Klotho/FGF23 and Wnt Signaling as Important Players in the Comorbidities Associated with Chronic Kidney Disease

 ,
,  , , and
, , and 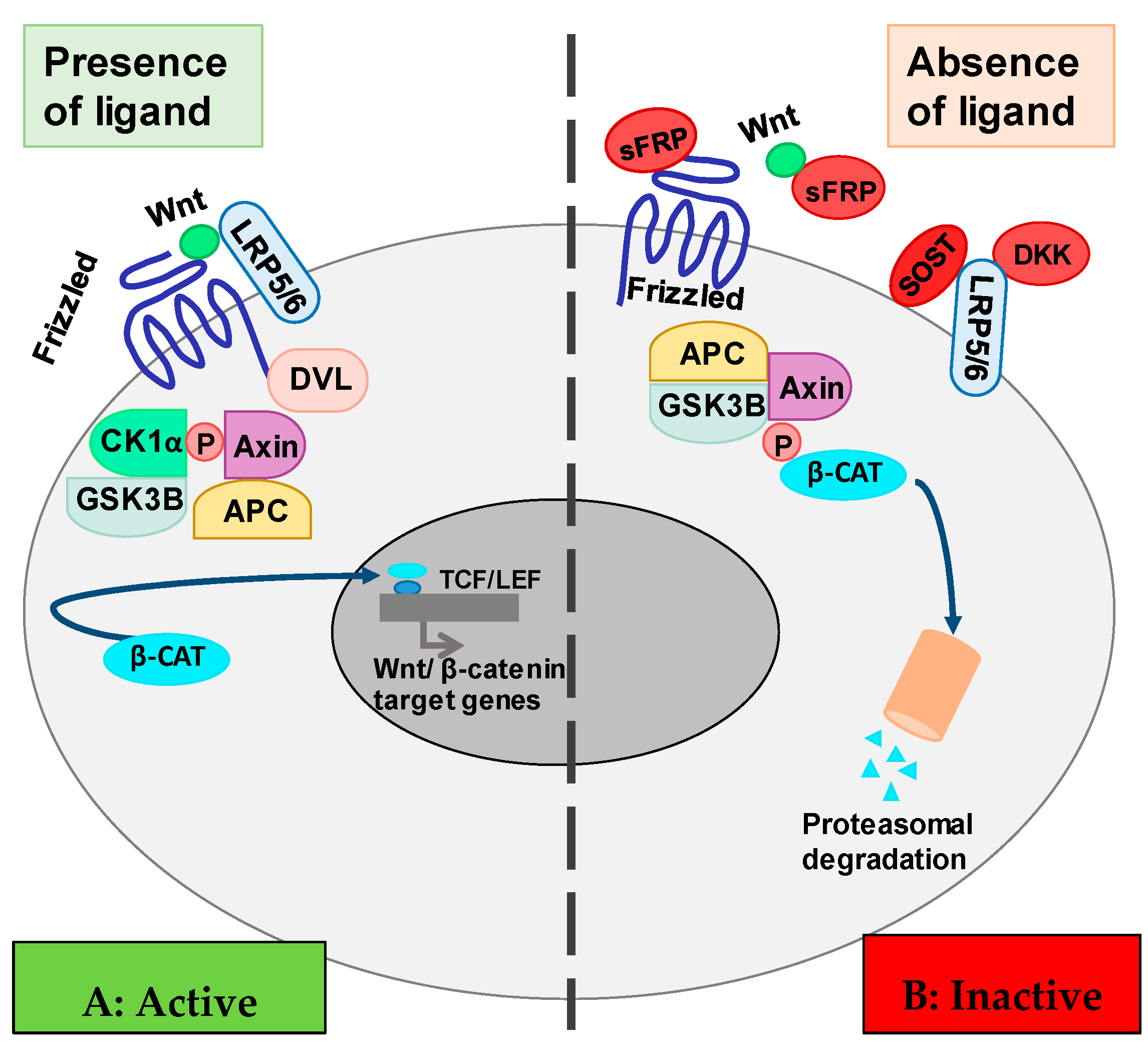{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Wnt/β-Catenin Cell Signaling Pathway
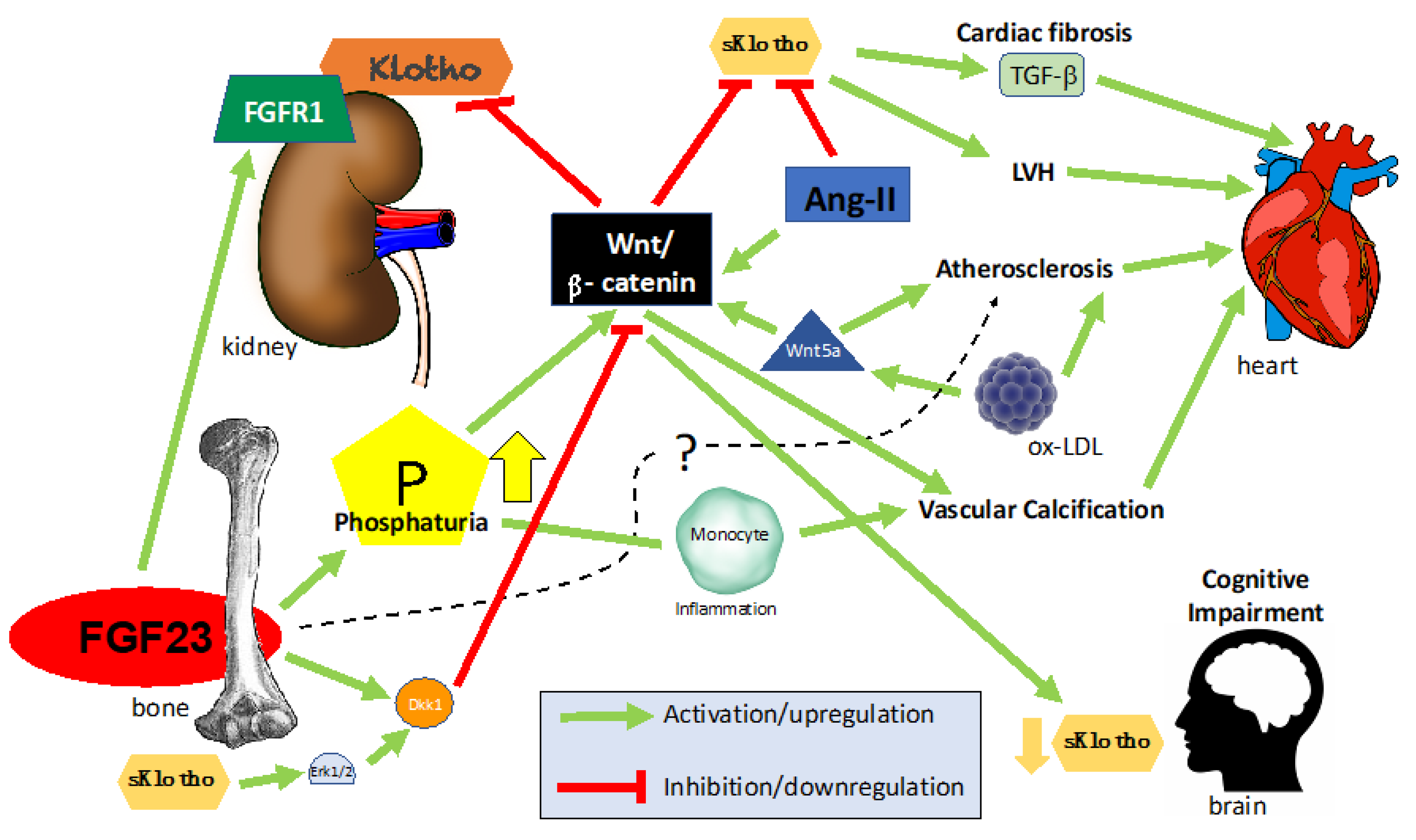
3. Klotho-FGF23 and Wnt in Chronic Kidney Disease
3.1. Regulation of Klotho Expression in the Kidney: The Effect of the Tubular Load of Phosphate
3.2. Klotho, Wnt/β-Catenin, and Renal Damage
3.3. Klotho, Wnt/β-Catenin, and Polycystic Kidney Disease (PKD)
3.4. Consequences of the Close Relationship between Wnt and Klotho
3.5. Kidney Fibrosis
3.6. Cardiorenal Syndrome
4. FGF23/Klotho/Wnt in Cardiovascular Disease (CVD)
4.1. Klotho, Wnt/β-Catenin, and RAS
4.2. FGF23/Klotho and Cardiac Hypertrophy
4.3. FGF23–Klotho–Wnt and Cardiac Fibrosis
4.4. FGF23/Klotho and Atherosclerosis
4.5. FGF23/Klotho and Vascular Calcification
5. FGF23-Klotho and Wnt in Bone
6. Wnt and the Central Nervous System
7. FGF23/Klotho and Wnt in Other Organs
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FGF23 | Fibroblast Growth Factor 23 |
| CKD | Chronic kidney disease |
| FGFR1 | Fibroblast Growth Factor Receptor-1 |
| 1,25(OH)2D | 1,25 hydroxyvitamin D |
| PTH | Parathyroid Hormone |
| Dkk1 | Dickkopf-related protein-1 |
| TCF/LEF | T-cell factor/lymphoid enhancer-binding factor |
| LRP5 | Lipoprotein-receptor related protein 5 |
| APC | Adenomatous Polyposis Coli |
| CK1α | Casein kinase 1 isoform α |
| GSK3β | Glycogen Synthase Kinase 3β |
| sFRP | Secreted Frizzled-Related Protein |
| SOST | Sclerostin |
| DVL | Disheveled |
| TRPC6 | Transient Receptor Potential Cation Channel |
| CRS | Cardiorenal Syndrome |
| PKD | Polycystic kidney disease |
| ADPKD | Autosomal Polycystic kidney disease |
| CVD | Cardiovascular disease |
| LVH | Left Ventricular Hypertrophy |
| HF | Heart Failure |
| ACE | Angiotensin-Converting Enzyme |
| AT1 | Angiotensin I |
| PRR | Pro-Renin Receptor |
| RAS | Renin-angiotensin system |
| VSMC | Vascular Smooth Muscle Cells |
| HUVEC | Human Umbilical Vein Endothelial Cell |
| CIMT | Carotid Intima-Media Thickness |
| LPS | Lipopolysaccharide |
| COPD | Chronic Obstructive Pulmonary Disease |
References
- Drüeke, T.B.; Massy, Z.A. Changing bone patterns with progression of chronic kidney disease. Kidney Int. 2016, 89, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ. Res. 2018, 123, 825–848. [Google Scholar] [CrossRef]
- Komaba, H.; Kaludjerovic, J.; Hu, D.Z.; Nagano, K.; Amano, K.; Ide, N.; Sato, T.; Densmore, M.J.; Hanai, J.-I.; Olauson, H.; et al. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int. 2017, 92, 599–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, I.; Levanon-Cohen, S.; Bose, S.; Ligumsky, H.; Sredni, B.; Kanety, H.; Kuro-o, M.; Karlan, B.; Kaufman, B.; Koeffler, H.P.; et al. Klotho: A tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 2008, 27, 7094–7105. [Google Scholar] [CrossRef]
- Abramovitz, L.; Rubinek, T.; Ligumsky, H.; Bose, S.; Barshack, I.; Avivi, C.; Kaufman, B.; Wolf, I. KL1 internal repeat mediates klotho tumor suppressor activities and inhibits bFGF and IGF-I signaling in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 4254–4266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Castañeda, J.R.; Herencia, C.; Pendón-Ruiz de Mier, M.V.; Rodriguez-Ortiz, M.E.; Diaz-Tocados, J.M.; Vergara, N.; Martínez-Moreno, J.M.; Salmerón, M.D.; Richards, W.G.; Felsenfeld, A.; et al. Differential regulation of renal Klotho and FGFR1 in normal and uremic rats. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 3858–3867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacchetta, J.; Bardet, C.; Prié, D. Physiology of FGF23 and overview of genetic diseases associated with renal phosphate wasting. Metabolism 2020, 103S, 153865. [Google Scholar] [CrossRef] [PubMed]
- John, G.B.; Cheng, C.-Y.; Kuro-o, M. Role of Klotho in aging, phosphate metabolism, and CKD. Am. J. Kidney Dis. 2011, 58, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Krishnan, V.; Bryant, H.U.; Macdougald, O.A. Regulation of bone mass by Wnt signaling. J. Clin. Investig. 2006, 116, 1202–1209. [Google Scholar] [CrossRef]
- Semënov, M.; Tamai, K.; He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 2005, 280, 26770–26775. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, B.; Wu, W.; Li, Y.; Hoppe, D.; Stannek, P.; Glinka, A.; Niehrs, C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 2001, 411, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z. Current understanding of klotho. Ageing Res. Rev. 2009, 8, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruska, K.A.; Sugatani, T.; Agapova, O.; Fang, Y. The chronic kidney disease—Mineral bone disorder (CKD-MBD): Advances in pathophysiology. Bone 2017, 100, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin downregulates Klotho in tubular cells. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2018, 33, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Stolz, D.B.; Kiss, L.P.; Monga, S.P.; Holzman, L.B.; Liu, Y. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J. Am. Soc. Nephrol. JASN 2009, 20, 1997–2008. [Google Scholar] [CrossRef] [Green Version]
- Matsui, I.; Ito, T.; Kurihara, H.; Imai, E.; Ogihara, T.; Hori, M. Snail, a transcriptional regulator, represses nephrin expression in glomerular epithelial cells of nephrotic rats. Lab. Investig. J. Tech. Methods Pathol. 2007, 87, 273–283. [Google Scholar] [CrossRef]
- Kim, M.S.; Yoon, S.K.; Bollig, F.; Kitagaki, J.; Hur, W.; Whye, N.J.; Wu, Y.-P.; Rivera, M.N.; Park, J.Y.; Kim, H.-S.; et al. A novel Wilms tumor 1 (WT1) target gene negatively regulates the WNT signaling pathway. J. Biol. Chem. 2010, 285, 14585–14593. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-L.; Wang, J.-Y.; Huang, Y.-T.; Kuo, Y.-H.; Surendran, K.; Wang, F.-S. Wnt/beta-catenin signaling modulates survival of high glucose-stressed mesangial cells. J. Am. Soc. Nephrol. JASN 2006, 17, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.J.; Nam, B.Y.; Wu, M.; Kim, S.; Park, J.; Kang, S.; Park, J.T.; Yoo, T.-H.; Kang, S.-W.; Han, S.H. Klotho plays a protective role against glomerular hypertrophy in a cell cycle-dependent manner in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2018, 315, F791–F805. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Xie, J.; Hwang, K.-H.; Wu, Y.-L.; Oliver, N.; Eom, M.; Park, K.-S.; Barrezueta, N.; Kong, I.-D.; Fracasso, R.P.; et al. Klotho May Ameliorate Proteinuria by Targeting TRPC6 Channels in Podocytes. J. Am. Soc. Nephrol. JASN 2017, 28, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Cha, S.-K.; An, S.-W.; Kuro-O, M.; Birnbaumer, L.; Huang, C.-L. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat. Commun. 2012, 3, 1238. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhou, C.J.; Liu, Y. Wnt Signaling in Kidney Development and Disease. Prog. Mol. Biol. Transl. Sci. 2018, 153, 181–207. [Google Scholar]
- Wuebken, A.; Schmidt-Ott, K.M. WNT/β-catenin signaling in polycystic kidney disease. Kidney Int. 2011, 80, 135–138. [Google Scholar] [CrossRef] [Green Version]
- Pavik, I.; Jaeger, P.; Kistler, A.D.; Poster, D.; Krauer, F.; Cavelti-Weder, C.; Rentsch, K.M.; Wüthrich, R.P.; Serra, A.L. Patients with autosomal dominant polycystic kidney disease have elevated fibroblast growth factor 23 levels and a renal leak of phosphate. Kidney Int. 2011, 79, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Pavik, I.; Jaeger, P.; Ebner, L.; Poster, D.; Krauer, F.; Kistler, A.D.; Rentsch, K.; Andreisek, G.; Wagner, C.A.; Devuyst, O.; et al. Soluble klotho and autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. CJASN 2012, 7, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Perrone, R.D. Extrarenal manifestations of ADPKD. Kidney Int. 1997, 51, 2022–2036. [Google Scholar] [CrossRef] [Green Version]
- Perrone, R.D.; Malek, A.M.; Watnick, T. Vascular complications in autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Krishnappa, V.; Vinod, P.; Deverakonda, D.; Raina, R. Autosomal dominant polycystic kidney disease and the heart and brain. Cleve. Clin. J. Med. 2017, 84, 471–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Li, Y.; Zhou, D.; Tan, R.J.; Liu, Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/β-catenin signaling. J. Am. Soc. Nephrol. JASN 2013, 24, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, M.; Nagasu, H.; Morita, Y.; Yamaguchi, T.P.; Kanwar, Y.S.; Kashihara, N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am. J. Physiol. Renal Physiol. 2012, 303, F1641–F1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kida, Y.; Duffield, J.S. Pivotal role of pericytes in kidney fibrosis. Clin. Exp. Pharmacol. Physiol. 2011, 38, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Dai, C.; Li, Y.; Zeng, G.; Monga, S.P.; Liu, Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. JASN 2009, 20, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardio-renal syndromes: Report from the consensus conference of the acute dialysis quality initiative. Eur. Heart J. 2010, 31, 703–711. [Google Scholar] [CrossRef]
- Ishigami, J.; Cowan, L.T.; Demmer, R.T.; Grams, M.E.; Lutsey, P.L.; Carrero, J.-J.; Coresh, J.; Matsushita, K. Incident Hospitalization with Major Cardiovascular Diseases and Subsequent Risk of ESKD: Implications for Cardiorenal Syndrome. J. Am. Soc. Nephrol. JASN 2020. [Google Scholar] [CrossRef]
- Tanaka, S.; Fujita, S.-I.; Kizawa, S.; Morita, H.; Ishizaka, N. Association between FGF23, α-Klotho, and Cardiac Abnormalities among Patients with Various Chronic Kidney Disease Stages. PLoS ONE 2016, 11, e0156860. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Hong, X.; Miao, J.; Liao, Y.; Hou, F.F.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling mediates both heart and kidney injury in type 2 cardiorenal syndrome. Kidney Int. 2019, 95, 815–829. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Cho, H.J.; Adams-Huet, B.; Paek, J.; Hill, K.; Shelton, J.; Amaral, A.P.; Faul, C.; Taniguchi, M.; et al. Klotho and Phosphate Are Modulators of Pathologic Uremic Cardiac Remodeling. J. Am. Soc. Nephrol. 2015, 26, 1290–1302. [Google Scholar] [CrossRef] [Green Version]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharmacol. Rev. 2018, 70, 68–141. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, L.; Wang, Y.; Miao, J.; Hong, X.; Hou, F.F.; Liu, Y. (Pro)renin Receptor Is an Amplifier of Wnt/β-Catenin Signaling in Kidney Injury and Fibrosis. J. Am. Soc. Nephrol. JASN 2017, 28, 2393–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramkumar, N.; Stuart, D.; Mironova, E.; Bugay, V.; Wang, S.; Abraham, N.; Ichihara, A.; Stockand, J.D.; Kohan, D.E. Renal tubular epithelial cell prorenin receptor regulates blood pressure and sodium transport. Am. J. Physiol. Renal Physiol. 2016, 311, F186–F194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Li, Y.; Hao, S.; Zhou, D.; Tan, R.J.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Multiple genes of the renin-angiotensin system are novel targets of Wnt/β-catenin signaling. J. Am. Soc. Nephrol. JASN 2015, 26, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Andrukhova, O.; Slavic, S.; Smorodchenko, A.; Zeitz, U.; Shalhoub, V.; Lanske, B.; Pohl, E.E.; Erben, R.G. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol. Med. 2014, 6, 744–759. [Google Scholar] [CrossRef]
- Rodelo-Haad, C.; Santamaria, R.; Muñoz-Castañeda, J.R.; Pendón-Ruiz de Mier, M.V.; Martin-Malo, A.; Rodriguez, M. FGF23, Biomarker or Target? Toxins 2019, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.E.; de Las Fuentes, L.; Ginsberg, C.; Rothstein, M.; Dietzen, D.J.; Cheng, S.C.; Ross, W.; Windus, D.; Dávila-Román, V.G.; Hruska, K.A. Left ventricular mass progression despite stable blood pressure and kidney function in stage 3 chronic kidney disease. Am. J. Nephrol. 2014, 39, 392–399. [Google Scholar] [CrossRef] [Green Version]
- Memon, F.; El-Abbadi, M.; Nakatani, T.; Taguchi, T.; Lanske, B.; Razzaque, M.S. Does Fgf23-klotho activity influence vascular and soft tissue calcification through regulating mineral ion metabolism? Kidney Int. 2008, 74, 566–570. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Giachelli, C.M. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017, 100, 87–93. [Google Scholar] [CrossRef]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.-C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Invest. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, M.W. WNT signaling in adult cardiac hypertrophy and remodeling: Lessons learned from cardiac development. Circ. Res. 2010, 107, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Meng, W.; Ding, J.; Cheng, M. Klotho inhibits angiotensin II-induced cardiomyocyte hypertrophy through suppression of the AT1R/beta catenin pathway. Biochem. Biophys. Res. Commun. 2016, 473, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Milovanova, L.Y.; Dobrosmyslov, I.A.; Milovanov, Y.S.; Taranova, M.V.; Kozlov, V.V.; Milovanova, S.Y.; Kozevnikova, E.I. Fibroblast growth factor-23 (FGF-23) / soluble Klotho protein (sKlotho) / sclerostin glycoprotein ratio disturbance is a novel risk factor for cardiovascular complications in ESRD patients receiving treatment with regular hemodialysis or hemodiafiltration. Ter. Arkh. 2018, 90, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef]
- Ye, B.; Ge, Y.; Perens, G.; Hong, L.; Xu, H.; Fishbein, M.C.; Li, F. Canonical Wnt/β-catenin signaling in epicardial fibrosis of failed pediatric heart allografts with diastolic dysfunction. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2013, 22, 54–57. [Google Scholar] [CrossRef] [Green Version]
- Działo, E.; Tkacz, K.; Błyszczuk, P. Crosstalk between the TGF-β and WNT signalling pathways during cardiac fibrogenesis. Acta Biochim. Pol. 2018, 65, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhu, L.-J.; Waaga-Gasser, A.M.; Ding, Y.; Cao, M.; Jadhav, S.J.; Kirollos, S.; Shekar, P.S.; Padera, R.F.; Chang, Y.-C.; et al. The axis of local cardiac endogenous Klotho-TGF-β1-Wnt signaling mediates cardiac fibrosis in human. J. Mol. Cell. Cardiol. 2019, 136, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.; Li, X.; Li, Q.; Lin, H.; Chen, Z.; Xie, J.; Xuan, W.; Liao, W.; Bin, J.; Huang, X.; et al. FGF23 promotes myocardial fibrosis in mice through activation of β-catenin. Oncotarget 2016, 7, 64649–64664. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, D.; Alampour-Rajabi, S.; Ponomariov, V.; Curaj, A.; Wu, Z.; Staudt, M.; Rusu, M.; Jankowski, V.; Marx, N.; Jankowski, J.; et al. Cardiac FGF23: New insights into the role and function of FGF23 after acute myocardial infarction. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2019, 40, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Christman, M.A.; Goetz, D.J.; Dickerson, E.; McCall, K.D.; Lewis, C.J.; Benencia, F.; Silver, M.J.; Kohn, L.D.; Malgor, R. Wnt5a is expressed in murine and human atherosclerotic lesions. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2864–H2870. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J. Am. Soc. Nephrol. JASN 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Wang, Y.; Zhang, Y.; Liu, C. Klotho ameliorates oxidized low density lipoprotein (ox-LDL)-induced oxidative stress via regulating LOX-1 and PI3K/Akt/eNOS pathways. Lipids Health Dis. 2017, 16, 77. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ortiz, M.E.; Alcalá-Díaz, J.F.; Canalejo, A.; Torres-Peña, J.D.; Gómez-Delgado, F.; Muñoz-Castañeda, J.R.; Delgado-Lista, J.; Rodríguez, M.; López-Miranda, J.; Almadén, Y. Fibroblast growth factor 23 predicts carotid atherosclerosis in individuals without kidney disease. The CORDIOPREV study. Eur. J. Intern. Med. 2019. [Google Scholar] [CrossRef]
- Chen, A.; Sun, Y.; Cui, J.; Zhao, B.; Wang, H.; Chen, X.; Mao, Y. Associations of sclerostin with carotid artery atherosclerosis and all-cause mortality in Chinese patients undergoing maintenance hemodialysis. BMC Nephrol. 2018, 19, 264. [Google Scholar] [CrossRef] [Green Version]
- London, G.M.; Marchais, S.J.; Guérin, A.P.; Métivier, F. Arteriosclerosis, vascular calcifications and cardiovascular disease in uremia. Curr. Opin. Nephrol. Hypertens. 2005, 14, 525–531. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Dusso, A.S.; Slatopolsky, E. Role of calcium-phosphate product and bone-associated proteins on vascular calcification in renal failure. J. Am. Soc. Nephrol. 2001, 12, 2511–2516. [Google Scholar]
- Montes de Oca, A.; Guerrero, F.; Martinez-Moreno, J.M.; Madueño, J.A.; Herencia, C.; Peralta, A.; Almaden, Y.; Lopez, I.; Aguilera-Tejero, E.; Gundlach, K.; et al. Magnesium inhibits Wnt/β-catenin activity and reverses the osteogenic transformation of vascular smooth muscle cells. PLoS ONE 2014, 9, e89525. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, J.; Lang, F.; Eckardt, K.-U.; Amann, K.; Kuro-O, M.; Pasch, A.; Pieske, B.; Alesutan, I. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell. Mol. Life Sci. CMLS 2019, 76, 2077–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Mao, H.; Chen, C.; Wu, L.; Wang, N.; Zhao, X.; Qian, J.; Xing, C. The Role and Mechanism of α-Klotho in the Calcification of Rat Aortic Vascular Smooth Muscle Cells. Available online: https://www.hindawi.com/journals/bmri/2015/194362/abs/ (accessed on 2 January 2020).
- Rodríguez-Ortiz, M.E.; Díaz-Tocados, J.M.; Muñoz-Castañeda, J.R.; Herencia, C.; Pineda, C.; Martínez-Moreno, J.M.; Montes de Oca, A.; López-Baltanás, R.; Alcalá-Díaz, J.; Ortiz, A.; et al. Inflammation both increases and causes resistance to FGF23 in normal and uremic rats. Clin. Sci. Lond. Engl. 1979 2020, 134, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Scialla, J.J.; Lau, W.L.; Reilly, M.P.; Isakova, T.; Yang, H.-Y.; Crouthamel, M.H.; Chavkin, N.W.; Rahman, M.; Wahl, P.; Amaral, A.P.; et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013, 83, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Mackenzie, N.C.W.; Millan, J.L.; Farquharson, C.; MacRae, V.E. A protective role for FGF-23 in local defence against disrupted arterial wall integrity? Mol. Cell. Endocrinol. 2013, 372, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Jimbo, R.; Kawakami-Mori, F.; Mu, S.; Hirohama, D.; Majtan, B.; Shimizu, Y.; Yatomi, Y.; Fukumoto, S.; Fujita, T.; Shimosawa, T. Fibroblast growth factor 23 accelerates phosphate-induced vascular calcification in the absence of Klotho deficiency. Kidney Int. 2014, 85, 1103–1111. [Google Scholar] [CrossRef] [Green Version]
- Verkaik, M.; Juni, R.P.; van Loon, E.P.M.; van Poelgeest, E.M.; Kwekkeboom, R.F.J.; Gam, Z.; Richards, W.G.; Ter Wee, P.M.; Hoenderop, J.G.; Eringa, E.C.; et al. FGF23 impairs peripheral microvascular function in renal failure. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1414–H1424. [Google Scholar] [CrossRef]
- Evenepoel, P.; D’Haese, P.; Brandenburg, V. Sclerostin and DKK1: New players in renal bone and vascular disease. Kidney Int. 2015, 88, 235–240. [Google Scholar] [CrossRef]
- Cejka, D.; Herberth, J.; Branscum, A.J.; Fardo, D.W.; Monier-Faugere, M.-C.; Diarra, D.; Haas, M.; Malluche, H.H. Sclerostin and Dickkopf-1 in renal osteodystrophy. Clin. J. Am. Soc. Nephrol. CJASN 2011, 6, 877–882. [Google Scholar] [CrossRef] [Green Version]
- Behets, G.J.; Viaene, L.; Meijers, B.; Blocki, F.; Brandenburg, V.M.; Verhulst, A.; D’Haese, P.C.; Evenepoel, P. Circulating levels of sclerostin but not DKK1 associate with laboratory parameters of CKD-MBD. PLoS ONE 2017, 12, e0176411. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, Y.; Graciolli, F.G.; O’Brien, S.; Tang, W.; dos Reis, L.M.; Ryan, S.; Phillips, L.; Boulanger, J.; Song, W.; Bracken, C.; et al. Repression of osteocyte Wnt/β-catenin signaling is an early event in the progression of renal osteodystrophy. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2012, 27, 1757–1772. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, S.; Dubourg, L.; Carlier, M.-C.; Hadj-Aissa, A.; Fouque, D. The relation between renal function and serum sclerostin in adult patients with CKD. Clin. J. Am. Soc. Nephrol. CJASN 2013, 8, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Cejka, D.; Jäger-Lansky, A.; Kieweg, H.; Weber, M.; Bieglmayer, C.; Haider, D.G.; Diarra, D.; Patsch, J.M.; Kainberger, F.; Bohle, B.; et al. Sclerostin serum levels correlate positively with bone mineral density and microarchitecture in haemodialysis patients. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2012, 27, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Chen, N.X.; Newman, C.L.; Organ, J.M.; Kneissel, M.; Kramer, I.; Gattone, V.H.; Allen, M.R. Anti-sclerostin antibody treatment in a rat model of progressive renal osteodystrophy. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2015, 30, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo-López, N.; Panizo, S.; Alonso-Montes, C.; Román-García, P.; Rodríguez, I.; Martínez-Salgado, C.; Dusso, A.S.; Naves, M.; Cannata-Andía, J.B. Direct inhibition of osteoblastic Wnt pathway by fibroblast growth factor 23 contributes to bone loss in chronic kidney disease. Kidney Int 2016, 90, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Fayed, A.; Elgohary, R.; Fawzy, M. Evaluating the role of serum sclerostin as an indicator of activity and damage in Egyptian patients with rheumatoid arthritis: University hospital experience. Clin. Rheumatol. 2019. [Google Scholar] [CrossRef]
- Raimann, A.; Ertl, D.A.; Helmreich, M.; Sagmeister, S.; Egerbacher, M.; Haeusler, G. Fibroblast growth factor 23 and Klotho are present in the growth plate. Connect. Tissue Res. 2013, 54, 108–117. [Google Scholar] [CrossRef]
- Yuan, Q.; Sato, T.; Densmore, M.; Saito, H.; Schüler, C.; Erben, R.G.; Lanske, B. Deletion of PTH rescues skeletal abnormalities and high osteopontin levels in Klotho-/- mice. PLoS Genet. 2012, 8, e1002726. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Gao, M.; Wu, L.; Zhao, X.; Mao, H.; Xing, C. The suppressive effect of soluble Klotho on fibroblastic growth factor 23 synthesis in UMR-106 osteoblast-like cells. Cell Biol. Int. 2018, 42, 1270–1274. [Google Scholar] [CrossRef]
- Zhang, W.; Xue, D.; Hu, D.; Xie, T.; Tao, Y.; Zhu, T.; Chen, E.; Pan, Z. Secreted klotho protein attenuates osteogenic differentiation of human bone marrow mesenchymal stem cells in vitro via inactivation of the FGFR1/ERK signaling pathway. Growth Factors Chur Switz. 2015, 33, 356–365. [Google Scholar] [CrossRef]
- Shalhoub, V.; Ward, S.C.; Sun, B.; Stevens, J.; Renshaw, L.; Hawkins, N.; Richards, W.G. Fibroblast growth factor 23 (FGF23) and alpha-klotho stimulate osteoblastic MC3T3.E1 cell proliferation and inhibit mineralization. Calcif. Tissue Int. 2011, 89, 140–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffner, D.; Leifheit-Nestler, M. Extrarenal effects of FGF23. Pediatr. Nephrol. 2017, 32, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Faul, C. FGF23 Actions on Target Tissues—With and Without Klotho. Front. Endocrinol. 2018, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Shardell, M.; Semba, R.D.; Rosano, C.; Kalyani, R.R.; Bandinelli, S.; Chia, C.W.; Ferrucci, L. Plasma Klotho and Cognitive Decline in Older Adults: Findings From the InCHIANTI Study. J. Gerontol. A. Biol. Sci. Med. Sci. 2016, 71, 677–682. [Google Scholar] [CrossRef] [Green Version]
- Semba, R.D.; Moghekar, A.R.; Hu, J.; Sun, K.; Turner, R.; Ferrucci, L.; O’Brien, R. Klotho in the cerebrospinal fluid of adults with and without Alzheimer’s disease. Neurosci. Lett. 2014, 558, 37–40. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.; Sun, Z. Klotho Deficiency Accelerates Stem Cells Aging by Impairing Telomerase Activity. J. Gerontol. Ser. A 2019, 74, 1396–1407. [Google Scholar] [CrossRef]
- Kuro-o, M. Klotho and aging. Biochim. Biophys. Acta BBA Gen. Subj. 2009, 1790, 1049–1058. [Google Scholar] [CrossRef]
- Li, D.; Jing, D.; Liu, Z.; Chen, Y.; Huang, F.; Behnisch, T. Enhanced Expression of Secreted α-Klotho in the Hippocampus Alters Nesting Behavior and Memory Formation in Mice. Front. Cell. Neurosci. 2019, 13, 133. [Google Scholar] [CrossRef]
- Massó, A.; Sánchez, A.; Bosch, A.; Giménez-Llort, L.; Chillón, M. Secreted αKlotho isoform protects against age-dependent memory deficits. Mol. Psychiatry 2018, 23, 1937–1947. [Google Scholar] [CrossRef] [Green Version]
- Abraham, C.R.; Mullen, P.C.; Tucker-Zhou, T.; Chen, C.D.; Zeldich, E. Klotho Is a Neuroprotective and Cognition-Enhancing Protein. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2016; Volume 101, pp. 215–238. ISBN 978-0-12-804819-1. [Google Scholar]
- Barnes, J.W.; Duncan, D.; Helton, S.; Hutcheson, S.; Kurundkar, D.; Logsdon, N.J.; Locy, M.; Garth, J.; Denson, R.; Farver, C.; et al. Role of fibroblast growth factor 23 and klotho cross talk in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L141–L154. [Google Scholar] [CrossRef]
- Buendia-Roldan, I.; Machuca, N.; Mejía, M.; Maldonado, M.; Pardo, A.; Selman, M. Lower levels of α-Klotho in serum are associated with decreased lung function in individuals with interstitial lung abnormalities. Sci. Rep. 2019, 9, 10801. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Yuan, C.; Zhang, J.; Li, L.; Yu, L.; Wiegman, C.H.; Barnes, P.J.; Adcock, I.M.; Huang, M.; Yao, X. Klotho expression is reduced in COPD airway epithelial cells: Effects on inflammation and oxidant injury. Clin. Sci. Lond. Engl. 1979 2015, 129, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krick, S.; Grabner, A.; Baumlin, N.; Yanucil, C.; Helton, S.; Grosche, A.; Sailland, J.; Geraghty, P.; Viera, L.; Russell, D.W.; et al. Fibroblast growth factor 23 and Klotho contribute to airway inflammation. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baarsma, H.A.; Skronska-Wasek, W.; Mutze, K.; Ciolek, F.; Wagner, D.E.; John-Schuster, G.; Heinzelmann, K.; Günther, A.; Bracke, K.R.; Dagouassat, M.; et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J. Exp. Med. 2017, 214, 143–163. [Google Scholar] [CrossRef] [Green Version]
- Kumawat, K.; Gosens, R. WNT-5A: Signaling and functions in health and disease. Cell. Mol. Life Sci. CMLS 2016, 73, 567–587. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; Di Marco, G.S.; et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016, 90, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Perugorria, M.J.; Olaizola, P.; Labiano, I.; Esparza-Baquer, A.; Marzioni, M.; Marin, J.J.G.; Bujanda, L.; Banales, J.M. Wnt-β-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 121–136. [Google Scholar] [CrossRef]
- Rao, Z.; Landry, T.; Li, P.; Bunner, W.; Laing, B.T.; Yuan, Y.; Huang, H. Administration of alpha klotho reduces liver and adipose lipid accumulation in obese mice. Heliyon 2019, 5, e01494. [Google Scholar] [CrossRef] [Green Version]
- Somm, E.; Henry, H.; Bruce, S.J.; Bonnet, N.; Montandon, S.A.; Niederländer, N.J.; Messina, A.; Aeby, S.; Rosikiewicz, M.; Fajas, L.; et al. β-Klotho deficiency shifts the gut-liver bile acid axis and induces hepatic alterations in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E833–E847. [Google Scholar] [CrossRef]
- Beljaars, L.; Daliri, S.; Dijkhuizen, C.; Poelstra, K.; Gosens, R. WNT-5A regulates TGF-β-related activities in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G219–G227. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Castañeda, J.R.; Rodelo-Haad, C.; Pendon-Ruiz de Mier, M.V.; Martin-Malo, A.; Santamaria, R.; Rodriguez, M. Klotho/FGF23 and Wnt Signaling as Important Players in the Comorbidities Associated with Chronic Kidney Disease. Toxins 2020, 12, 185. https://doi.org/10.3390/toxins12030185
Muñoz-Castañeda JR, Rodelo-Haad C, Pendon-Ruiz de Mier MV, Martin-Malo A, Santamaria R, Rodriguez M. Klotho/FGF23 and Wnt Signaling as Important Players in the Comorbidities Associated with Chronic Kidney Disease. Toxins. 2020; 12(3):185. https://doi.org/10.3390/toxins12030185
Chicago/Turabian StyleMuñoz-Castañeda, Juan Rafael, Cristian Rodelo-Haad, Maria Victoria Pendon-Ruiz de Mier, Alejandro Martin-Malo, Rafael Santamaria, and Mariano Rodriguez. 2020. "Klotho/FGF23 and Wnt Signaling as Important Players in the Comorbidities Associated with Chronic Kidney Disease" Toxins 12, no. 3: 185. https://doi.org/10.3390/toxins12030185
APA StyleMuñoz-Castañeda, J. R., Rodelo-Haad, C., Pendon-Ruiz de Mier, M. V., Martin-Malo, A., Santamaria, R., & Rodriguez, M. (2020). Klotho/FGF23 and Wnt Signaling as Important Players in the Comorbidities Associated with Chronic Kidney Disease. Toxins, 12(3), 185. https://doi.org/10.3390/toxins12030185