Ultra-High-Performance Liquid Chromatography Coupled with Quadrupole Orbitrap High-Resolution Mass Spectrometry for Multi-Residue Analysis of Mycotoxins and Pesticides in Botanical Nutraceuticals

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
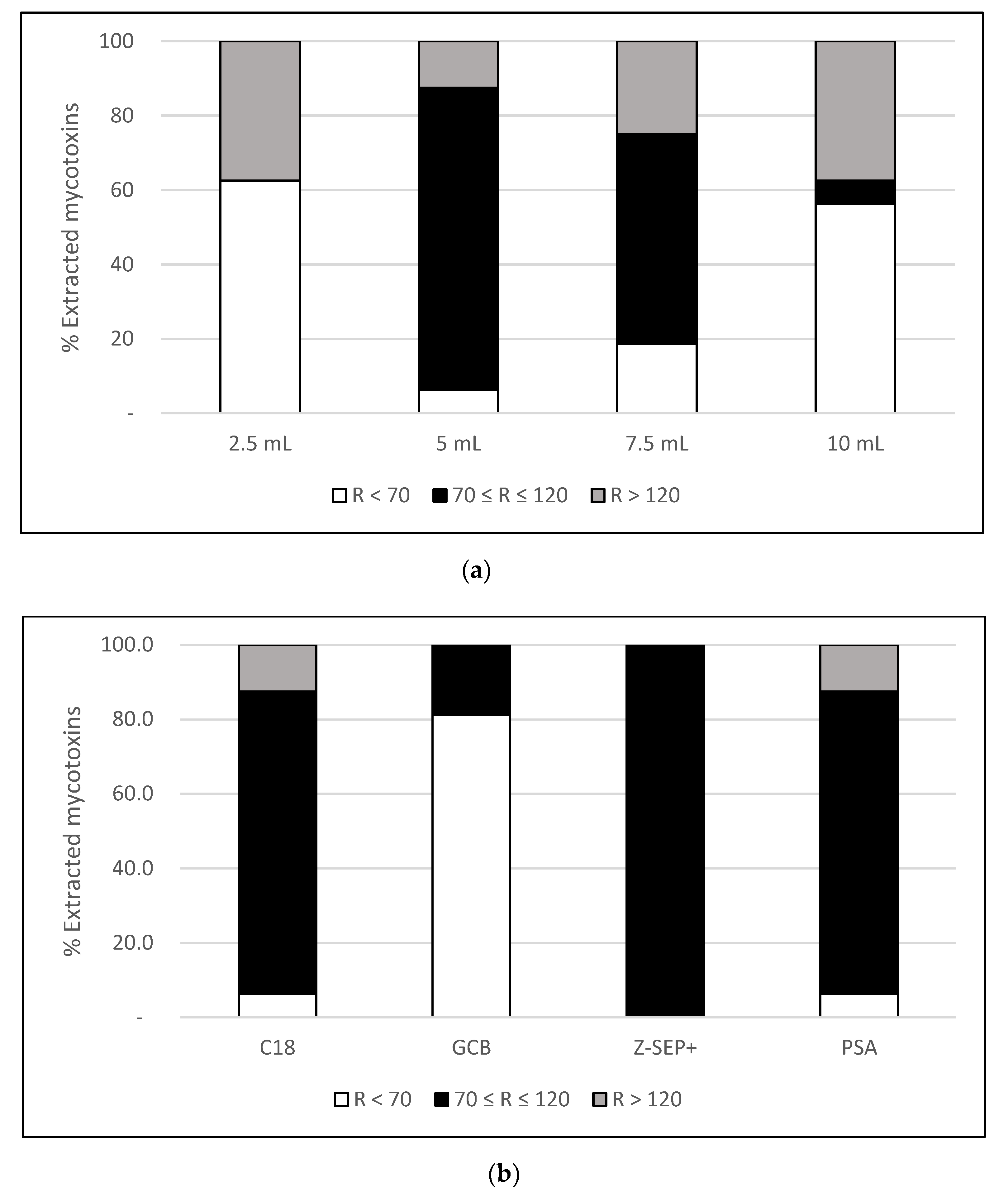
2.1. Optimization of Extraction Procedure
2.1.1. Evaluation of the Volume of Extraction Solvent
2.1.2. Evaluation of the Type of Sorbent for Clean-Up
2.2. Analytical Method Validation
2.3. Application to Commercial CBD-Based Products
2.4. Identification of Non-Target Compounds through Retrospective Analysis in Studied Samples
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Sampling
4.3. Sample Preparation
4.4. UHPLC-Q-Orbitrap HRMS Analysis
4.5. Validation Parameters
4.6. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Afshin, A.; Sur, P.J.; Fay, K.A.; Cornaby, L.; Ferrara, G.; Salama, J.S.; Mullany, E.C.; Abate, K.H.; Abbafati, C.; Abebe, Z.; et al. Health effects of dietary risks in 195 countries, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2019, 393, 1958–1972. [Google Scholar] [CrossRef] [Green Version]
- Almada, A.L. Nutraceuticals and functional foods: Innovation, insulation, evangelism, and evidence. In Nutraceutical and Functional Food Regulations in the United States and around the World, 3rd ed.; Academic Press: Dana Point, CA, USA, 2019; pp. 3–11. [Google Scholar]
- Binns, C.W.; Lee, M.K.; Lee, A.H. Problems and Prospects: Public Health Regulation of Dietary Supplements. Annu. Rev. Public Health 2018, 39, 403–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuardi, A. Cannabidiol: From an inactive cannabinoid to a drug with wide spectrum of action. Rev. Bras. Psiquiatr. 2008, 30, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casares, L.; García, V.; Garrido-Rodríguez, M.; Millán, E.; Collado, J.A.; García-Martín, A.; Peñarando, J.; Calzado, M.A.; de la Vega, L.; Muñoz, E. Cannabidiol induces antioxidant pathways in keratinocytes by targeting BACH1. Redox Biol. 2019, 28, 101321. [Google Scholar] [CrossRef]
- Maroon, J.; Bost, J. Review of the neurological benefits of phytocannabinoids. J. Surg. Neurol. Int. 2018, 9, 91. [Google Scholar] [CrossRef]
- Regulation (EU) 2015/2283 of the European Parliament and of the Council of 25 November 2015 on Novel Foods, Amending Regulation (EU) No 1169/2011 of the European Parliament and of the Council and Repealing Regulation (EC) No 258/97 of the European Parliament and of the Council and Commission Regulation (EC) No 1852/2001; European Parliament and the Council: Strasbourg, France, 2015.
- International CBD and Cannabis Market Landscape; BrightField Group Database; Brightfield Group: Chicago, IL, USA, 2019.
- Santini, A.; Cammarata, S.M.; Capone, G.; Ianaro, A.; Tenore, G.C.; Pani, L.; Novellino, E. Nutraceuticals: Opening the debate for a regulatory framework. Br. J. Clin. Pharmacol. 2018, 84, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Gulati, O.P.; Ottaway, P.B.; Jennings, S.; Coppens, P.; Gulati, N. Botanical nutraceuticals (food supplements and fortified and functional foods) and novel foods in the EU, with a main focus on legislative controls on safety aspects. In Nutraceutical and Functional Food Regulations in the United States and around the World, 3rd ed.; Academic Press: Brussels, Belgium, 2019; pp. 277–321. [Google Scholar]
- Rodríguez-Carrasco, Y.; Fattore, M.; Albrizio, S.; Berrada, H.; Mañes, J. Occurrence of Fusarium mycotoxins and their dietary intake through beer consumption by the European population. Food Chem. 2015, 178, 149–155. [Google Scholar] [CrossRef]
- Ostry, V.; Malir, F.; Toman, J.; Grosse, Y. Mycotoxins as human carcinogens—The IARC Monographs classification. J. Mycotoxin Res. 2017, 33, 65–73. [Google Scholar] [CrossRef]
- McKernan, K.; Spangler, J.; Zhang, L.; Tadigotla, V.; Helbert, Y.; Foss, T.; Smith, D. Cannabis microbiome sequencing reveals several mycotoxic fungi native to dispensary grade Cannabis flowers. F1000Res 2015, 4, 1422. [Google Scholar] [CrossRef] [Green Version]
- McHardy, I.; Romanelli, A.; Harris, L.J.; Opp, G.; Gaudino, R.; Torres, A.; Polage, C.R.; Tuscano, J.M.; Thompson, G.R. Infectious risks associated with medicinal Cannabis: Potential implications for immunocompromised patients? J. Infect. 2018, 76, 500–501. [Google Scholar] [CrossRef]
- Atapattu, S.N.; Johnson, K.R.D. Pesticide analysis in cannabis products. J. Chromatogr. A 2019, 1612, 460656. [Google Scholar] [CrossRef] [PubMed]
- Choudri, B.S.; Charabi, Y. Pesticides and herbicides. Water Environ. Res. 2019, 91, 1342–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafalou, S.; Abdollahi, M. Pesticides: An update of human exposure and toxicity. Arch. Toxicol. 2017, 91, 549–599. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Beach, J.; Martin, J.W.; Senthilselvan, A. Pesticide exposures and respiratory health in general populations. J. Environ. Sci. 2017, 51, 361–370. [Google Scholar] [CrossRef]
- Regulation (EC) No. 396/2005 of the European Parliament and of the Council of 23 February 2005 on Maximum Residue Levels of Pesticides in or on Food and Feed of Plant and Animal Origin and Amending Council Directive 91/414/EEC; European Parliament and the Council: Strasbourg, France, 2005.
- Commission Regulation (EC) No. 1881/2006 of the European Parliament and the Council of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs; European Parliament and the Council: Brussels, Belgium, 2006.
- Jeong, M.L.; Zahn, M.; Trinh, T.; Brooke, F.A.; Ma, W. Pesticide residue analysis of a dietary ingredient by gas chromatography/selected-ion monitoring mass spectrometry using neutral alumina solid-phase extraction cleanup. J. AOAC Int. 2008, 91, 630–636. [Google Scholar] [CrossRef] [Green Version]
- González-Martín, M.I.; Revilla, I.; Betances-Salcedo, E.V.; Vivar-Quintana, A.M. Pesticide residues and heavy metals in commercially processed propolis. Microchem. J. 2018, 143, 423–429. [Google Scholar] [CrossRef]
- Vaclavik, L.; Vaclavikova, M.; Begley, T.; Krynitsky, A.; Rader, J. Determination of Multiple Mycotoxins in Dietary Supplements Containing Green Coffee Bean Extracts Using Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry (UHPLC-MS/MS). J. Agric. Food Chem. 2013, 61, 4822–4830. [Google Scholar] [CrossRef]
- Veprikova, Z.; Zachariasova, M.; Dzuman, Z.; Zachariasova, A.; Fenclova, M.; Slavikova, P.; Vaclavikova, M.; Mastovska, K.; Hengst, D.; Hajslova, J. Mycotoxins in Plant-Based Dietary Supplements: Hidden Health Risk for Consumers. J. Agric. Food Chem. 2015, 63, 6633–6643. [Google Scholar] [CrossRef]
- Martínez-Domínguez, G.; Romero-González, R.; Garrido Frenich, A. Determination of toxic substances, pesticides and mycotoxins, in ginkgo biloba nutraceutical products by liquid chromatography Orbitrap-mass spectrometry. Microchem. J. 2015, 118, 124–130. [Google Scholar] [CrossRef]
- Martínez-Domínguez, G.; Romero-González, R.; Garrido Frenich, A. Multi-class methodology to determine pesticides and mycotoxins in green tea and royal jelly supplements by liquid chromatography coupled with Orbitrap high resolution mass spectrometry. Food Chem. 2016, 197, 907–915. [Google Scholar] [CrossRef]
- Martínez-Domínguez, G.; Romero-González, R.; Arrebola, F.J.; Garrido Frenich, A. Multi-class determination of pesticides and mycotoxins in isoflavones supplements obtained from soy by liquid chromatography coupled with Orbitrap high resolution mass spectrometry. Food Control 2016, 59, 218–224. [Google Scholar] [CrossRef]
- Santos, L.; Marín, S.; Sanchis, V.; Ramos, A.J. Screening of mycotoxin multicontamination in medicinal and aromatic herbs sampled in Spain. J. Sci. Food Agric. 2009, 89, 1802–1807. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, X.; Shen, C.; Qu, B. Determination of 16 mycotoxins in vegetable oils using a QuEChERS method combined with high-performance liquid chromatography-tandem mass spectrometry. Food Addit. Contam. 2017, 34, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Ruiz, J.L.; Romero-González, R.; Martínez Vidal, J.L.; Garrido Frenich, A. A rapid method for the determination of mycotoxins in edible vegetable oils by ultra-high performance liquid chromatography-tandem mass spectrometry. Food Chem. 2019, 288, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Myresiotis, C.K.; Testempasis, S.; Vryzas, Z.; Karaoglanidis, G.S.; Papadopoulou-Mourkidou, E. Determination of mycotoxins in pomegranate fruits and juices using a QuEChERS-based method. Food Chem. 2015, 182, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Matarrita, J.; Sapozhnikova, Y.; Lehotay, S.J. Evaluation of a recent product to remove lipids and other matrix co-extractives in the analysis of pesticide residues and environmental contaminants in foods. J. Chromatogr. A 2016, 1449, 17–29. [Google Scholar] [CrossRef]
- Rajski, Ł.; Lozano, A.; Uclés, A.; Ferrer, C.; Fernández-Alba, A.R. Determination of pesticide residues in high oil vegetal commodities by using various multi-residue methods and clean-ups followed by liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2013, 1304, 109–120. [Google Scholar] [CrossRef]
- Lozano, A.; Rajski, Ł.; Uclés, S.; Belmonte-Valles, N.; Mezcua, M.; Fernández-Alba, A.R. Evaluation of zirconium dioxide-based sorbents to decrease the matrix effect in avocado and almond multiresidue pesticide analysis followed by gas chromatography tandem mass spectrometry. Talanta 2014, 118, 68–83. [Google Scholar] [CrossRef]
- Tuzimski, T.; Szubartowski, S. Method Development for Selected Bisphenols Analysis in Sweetened Condensed Milk from a Can and Breast Milk Samples by HPLC-DAD and HPLC-QqQ-MS: Comparison of Sorbents (Z-SEP, Z-SEP Plus, PSA, C18, Chitin and EMR-Lipid) for Clean-Up of QuEChERS Extract. Molecules 2019, 24, 2093. [Google Scholar] [CrossRef] [Green Version]
- Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results (Text with EEA Relevance); Brussels, Belgium, 2002; Available online: https://op.europa.eu/en/publication-detail/-/publication/ed928116-a955-4a84-b10a-cf7a82bad858/language-en (accessed on 12 February 2020).
- Han, Z.; Ren, Y.; Zhu, J.; Cai, Z.; Chen, Y.; Luan, L.; Wu, Y. Multianalysis of 35 Mycotoxins in Traditional Chinese Medicines by Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry Coupled with Accelerated Solvent Extraction. J. Agric. Food Chem. 2012, 60, 8233–8247. [Google Scholar] [CrossRef]
- Arroyo-Manzanares, N.; García-Campaña, A.M.; Gámiz-Gracia, L. Multiclass mycotoxin analysis in Silybum marianum by ultra high performance liquid chromatography–tandem mass spectrometry using a procedure based on QuEChERS and dispersive liquid–liquid microextraction. J. Chromatogr. A 2013, 1282, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Ren, Y.; Zhou, H.; Luan, L.; Cai, Z.; Wu, Y. A rapid method for simultaneous determination of zearalenone, α-zearalenol, β-zearalenol, zearalanone, α-zearalanol and β-zearalanol in traditional Chinese medicines by ultra-high-performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2011, 879, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.-C.; Madec, S.; Coton, E.; Hymery, N. Natural Co-Occurrence of Mycotoxins in Foods and Feeds and Their in vitro Combined Toxicological Effects. Toxins 2016, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Tournas, V.H.; Sapp, C.; Trucksess, M.W. Occurrence of aflatoxins in milk thistle herbal supplements. Food Addit. Contam. 2012, 29, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Pesticides Properties DataBase (PPDB). University of Hertfordshire, United Kingdom. Available online: http://sitem.herts.ac.uk/aeru/ppdb/en/ (accessed on 22 December 2019).
- Regulation (EC) No. 1107/2009 of the European Parliament and of the Council of 21 October 2009 Concerning the Placing of Plant Protection Products on the Market and Repealing Council Directives 79/117/EEC and 91/414/EEC; European Parliament and the Council: Strasbourg, France, 2009.
- Commission Decision 2004/247/EC of 10 March 2004 Concerning the Non-Inclusion of Simazine in Annex I to Council Directive 91/414/EEC and the Withdrawal of Authorisations for Plant Protection Products Containing this Active Substance; Brussels, Belgium, 2004. Available online: https://www.legislation.gov.uk/eur/2009/1107/contents (accessed on 12 February 2020).
- Castaldo, L.; Graziani, G.; Gaspari, A.; Izzo, L.; Tolosa, J.; Rodríguez-Carrasco, Y.; Ritieni, A. Target Analysis and Retrospective Screening of Multiple Mycotoxins in Pet Food Using UHPLC-Q-Orbitrap HRMS. Toxins 2019, 11, 434. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Analyte | Linearity (r2) | SSE (%) | Recovery (%) | Precision (%) [RSDr, (RSDR)] | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 ng/g 1 | 10 ng/g | 20 ng/g | 50 ng/g | 2 ng/g 1 | 10 ng/g | 20 ng/g | 50 ng/g | LOQ (ng/g) | |||
| AFG2 | 0.9975 | 111 | 78 | 77 | 81 | 98 | 16 (19) | 5 (6) | 6 (6) | 5 (4) | 0.78 |
| AFG1 | 0.9982 | 106 | 81 | 86 | 86 | 105 | 12 (9) | 16 (19) | 7 (6) | 11 (10) | 1.56 |
| AFB1 | 0.9984 | 115 | 71 | 91 | 98 | 107 | 14 (13) | 10 (8) | 4 (4) | 4 (3) | 0.20 |
| AFB2 | 0.9998 | 111 | 86 | 88 | 91 | 103 | 18 (15) | 10 (8) | 7 (5) | 5 (4) | 0.20 |
| NEO | 0.9988 | 112 | 88 | 93 | 104 | 18 (14) | 16 (18) | 17 (18) | 0.78 | ||
| HT-2 | 0.9984 | 108 | 113 | 101 | 92 | 12 (14) | 16 (11) | 12 (15) | 6.25 | ||
| T-2 | 0.9990 | 83 | 89 | 98 | 110 | 19 (13) | 9 (7) | 7 (10) | 0.78 | ||
| α-ZEL | 0.9943 | 81 | 81 | 94 | 100 | 11 (11) | 10 (14) | 5 (16) | 6.25 | ||
| β-ZEL | 0.9985 | 84 | 106 | 103 | 89 | 8 (18) | 15 (16) | 9 (11) | 3.13 | ||
| ZAN | 0.9992 | 108 | 111 | 100 | 105 | 15 (13) | 18 (11) | 5 (13) | 1.56 | ||
| ZEN | 0.9991 | 109 | 104 | 103 | 93 | 5 (16) | 15 (14) | 10 (19) | 3.13 | ||
| ENN B | 0.9998 | 102 | 63 | 63 | 65 | 18 (19) | 18 (18) | 6 (7) | 6.25 | ||
| ENN B1 | 0.9982 | 99 | 83 | 89 | 85 | 12 (11) | 8 (6) | 8 (8) | 1.56 | ||
| ENN A | 0.9942 | 84 | 96 | 91 | 80 | 11 (9) | 14 (17) | 11 (12) | 3.13 | ||
| ENN A1 | 0.9972 | 87 | 92 | 101 | 90 | 12 (14) | 9 (6) | 7 (14) | 1.56 | ||
| BEA | 0.9971 | 119 | 80 | 71 | 63 | 18 (17) | 10 (18) | 10 (19) | 6.25 | ||
| Samples Procedence (no.) | Positives Samples (%) | Major Analytes Detected | Concentration Reported (ng/g) | Determination | ||
|---|---|---|---|---|---|---|
| Sensitivity (LOQ, ng/g) | Detection Method | Reference | ||||
| Medicinal or aromatic herbs (84) | 99 | ZEN | 1.0–44.1 | 0.14 | ELISA detection (EIA reader, SIRIO S) | [28] |
| T-2 | 0.6–256.9 | 0.28 | ||||
| DON3 | 20.5–343.5 | 14.8 | ||||
| CIT3 | 14.9–354.8 | 16.5 | ||||
| Traditional Chinese herbs (60) | 83 | ZEN | 2.1–15.5 | 0.4 | QQQ (Applied Biosystems) ESI+ MRM mode | [37] |
| AFs3 | 0.2–19.5 | 0.1 | ||||
| MPA3 | 0.2–22.7 | 0.02 | ||||
| Milk thistle (83) | 19 | AFB1 | 0.04–1.9 | 0.03 | LC-FLD (Waters) | [41] |
| Green coffee bean (50) | 36 | OTA3 | 1–136.9 | 2.5 | QQQ (AB SCIEX) ESI+ and ESI- MRM mode | [23] |
| OTB3 | 1–20.2 | 2.5 | ||||
| FB13 | 50–415 | 100 | ||||
| MPA | 5–395 | 10 | ||||
| Milk thistle (7) | 29 | T-2 | 363–453.9 | 30.5 | QQQ (AB SCIEX) ESI+ MRM mode | [38] |
| HT-2 | 826.9–943.7 | 43.8 | ||||
| Herbals (69) | 96 | ZEN | 5–824 | 10 | QQQ (AB SCIEX) ESI+ and ESI- MRM mode | [24] |
| T-2 | 69–1870 | 10 | ||||
| HT-2 | 59–1530 | 50 | ||||
| ENNB | 5–9260 | 5 | ||||
| ENNB1 | 5–10,900 | 5 | ||||
| ENNA | 5–8340 | 5 | ||||
| ENNA1 | 5–2340 | 5 | ||||
| Gingko biloba (8) | 50 | AFB1 | 5.0–54 | 5 | Q-Orbitrap (Exactive, Thermo FisherScientific) ESI+ and ESI- HRMS | [25] |
| AFB2 | 4–300 | 10 | ||||
| T-2 | 18–20 | 30.5 | ||||
| Green tea (10) | 10 | AFB1 | 5.4 | 5 | Q-Orbitrap (Exactive, Thermo FisherScientific) ESI+ and ESI- HRMS | [26] |
| Royal jelly (8) | 0 | |||||
| Soy (11) | 27 | AFB1 | 8.2–17.1 | 5 | Q-Orbitrap (Exactive, Thermo FisherScientific) ESI+ and ESI- HRMS | [27] |
| AFG2 | 6.4 | 5 | ||||
| Cannabis sativa (10) | 70 | ZEN | 4.2–11.6 | 3.13 | Q-Orbitrap (Exactive, Thermo FisherScientific) ESI+ and ESI- HRMS | Current study |
| ENNB1 | <LOQ–11.6 | 1.56 | ||||
| Sample | Mycotoxin (ng/g) | |||||
|---|---|---|---|---|---|---|
| T-2 | ZAN | ZEN | ENN B1 | ENN A | ENN A1 | |
| 1 | 11.6 | 11.6 | 4.2 | 5.8 | ||
| 4 | 6.5 | |||||
| 5 | <LOQ | |||||
| 7 | 8.1 | |||||
| 8 | 1.9 | 4.7 | ||||
| 9 | 4.2 | <LOQ | ||||
| 10 | 2.0 | 6.3 | ||||
| Analyte | Retention Time (min) | Elemental Composition | Adduct Ion | Theoretical Mass (m/z) | Measured Mass (m/z) | Accuracy (Δ ppm) |
|---|---|---|---|---|---|---|
| NEO | 4.25 | C19H26O8 | [M+NH4]+ | 400.1966 | 400.1963 | −0.67 |
| AFG2 | 4.50 | C17H14O7 | [M+H]+ | 331.0812 | 331.0808 | −1.36 |
| AFG1 | 4.52 | C17H12O7 | [M+H]+ | 329.0656 | 329.0655 | −0.27 |
| AFB2 | 4.58 | C17H14O6 | [M+H]+ | 315.0863 | 315.0862 | −0.51 |
| AFB1 | 4.62 | C17H12O6 | [M+H]+ | 313.0707 | 313.0705 | −0.42 |
| HT-2 | 4.74 | C22H32O8 | [M+NH4]+ | 442.2435 | 442.2432 | −0.7 |
| α-ZEL | 4.83 | C18H24O5 | [M-H]- | 319.1551 | 319.1550 | −0.31 |
| T-2 | 4.85 | C24H34O9 | [M+NH4]+ | 484.2541 | 484.2543 | 0.39 |
| β-ZEL | 4.97 | C18H24O5 | [M-H]- | 319.1551 | 319.1550 | −0.31 |
| ZAN | 4.98 | C18H24O5 | [M-H]- | 319.1551 | 319.1549 | −0.6 |
| ZEN | 5.01 | C18H22O5 | [M+H]+ | 317.1395 | 317.1393 | −0.54 |
| ENN B | 5.56 | C33H57N3O9 | [M+NH4]+ | 657.4433 | 657.4435 | 0.26 |
| ENN B1 | 5.68 | C34H59N3O9 | [M+NH4]+ | 671.4599 | 671.4594 | −0.76 |
| BEA | 5.73 | C45H57N3O9 | [M+NH4]+ | 801.4433 | 801.4432 | −0.16 |
| ENN A1 | 5.82 | C35H61N3O9 | [M+NH4]+ | 685.4746 | 685.4745 | −0.18 |
| ENN A | 5.99 | C36H63N3O9 | [M+NH4]+ | 699.4903 | 699.4899 | −0.56 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narváez, A.; Rodríguez-Carrasco, Y.; Castaldo, L.; Izzo, L.; Ritieni, A. Ultra-High-Performance Liquid Chromatography Coupled with Quadrupole Orbitrap High-Resolution Mass Spectrometry for Multi-Residue Analysis of Mycotoxins and Pesticides in Botanical Nutraceuticals. Toxins 2020, 12, 114. https://doi.org/10.3390/toxins12020114
Narváez A, Rodríguez-Carrasco Y, Castaldo L, Izzo L, Ritieni A. Ultra-High-Performance Liquid Chromatography Coupled with Quadrupole Orbitrap High-Resolution Mass Spectrometry for Multi-Residue Analysis of Mycotoxins and Pesticides in Botanical Nutraceuticals. Toxins. 2020; 12(2):114. https://doi.org/10.3390/toxins12020114
Chicago/Turabian StyleNarváez, Alfonso, Yelko Rodríguez-Carrasco, Luigi Castaldo, Luana Izzo, and Alberto Ritieni. 2020. "Ultra-High-Performance Liquid Chromatography Coupled with Quadrupole Orbitrap High-Resolution Mass Spectrometry for Multi-Residue Analysis of Mycotoxins and Pesticides in Botanical Nutraceuticals" Toxins 12, no. 2: 114. https://doi.org/10.3390/toxins12020114
APA StyleNarváez, A., Rodríguez-Carrasco, Y., Castaldo, L., Izzo, L., & Ritieni, A. (2020). Ultra-High-Performance Liquid Chromatography Coupled with Quadrupole Orbitrap High-Resolution Mass Spectrometry for Multi-Residue Analysis of Mycotoxins and Pesticides in Botanical Nutraceuticals. Toxins, 12(2), 114. https://doi.org/10.3390/toxins12020114