The Influence of Calcium toward Order/Disorder Conformation of Repeat-in-Toxin (RTX) Structure of Family I.3 Lipase from Pseudomonas fluorescens AMS8

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
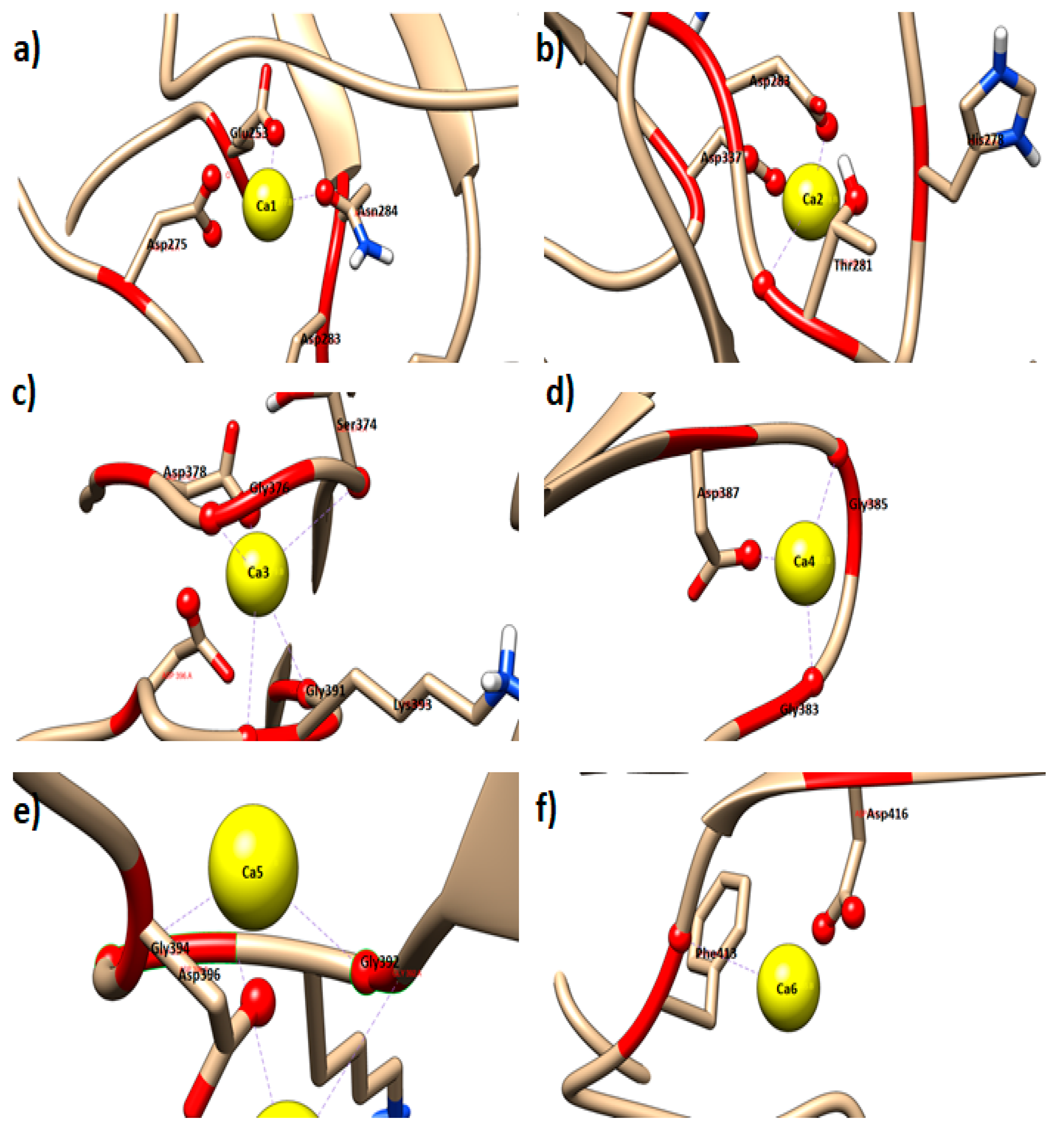
2.1. Molecular Docking (Metal Ion) of AMS8 Lipase Structure
2.2. Molecular Dynamics (MD) Simulation of AMS8 Lipase Structure
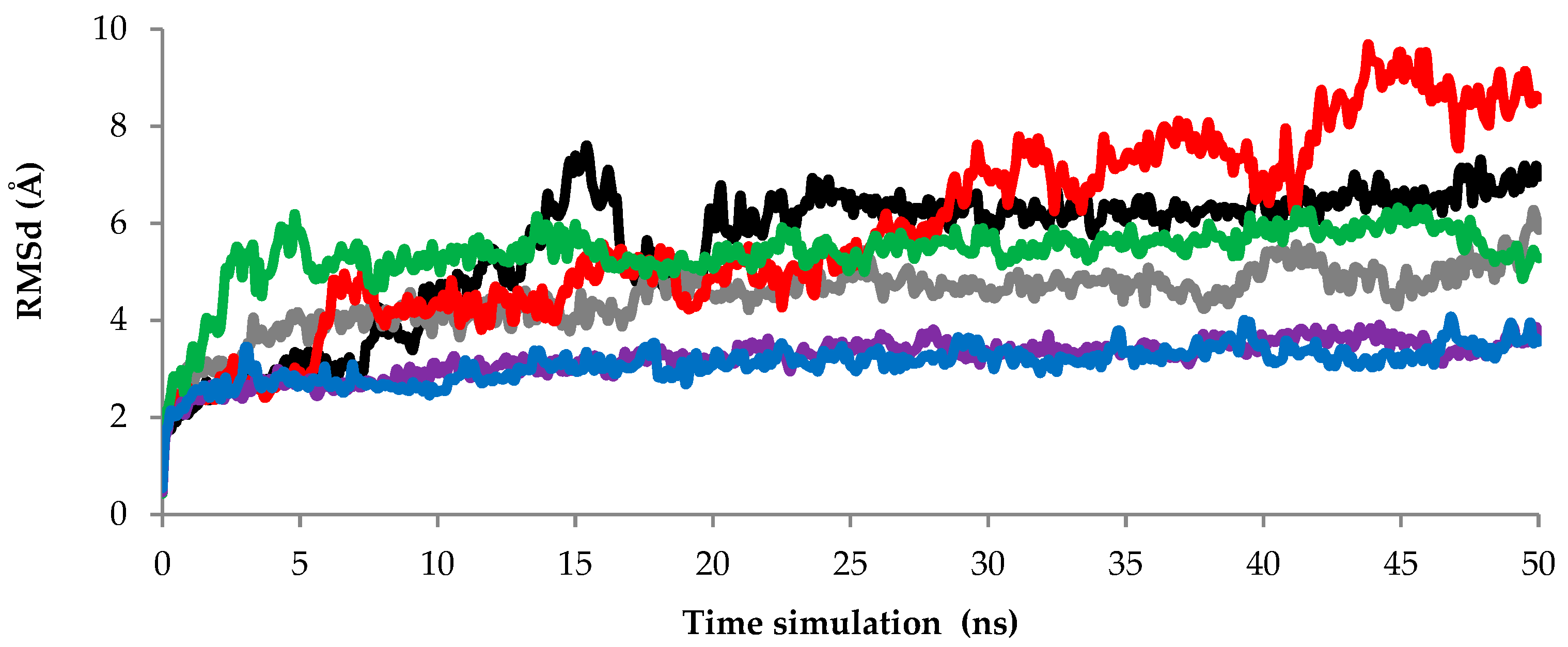
2.2.1. Root Mean Square Deviation (RMSd) Values of Backbone Atoms Analysis
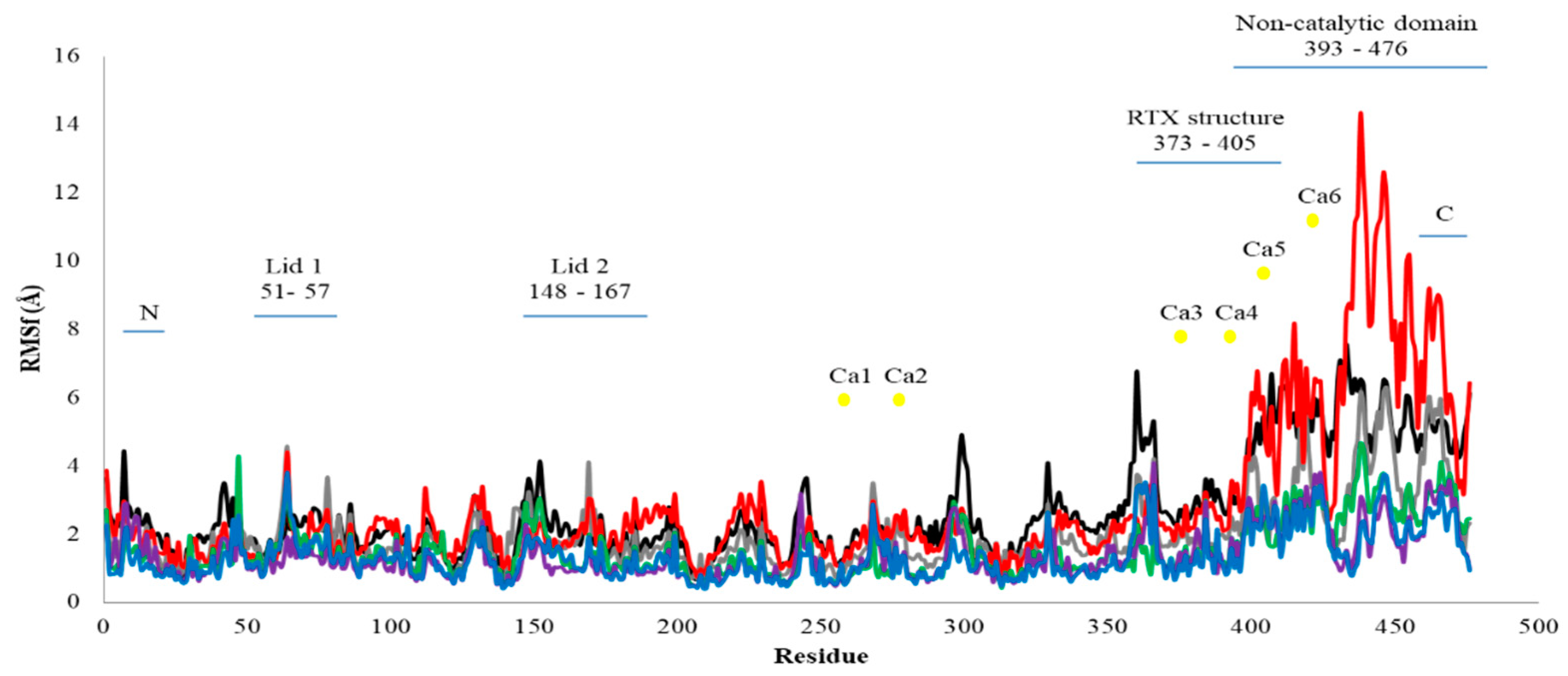
2.2.2. Root Mean Square Fluctuation (RMSf) Value of Residue Analysis
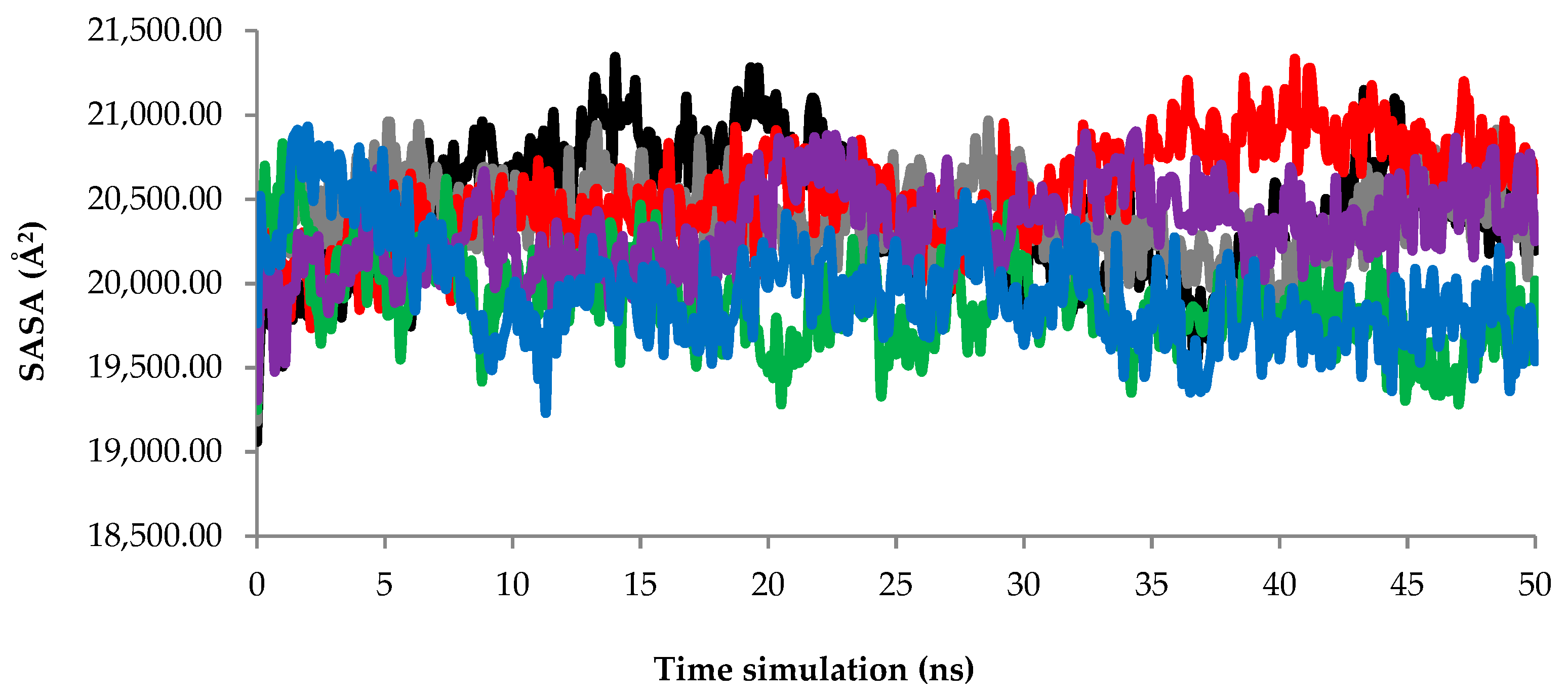
2.2.3. Solvent Accessible Surface Area (SASA) Analysis
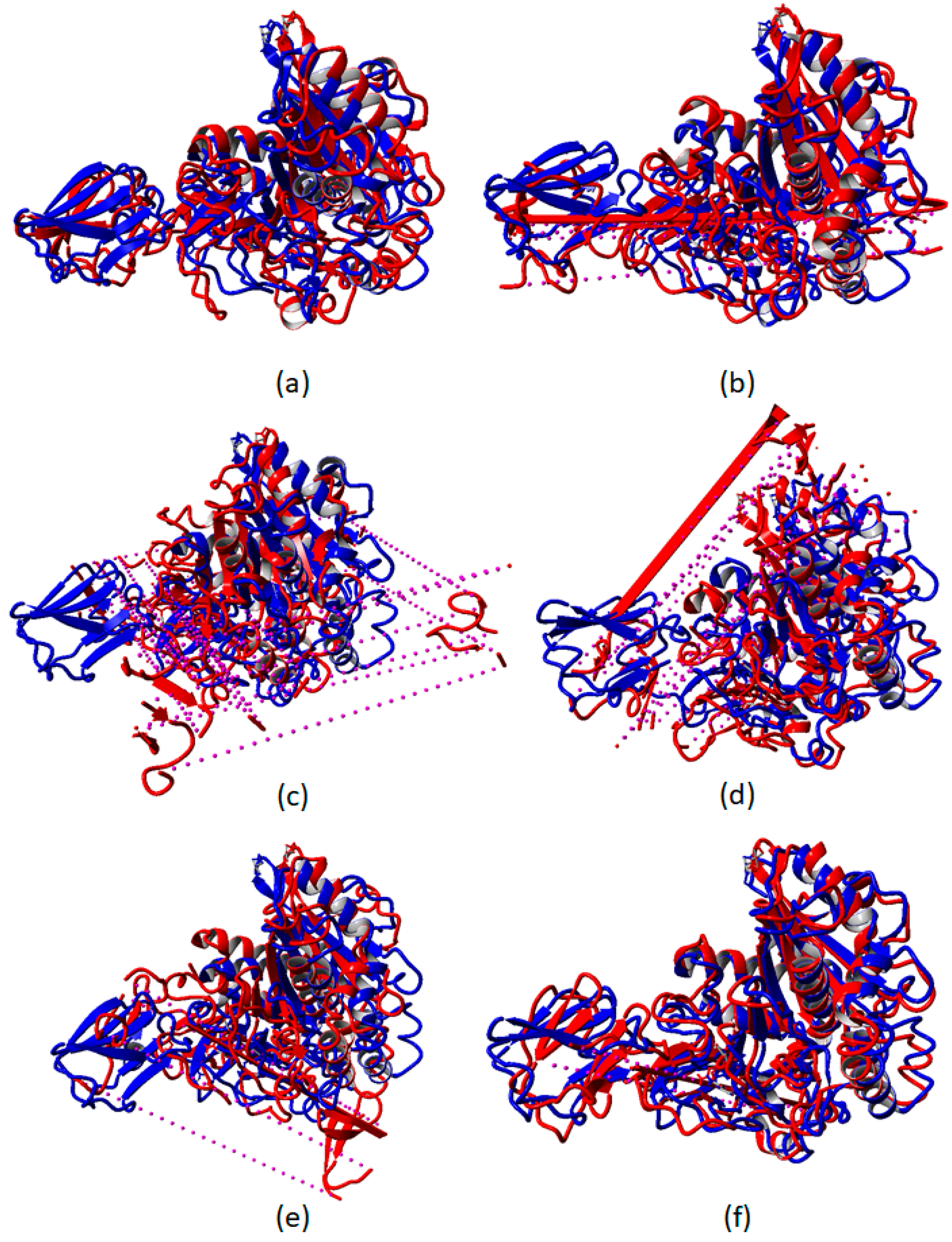
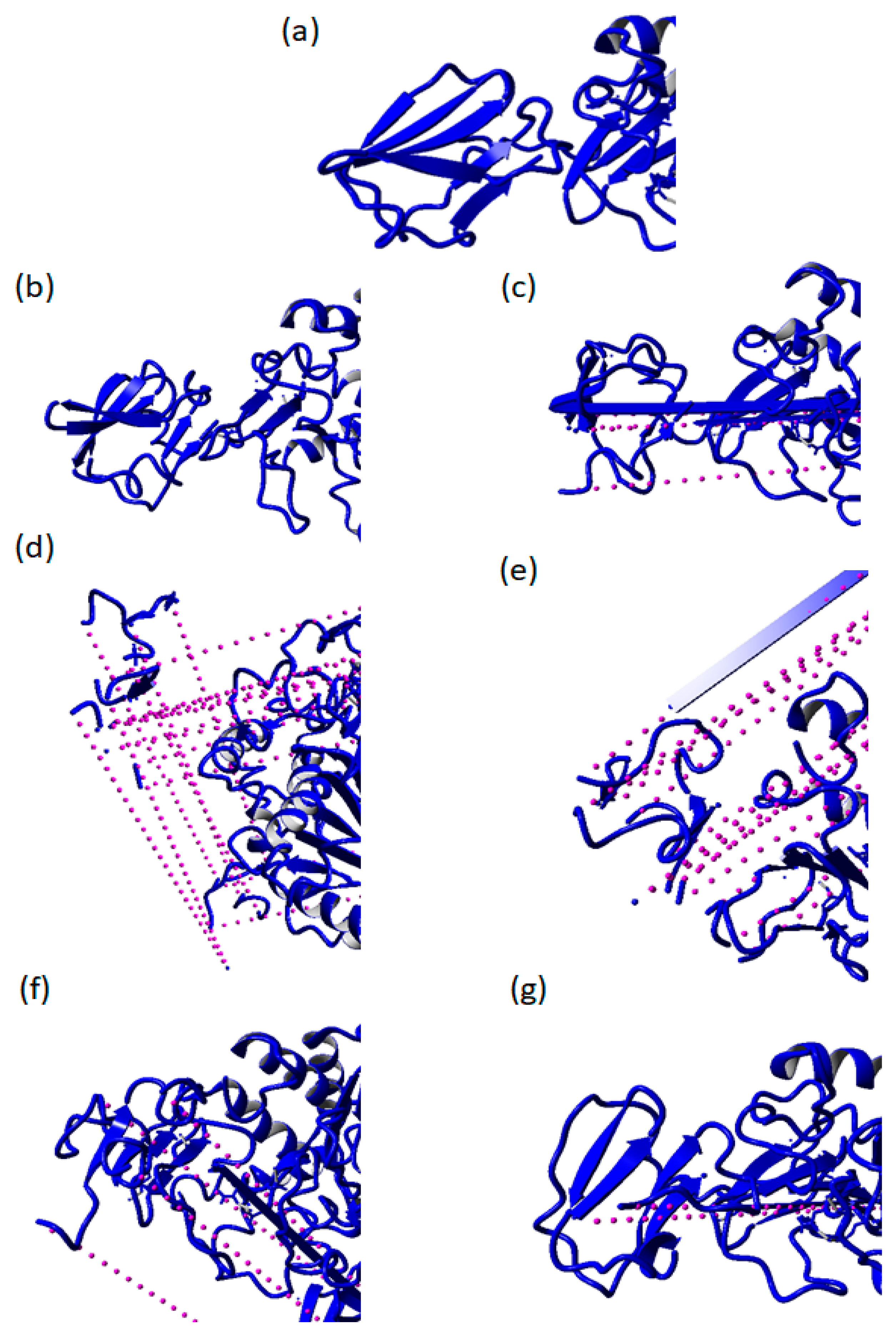
2.3. Destabilization of RTX Parallel β-Roll Motif Repeat the Structure of AMS8 Lipase Structure after 50 ns MD Simulations
3. Conclusions
4. Materials and Methods
4.1. AMS8 Lipase Sequence and Predicted Three-Dimensional (3D) Structure
4.2. Molecular Docking of AMS8 Lipase Structure
4.3. Molecular Dynamics (MD) Simulation Parameter of AMS8 Lipase
4.4. Molecular Dynamics (MD) Simulation Analysis of AMS8 Lipase
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sotomayor-Pérez, A.-C.; Ladant, D.; Chenal, A. Disorder-to-Order transition in the CyaA toxin RTX domain: Implications for toxin secretion. Toxins 2014, 7, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, P.K. Enzymes: An integrated view of structure, dynamics and function. Microb. Cell Fact. 2006, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromiha, M.M. Protein Stability; Academic Press: Singapore, 2010; pp. 209–245. [Google Scholar]
- Murphy, K.P. Stabilization of protein structure. In Protein Structure, Stability, and Folding; Murphy, K.P., Ed.; Humana Press: Tortowa, NJ, USA, 2001; pp. 1–16. [Google Scholar]
- Stigler, J.; Ziegler, F.; Gieseke, A.; Gebhardt, J.C.M.; Rief, M. The complex folding network of single calmodulin molecules. Science 2011, 334, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Akcapinar, G.B.; Sezerman, O.U. Computational approaches for de novo design and redesign of metal-binding sites on proteins. Biosci. Rep. 2017, 37, BSR20160179. [Google Scholar] [CrossRef] [Green Version]
- Grzybowska, E.A. Calcium-binding proteins with disordered structure and their role in secretion, storage, and cellular signaling. Biomolecules 2018, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Blenner, M.A.; Shur, O.; Szilvay, G.R.; Cropek, D.M.; Banta, S. Calcium-induced folding of a beta roll motif requires C-terminal entropic stabilization. J. Mol. Biol. 2010, 400, 244–256. [Google Scholar] [CrossRef]
- Sergeev, Y.V.; Bowles, K.E.; Ziccardi, L.; Sieving, P.A. Molecular modeling of protein structure, biology of disease and clinical electroretinography in human X-Linked Retinoschisis (XLRS). In Electroretinograms; IntechOpen: London, UK, 2011; pp. 133–156. [Google Scholar]
- Rose, T.; Sebo, P.; Bellalou, J.; Ladant, D. Interaction of calcium with Bordetella Pertussis adenylate cyclase toxin characterization of multiple calcium-binding sites and calcium-induced conformational changes. J. Biol. Chem. 1995, 270, 26370–26376. [Google Scholar] [CrossRef] [Green Version]
- Farfel, Z.; Friedman, E.; Hanski, E. The invasive adenylate cyclase of Bordetella Pertussis. Intracellular localization and kinetics of penetration into various cells. Biochem. J. 1987, 243, 153–158. [Google Scholar]
- Lilie, H.; Haehnel, W.; Rudolph, R.; Baumann, U. Folding of a synthetic parallel L-roll protein. FEBS Lett. 2000, 2, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.S.M.; Fuzi, S.F.M.; Ganasen, M.; Rahman, R.N.Z.R.A.; Basri, M.; Salleh, A.B. Structural adaptation of cold-active RTX lipase from Pseudomonas sp. strain AMS8 revealed via homology and molecular dynamics simulation approaches. Biomed Res. Int. 2013, 2013, 925373. [Google Scholar]
- Ali, N.S.M.; Salleh, A.B.; Rahman, R.N.Z.R.A.; Leow, T.C.; Ali, M.S.M. Calcium-induced activity and folding of a repeat in toxin lipase from antarctic Pseudomonas fluorescens strain AMS8. Toxins 2020, 12, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA view—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. J. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Welch, R.A. Pore-forming cytolysins of Gram-negative bacteria. J. Mol. Biol. 1991, 5, 521–528. [Google Scholar] [CrossRef]
- Coote, J.G. Structural and functional relationships among the RTX toxin determinants of gram-negative bacteria. FEMS Microbiol. Rev. 1992, 8, 137–161. [Google Scholar] [CrossRef]
- Karshikoff, A.; Nilsson, L.; Ladenstein, R. Rigidity versus flexibility: The dilemma of understanding protein thermal stability. FEBS J. 2015, 282, 3899–3917. [Google Scholar] [CrossRef]
- Angkawidjaja, C.; Kanaya, S. Family I.3 lipase: Bacterial lipases secreted by the type I secretion system. Cell. Mol. Life Sci. 2006, 63, 2804–2817. [Google Scholar] [CrossRef]
- Yaacob, N.; Ali, M.S.M.; Salleh, A.B.; Rahman, R.N.Z.R.A.; Leow, A.T.C. Toluene promotes lid 2 interfacial activation of cold active solvent tolerant lipase from Pseudomonas fluorescens strain AMS8. J. Mol. Graph. Model. 2016, 68, 224–235. [Google Scholar] [CrossRef]
- Giovanola, M.; D’Antoni, F.; Santacroce, M.; Mari, S.A.; Cherubino, F.; Bossi, E.; Castagna, M. Role of a conserved glycine triplet in the NSS amino acid transporter KAAT1. BBA Biomembr. 2012, 1818, 1737–1744. [Google Scholar] [CrossRef] [Green Version]
- El Khattabi, M.; Van Gelder, P.; Bitter, W.; Tommassen, J. Role of the calcium ion and the disulfide bond in the Burkholderia glumae lipase. J. Mol. Catal. B Enzym. 2003, 22, 329–338. [Google Scholar] [CrossRef]
- Salleh, A.B.; Rahim, A.S.M.A.; Rahman, R.N.Z.R.A.; Leow, T.C.; Basri, M. The role of Arg157Ser in improving the compactness and stability of ARM lipase. J. Comput. Biol. 2012, 5, 39–46. [Google Scholar] [CrossRef]
- Bindreither, D.; Lackner, P. Structural diversity of calcium binding sites. Gen. Physiol. Biophys. 2009, 28, F82–F88. [Google Scholar]
- Chenal, A.; Guijarro, J.I.; Raynal, B.; Delepierre, M.; Ladant, D. RTX calcium binding motifs are intrinsically disordered in the absence of calcium: Implication for protein secretion. J. Biol. Chem. 2009, 284, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, D.P.; Perez, A.C.S.; Karst, J.; Cannella, S.E.; Enguéné, V.Y.N.; Hessel, A.; Raoux-Barbot, D.; Voegele, A.; Subrini, O.; Davi, M.; et al. Calcium-dependent Disorder-to-Order transitions are central to the secretion and folding of the CyaA toxin of Bordetella Pertussis, the causative agent of whooping cough. Toxicon 2018, 149, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Bauche, C.; Chenal, A.; Knapp, O.; Bodenreider, C.; Benz, R.; Chaffotte, A.; Ladant, D. Structural and functional characterization of an essential RTX subdomain of Bordetella Pertussis adenylate cyclase toxin. J. Biol. Chem. 2006, 281, 16914–16926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, R.; Drepper, T.; Svensson, V.; Jaeger, K.-E.; Baumann, U. A calcium-gated lid and a large β-Roll sandwich are revealed by the crystal structure of extracellular lipase from Serratia Marcescens. J. Biol. Chem. 2007, 282, 31477–31483. [Google Scholar] [CrossRef] [Green Version]
- Angkawidjaja, C.; You, D.; Matsumura, H.; Kuwahara, K.; Koga, Y.; Takano, K.; Kanaya, S. Crystal structure of a family I.3 Lipase from Pseudomonas sp. MIS38 in a Closed Conformation. FEBS Lett. 2007, 581, 5060–5064. [Google Scholar] [CrossRef] [Green Version]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Huang, C.C.; Meng, E.C.; Morris, J.H.; Pettersen, E.F.; Ferrin, T.E. Enhancing UCSF chimera through web services. Nucleic Acids Res. 2014, 42, W478–W484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ca2+ Ion | Dissociate Constant (pM) | Binding Energy [kcal/mol] | Number of Interacting Residues |
|---|---|---|---|
| Ca1 | 113,732,214,784.0 | 1.2980 | 4 |
| Ca2 | 202,227,302,400.0 | 0.9470 | 4 |
| Ca3 | 160,478,724,096.0 | 1.0840 | 5 |
| Ca4 | 110,701,961,216.0 | 1.3040 | 3 |
| Ca5 | 110,515,273,728.0 | 1.3050 | 3 |
| Ca6 | 185,235,095,552.0 | 0.9990 | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, N.S.M.; Salleh, A.B.; Leow, T.C.; Rahman, R.N.Z.R.A.; Ali, M.S.M. The Influence of Calcium toward Order/Disorder Conformation of Repeat-in-Toxin (RTX) Structure of Family I.3 Lipase from Pseudomonas fluorescens AMS8. Toxins 2020, 12, 579. https://doi.org/10.3390/toxins12090579
Ali NSM, Salleh AB, Leow TC, Rahman RNZRA, Ali MSM. The Influence of Calcium toward Order/Disorder Conformation of Repeat-in-Toxin (RTX) Structure of Family I.3 Lipase from Pseudomonas fluorescens AMS8. Toxins. 2020; 12(9):579. https://doi.org/10.3390/toxins12090579
Chicago/Turabian StyleAli, Nur Shidaa Mohd, Abu Bakar Salleh, Thean Chor Leow, Raja Noor Zaliha Raja Abd Rahman, and Mohd Shukuri Mohamad Ali. 2020. "The Influence of Calcium toward Order/Disorder Conformation of Repeat-in-Toxin (RTX) Structure of Family I.3 Lipase from Pseudomonas fluorescens AMS8" Toxins 12, no. 9: 579. https://doi.org/10.3390/toxins12090579
APA StyleAli, N. S. M., Salleh, A. B., Leow, T. C., Rahman, R. N. Z. R. A., & Ali, M. S. M. (2020). The Influence of Calcium toward Order/Disorder Conformation of Repeat-in-Toxin (RTX) Structure of Family I.3 Lipase from Pseudomonas fluorescens AMS8. Toxins, 12(9), 579. https://doi.org/10.3390/toxins12090579