Factors into Host Cells. Toxins. 2019; 11(6):369. https://doi.org/10.3390/toxins11060369
Chicago/Turabian StylePitard, Irène, and Thérèse E Malliavin. 2019. "Structural Biology and Molecular Modeling to Analyze the Entry of Bacterial Toxins and Virulence Factors into Host Cells" Toxins 11, no. 6: 369. https://doi.org/10.3390/toxins11060369
APA StylePitard, I., & Malliavin, T. E. (2019). Structural Biology and Molecular Modeling to Analyze the Entry of Bacterial Toxins and Virulence Factors into Host Cells. Toxins, 11(6), 369. https://doi.org/10.3390/toxins11060369