The Isolation of New Pore-Forming Toxins from the Sea Anemone Actinia fragacea Provides Insights into the Mechanisms of Actinoporin Evolution

 , ,
, ,
Abstract
:1. Introduction
2. Results
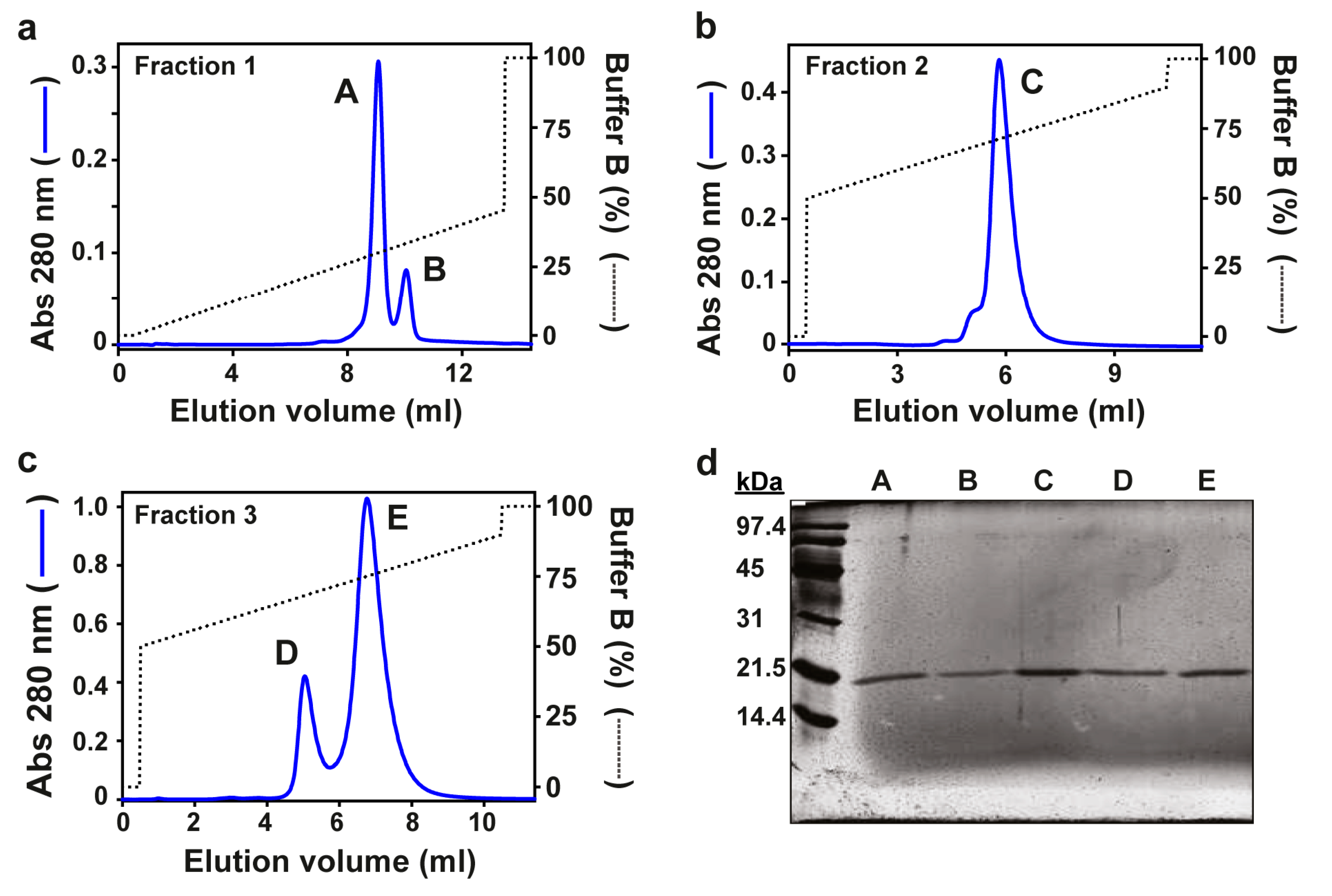
2.1. Purification of Fragaceatoxins
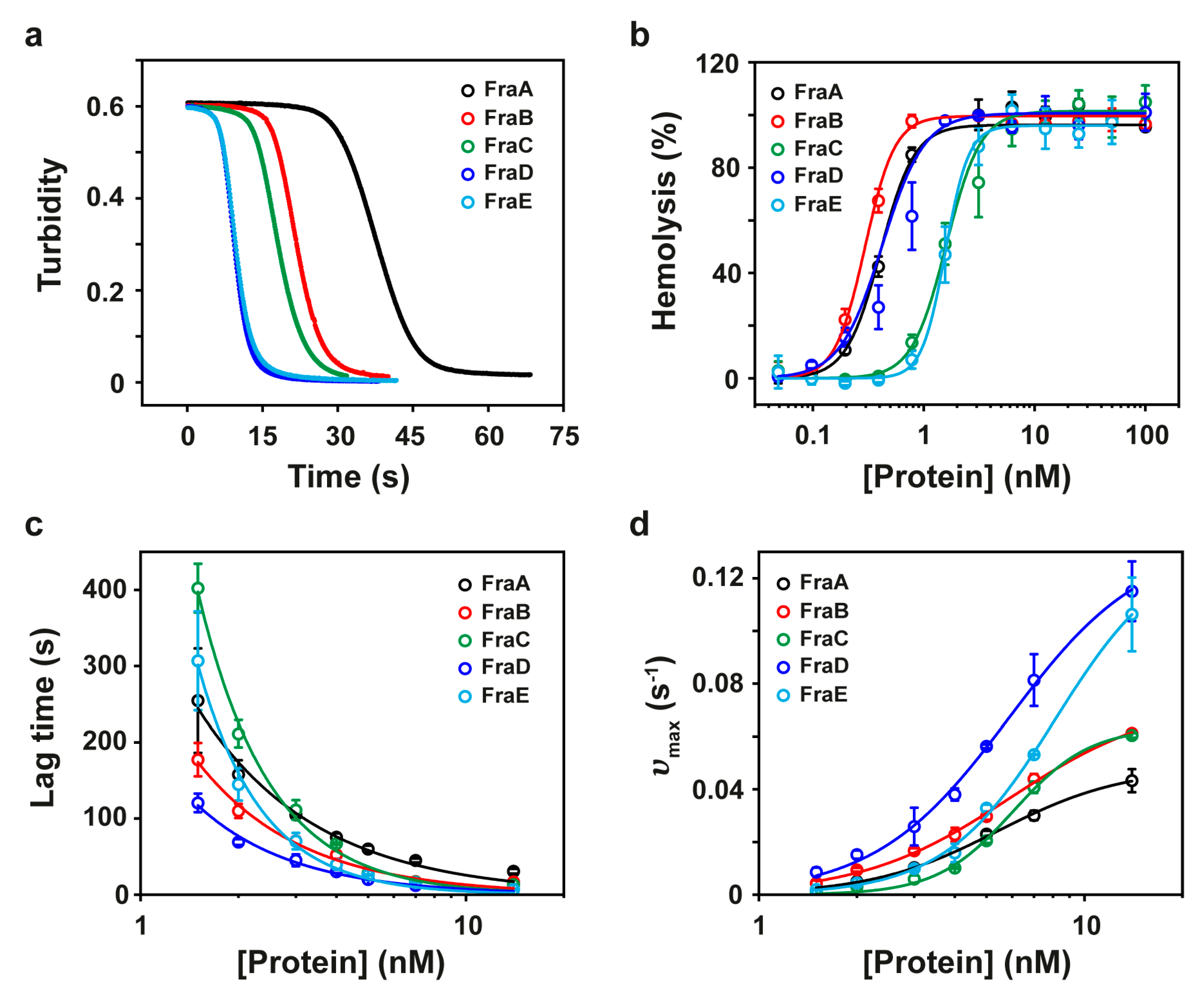
2.2. Hemolytic Activity
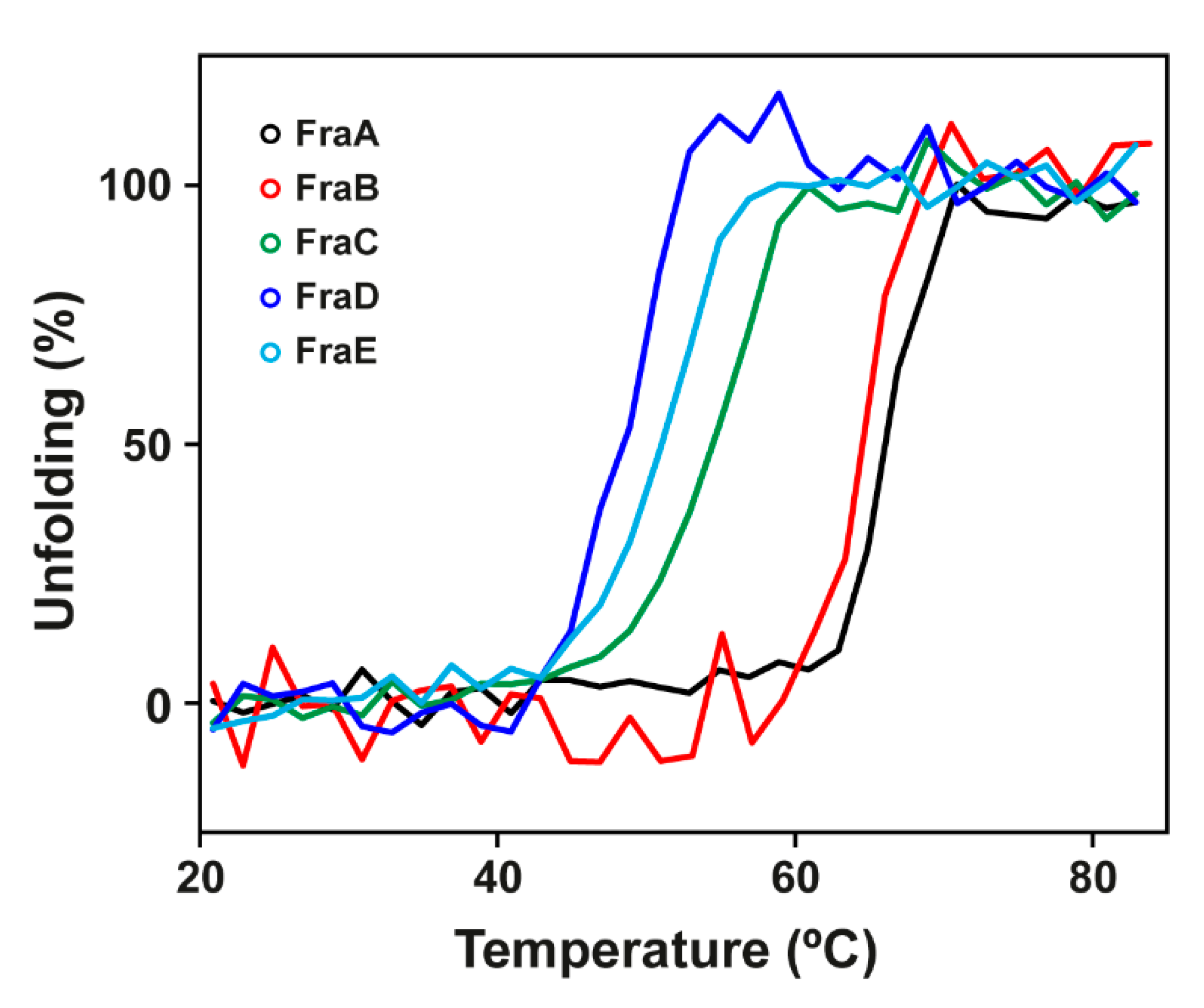
2.3. Thermal Stability
2.4. DNA Sequence of Fragaceatoxins
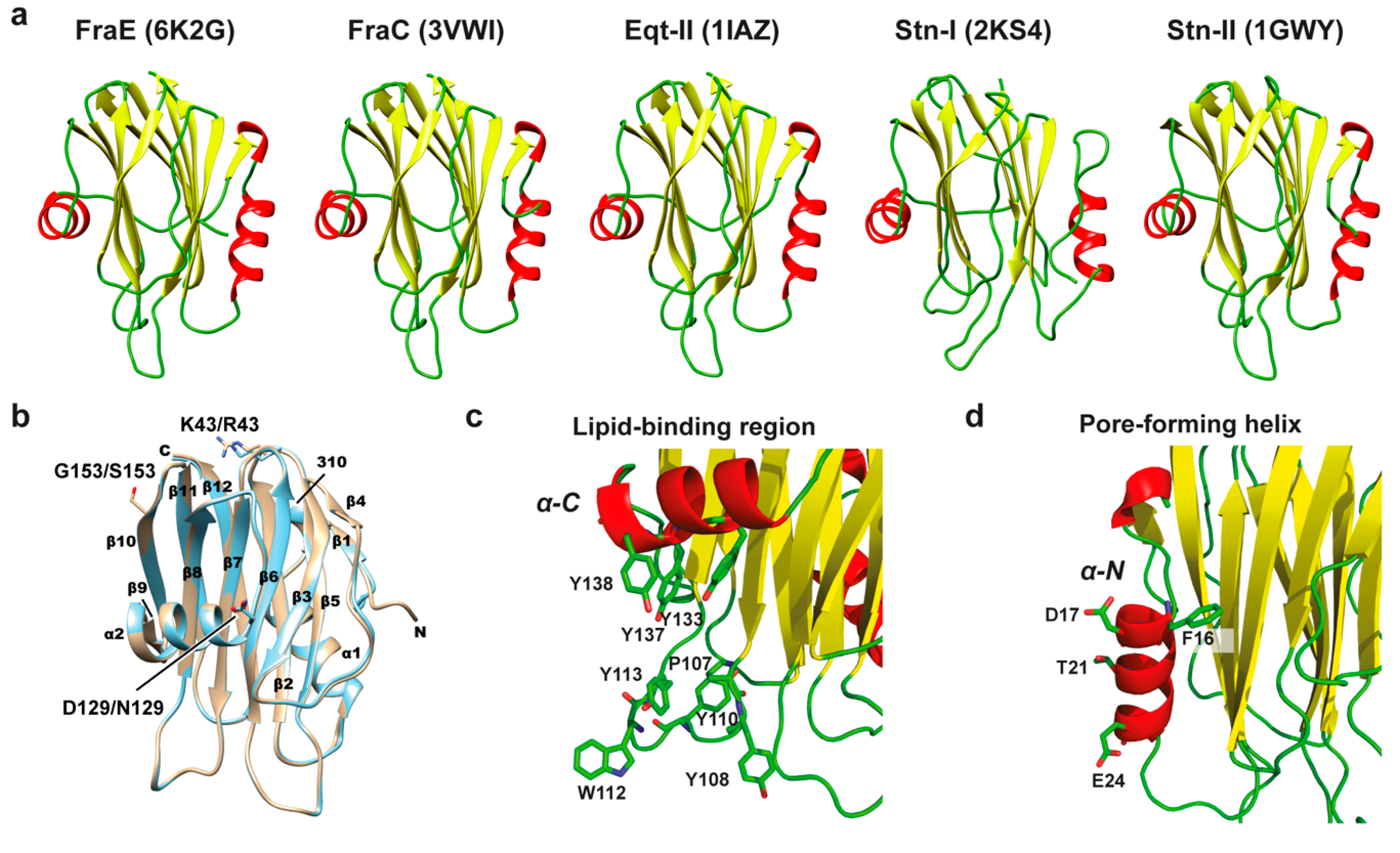
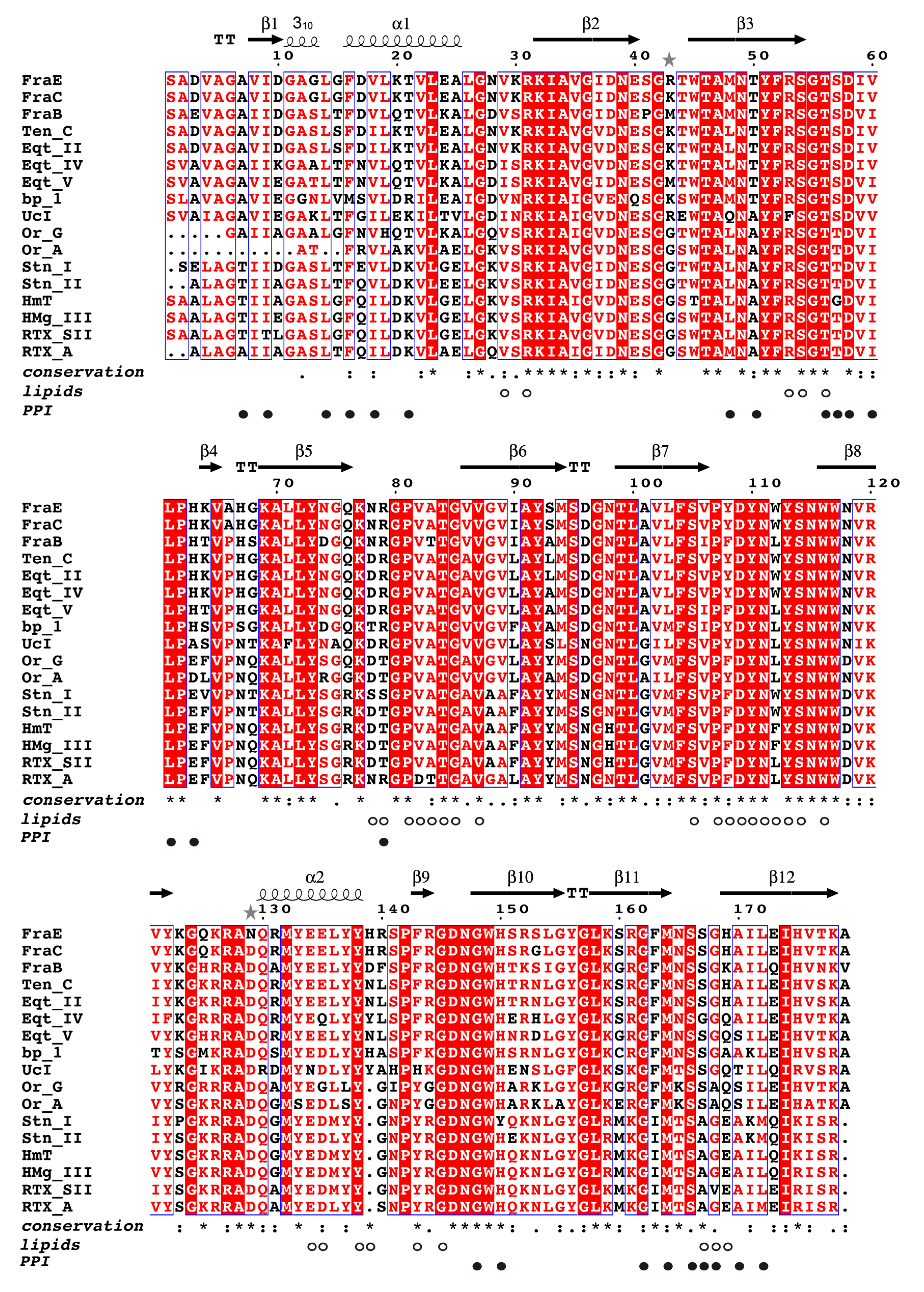
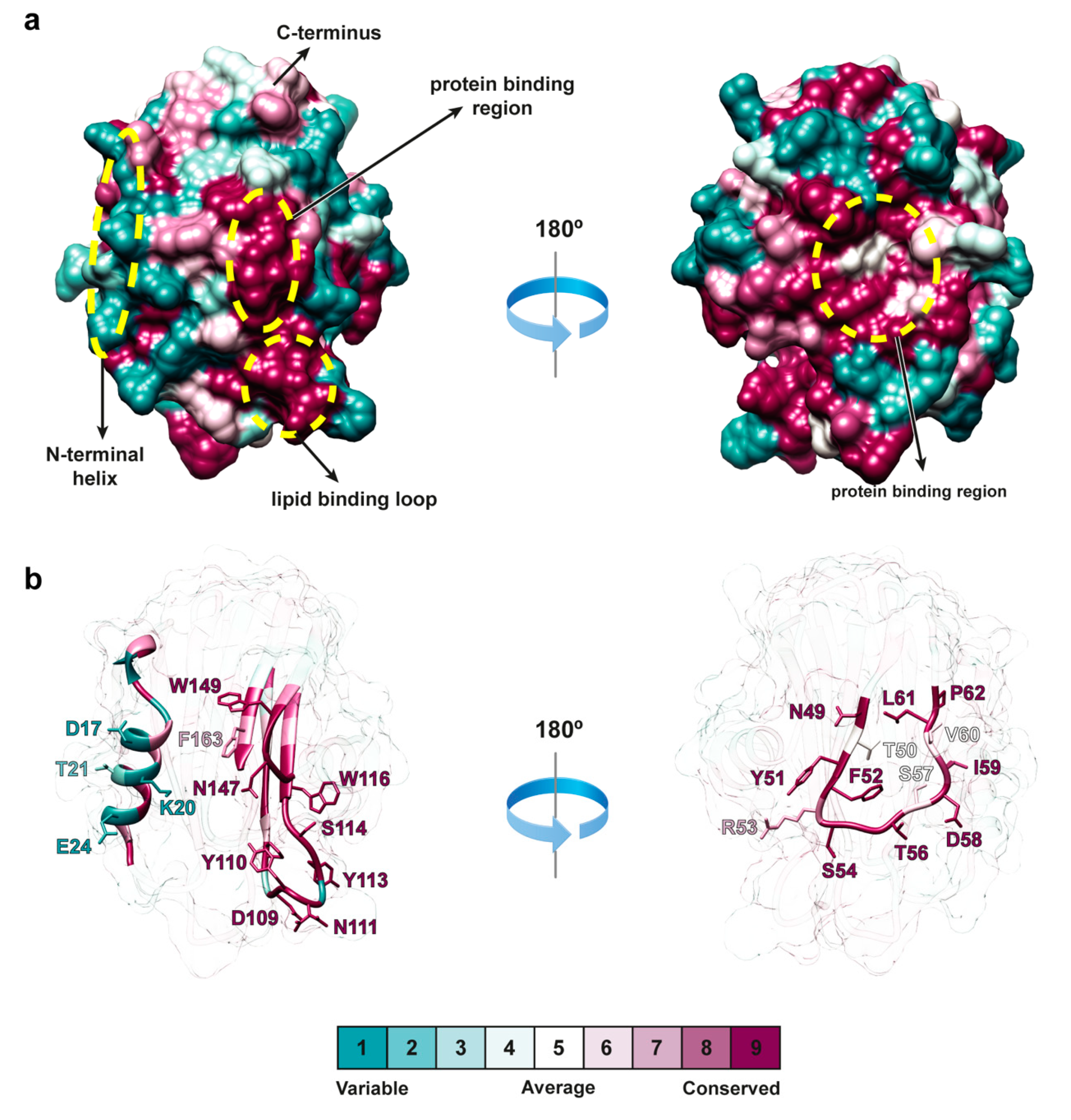
2.5. Structural Comparison of Actinoporins
2.6. Distribution of Actinoporin Variability
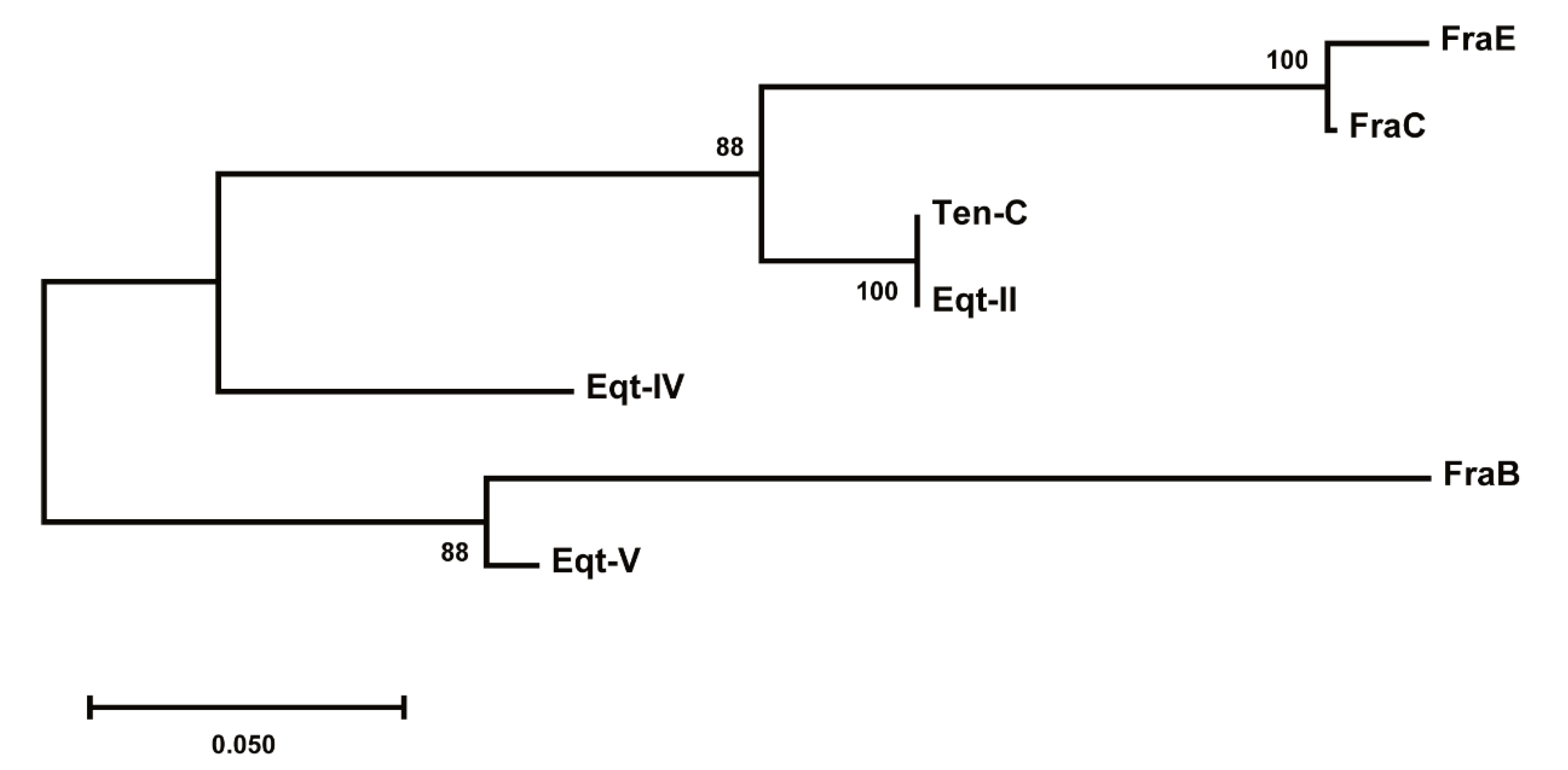
2.7. Evolutionary Divergence of Fragaceatoxins
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Purification of Fragaceatoxins
5.2. N-Terminal Sequencing
5.3. Hemolytic Activity
5.4. Protein Stability
5.5. Cloning and DNA Sequence Determination
5.6. Cloning, Expression, and Purification of FraE
5.7. Crystallization of FraE
5.8. Mass Spectrometry
5.9. Primary Structure Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anderluh, G.; Macek, P. Cytolytic peptide and protein toxins from sea anemones (Anthozoa: Actiniaria). Toxicon 2002, 40, 111–124. [Google Scholar] [CrossRef]
- Basulto, A.; Perez, V.M.; Noa, Y.; Varela, C.; Otero, A.J.; Pico, M.C. Immunohistochemical targeting of sea anemone cytolysins on tentacles, mesenteric filaments and isolated nematocysts of Stichodactyla helianthus. J. Exp. Zool. A Ecol. Genet. Physiol. 2006, 305, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Macrander, J.; Daly, M. Evolution of the cytolytic pore-forming proteins (actinoporins) in sea anemones. Toxins 2016, 8, 368. [Google Scholar] [CrossRef] [PubMed]
- Sher, D.; Fishman, Y.; Melamed-Book, N.; Zhang, M.L.; Zlotkin, E. Osmotically driven prey disintegration in the gastrovascular cavity of the green hydra by a pore-forming protein. FASEB J. 2008, 22, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Casewell, N.R.; Wuster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Duda, T.F. Extensive and continuous duplication facilitates rapid evolution and diversification of gene families. Mol. Biol. Evol. 2012, 29, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Jouiaei, M.; Sunagar, K.; Gross, A.F.; Scheib, H.; Alewood, P.F.; Moran, Y.; Fry, B.G. Evolution of an ancient venom: Recognition of a novel family of cnidarian toxins and the common evolutionary origin of sodium and potassium neurotoxins in sea anemone. Mol. Biol. Evol. 2015, 32, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Leychenko, E.; Isaeva, M.; Tkacheva, E.; Zelepuga, E.; Kvetkina, A.; Guzev, K.; Monastyrnaya, M.; Kozlovskaya, E. Multigene family of pore-forming toxins from sea anemone Heteractis crispa. Mar. Drugs 2018, 16, 183. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Sepcic, K.; Turk, T.; Macek, P. Cytolytic proteins from cnidarians-an Overview. Acta Chim. Slov. 2011, 58, 724–729. [Google Scholar]
- Tanaka, K.; Caaveiro, J.M.M.; Morante, K.; Gonzalez-Mañas, J.M.; Tsumoto, K. Structural basis for self-assembly of a cytolytic pore lined by protein and lipid. Nat. Commun. 2015, 6, 6337. [Google Scholar] [CrossRef]
- Tanaka, K.; Caaveiro, J.M.M.; Morante, K.; Tsumoto, K. Haemolytic actinoporins interact with carbohydrates using their lipid-binding module. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160216. [Google Scholar] [CrossRef] [PubMed]
- McCabe, T.M.; Mackessy, S.P. Evolution of Resistance to Toxins in Prey; Springer: Dordrecht, The Netherlands, 2017; pp. 47–65. [Google Scholar]
- Morante, K.; Caaveiro, J.M.M.; Viguera, A.R.; Tsumoto, K.; Gonzalez-Mañas, J.M. Functional characterization of Val60, a key residue involved in the membrane-oligomerization of fragaceatoxin C, an actinoporin from Actinia fragacea. FEBS Lett. 2015, 589, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Drummond, D.A.; Arnold, F.H.; Wilke, C.O. Structural determinants of the rate of protein evolution in yeast. Mol. Biol. Evol. 2006, 23, 1751–1761. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Ruiz, R.; Perez-Jimenez, R.; Ibarra-Molero, B.; Sanchez-Ruiz, J.M. Relation between protein stability, evolution and structure, as probed by carboxylic acid mutations. J. Mol. Biol. 2004, 336, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Larrea, D.; Perez-Jimenez, R.; Sanchez-Romero, I.; Delgado-Delgado, A.; Fernandez, J.M.; Sanchez-Ruiz, J.M. Role of conservative mutations in protein multi-property adaptation. Biochem. J. 2010, 429, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Stricher, F.; Schymkowitz, J.; Serrano, L.; Tawfik, D.S. The stability effects of protein mutations appear to be universally distributed. J. Mol. Biol. 2007, 369, 1318–1332. [Google Scholar] [CrossRef]
- Tokuriki, N.; Tawfik, D.S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009, 19, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Minasov, G.; Shoichet, B.K. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 2002, 320, 85–95. [Google Scholar] [CrossRef]
- Bloom, J.D.; Silberg, J.J.; Wilke, C.O.; Drummond, D.A.; Adami, C.; Arnold, F.H. Thermodynamic prediction of protein neutrality. Proc. Natl. Acad. Sci. USA 2005, 102, 606–611. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Weinreich, D.M.; Hartl, D.L. Missense meanderings in sequence space: A biophysical view of protein evolution. Nat. Rev. Genet. 2005, 6, 678–687. [Google Scholar] [CrossRef]
- Bellomio, A.; Morante, K.; Barlic, A.; Gutierrez-Aguirre, I.; Viguera, A.R.; Gonzalez-Manas, J.M. Purification, cloning and characterization of fragaceatoxin C, a novel actinoporin from the sea anemone Actinia fragacea. Toxicon 2009, 54, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Morante, K.; Caaveiro, J.M.M.; Tanaka, K.; Gonzalez-Mañas, J.M.; Tsumoto, K. A pore-forming toxin requires a specific residue for its activity in membranes with particular physicochemical properties. J. Biol. Chem. 2015, 290, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
- Morante, K.; Bellomio, A.; Gil-Carton, D.; Redondo-Morata, L.; Sot, J.; Scheuring, S.; Valle, M.; Gonzalez-Mañas, J.M.; Tsumoto, K.; Caaveiro, J.M.M. Identification of a membrane-bound prepore species clarifies the lytic mechanism of actinoporins. J. Biol. Chem. 2016, 291, 19210–19219. [Google Scholar] [CrossRef] [PubMed]
- Bischofberger, M.; Iacovache, I.; Boss, D.; Naef, F.; van der Goot, F.G.; Molina, N. Revealing assembly of a pore-forming complex using single-cell kinetic analysis and modeling. Biophys. J. 2016, 110, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Loytynoja, A.; Goldman, N. webPRANK: A phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinform. 2010, 11, 579. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.Z.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7. [Google Scholar] [CrossRef]
- Rivera-de-Torre, E.; Garcia-Linares, S.; Alegre-Cebollada, J.; Lacadena, J.; Gavilanes, J.G.; Martinez-del-Pozo, A. Synergistic action of actinoporin isoforms from the same sea anemone species assembled into functionally active heteropores. J. Biol. Chem. 2016, 291, 14109–14119. [Google Scholar] [CrossRef]
- Rivera-De-Torre, E.; Palacios-Ortega, J.; Garcia-Linares, S.; Gavilanes, J.G.; Martinez-Del-Pozo, A. One single salt bridge explains the different cytolytic activities shown by actinoporins sticholysin I and II from the venom of Stichodactyla helianthus. Arch. Biochem. Biophys. 2017, 636, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Armon, A.; Graur, D.; Ben-Tal, N. ConSurf: An algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 2001, 307, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Ruiz, R.; Perez-Jimenez, R.; Ibarra-Molero, B.; Sanchez-Ruiz, J.M. A stability pattern of protein hydrophobic mutations that reflects evolutionary structural optimization. Biophys. J. 2005, 89, 3320–3331. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. Evolutionary rate at the molecular level. Nature 1968, 217, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1983; p. 367. [Google Scholar]
- King, J.L.; Jukes, T.H. Non-darwinian evolution. Science 1969, 164, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T. Slightly deleterious mutant substitutions in evolution. Nature 1973, 246, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Linares, S.; Castrillo, I.; Bruix, M.; Menendez, M.; Alegre-Cebollada, J.; Martinez-del-Pozo, A.; Gavilanes, J.G. Three-dimensional structure of the actinoporin sticholysin I. Influence of long-distance effects on protein function. Arch. Biochem. Biophys. 2013, 532, 39–45. [Google Scholar] [CrossRef]
- Pardo-Cea, M.A.; Castrillo, I.; Alegre-Cebollada, J.; Martinez-del-Pozo, A.; Gavilanes, J.G.; Bruix, M. Intrinsic local disorder and a network of charge-charge interactions are key to actinoporin membrane disruption and cytotoxicity. FEBS J. 2011, 278, 2080–2089. [Google Scholar] [CrossRef]
- Wloka, C.; Mutter, N.L.; Soskine, M.; Maglia, G. Alpha-helical fragaceatoxin C nanopore engineered for double-stranded and single-stranded nucleic acid analysis. Angew. Chem. Int. Ed. Engl. 2016, 55, 12494–12498. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Edman, P.; Begg, G. A protein sequenator. Eur. J. Biochem. 1967, 1, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Signarvic, R.S.; DeGrado, W.F. Metal-binding dependent disruption of membranes by designed helices. J. Am. Chem. Soc. 2009, 131, 3377–3384. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate phenol chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W.; Sambrook, J. The Condensed Protocols from Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2006; p. 800. [Google Scholar]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 2011, 67, 235–242. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK-a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST plus: Architecture and applications. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Loytynoja, A.; Goldman, N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 2008, 320, 1632–1635. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Frazao, B.; Vasconcelos, V.; Antunes, A. Sea anemone (cnidaria, anthozoa, actiniaria) toxins: An overview. Mar. Drugs 2012, 10, 1812–1851. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Fraczkiewicz, R.; Braun, W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | N-Terminal Sequence | Mass a | HC50 (nM) b | Lag (s) c | υmax (s−1) c | TM (°C) |
|---|---|---|---|---|---|---|
| FraA | SAEVAGAVIEGAKLTFNVLQ | 19,728 ± 3 | 0.4 ± 0.02 | 31 ± 3 | 43 ± 4 | 65 ± 1 |
| FraB | SAEVAGAIIDGASLTFDVLQ | 19,672 ± 3 | 0.3 ± 0.02 | 17 ± 0.1 | 61 ± 1 | 62 ± 2 |
| FraC d | SADVAGAVIDGAGLG | 19,720 ± 3 | 1.6 ± 0.3 | 13 ± 0.4 | 60 ± 1 | 53 ± 3 |
| FraD | SVAVAGAVIKGAALTFNILQ | 19,721 ± 3 | 0.4 ± 0.3 | 7 ± 0.8 | 115 ± 11 | 47 ± 1 |
| FraE | SADVAGAVIDGAGLGFDVLK | 19,778 ± 3 | 1.6 ± 0.2 | 7 ± 1 | 106 ± 14 | 51 ± 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morante, K.; Bellomio, A.; Viguera, A.R.; González-Mañas, J.M.; Tsumoto, K.; Caaveiro, J.M.M. The Isolation of New Pore-Forming Toxins from the Sea Anemone Actinia fragacea Provides Insights into the Mechanisms of Actinoporin Evolution. Toxins 2019, 11, 401. https://doi.org/10.3390/toxins11070401
Morante K, Bellomio A, Viguera AR, González-Mañas JM, Tsumoto K, Caaveiro JMM. The Isolation of New Pore-Forming Toxins from the Sea Anemone Actinia fragacea Provides Insights into the Mechanisms of Actinoporin Evolution. Toxins. 2019; 11(7):401. https://doi.org/10.3390/toxins11070401
Chicago/Turabian StyleMorante, Koldo, Augusto Bellomio, Ana Rosa Viguera, Juan Manuel González-Mañas, Kouhei Tsumoto, and Jose M. M. Caaveiro. 2019. "The Isolation of New Pore-Forming Toxins from the Sea Anemone Actinia fragacea Provides Insights into the Mechanisms of Actinoporin Evolution" Toxins 11, no. 7: 401. https://doi.org/10.3390/toxins11070401
APA StyleMorante, K., Bellomio, A., Viguera, A. R., González-Mañas, J. M., Tsumoto, K., & Caaveiro, J. M. M. (2019). The Isolation of New Pore-Forming Toxins from the Sea Anemone Actinia fragacea Provides Insights into the Mechanisms of Actinoporin Evolution. Toxins, 11(7), 401. https://doi.org/10.3390/toxins11070401