An Investigation of Three-Finger Toxin—nAChR Interactions through Rosetta Protein Docking



Abstract
:1. Introduction
1.1. Nicotinic Acetylcholine Receptors (nAChR) Play Important Roles
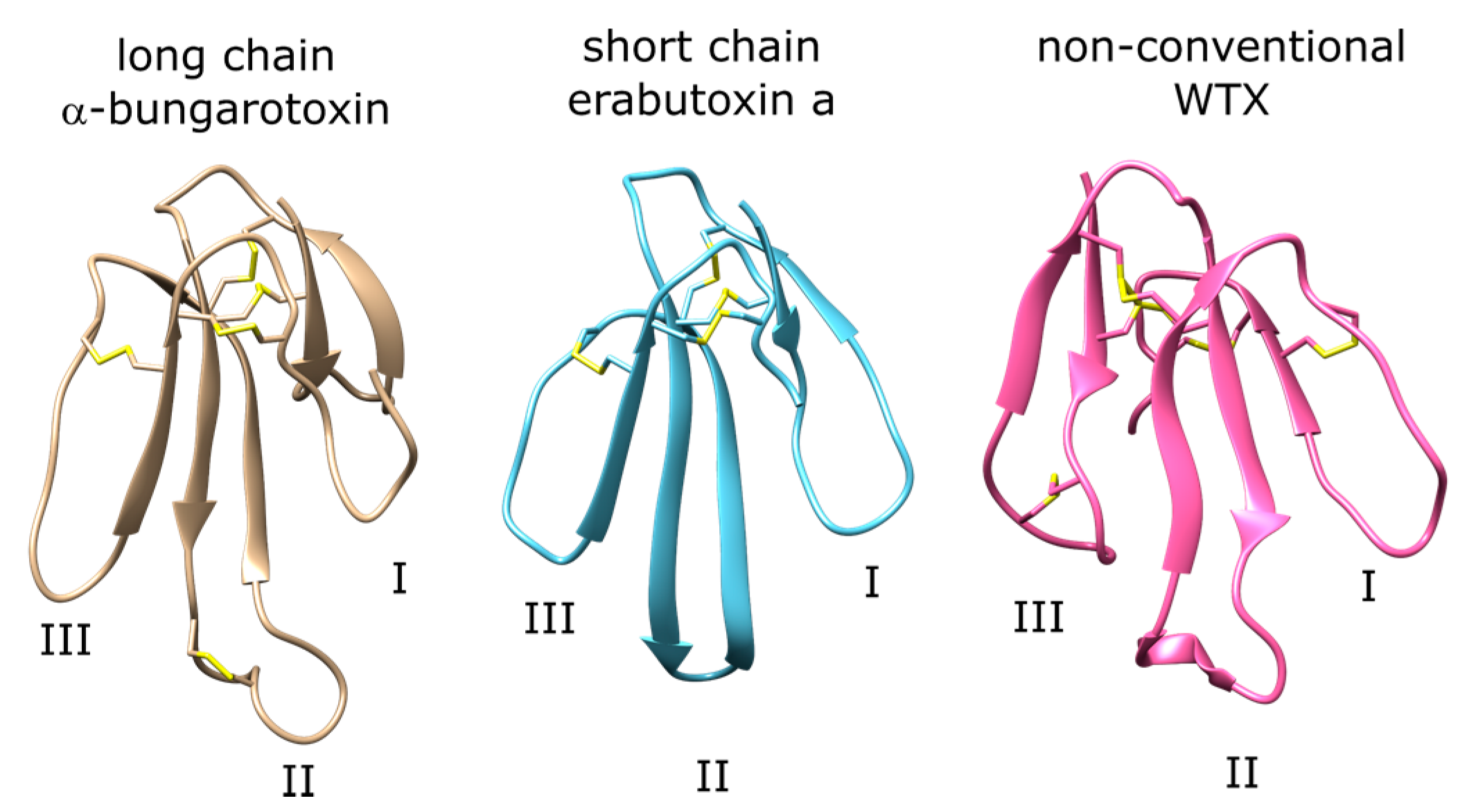
1.2. Three-Finger Toxins (3FTX) Target nAChR
1.3. 3FTX Interact with Different nAChR Subtypes with Varying Selectivity
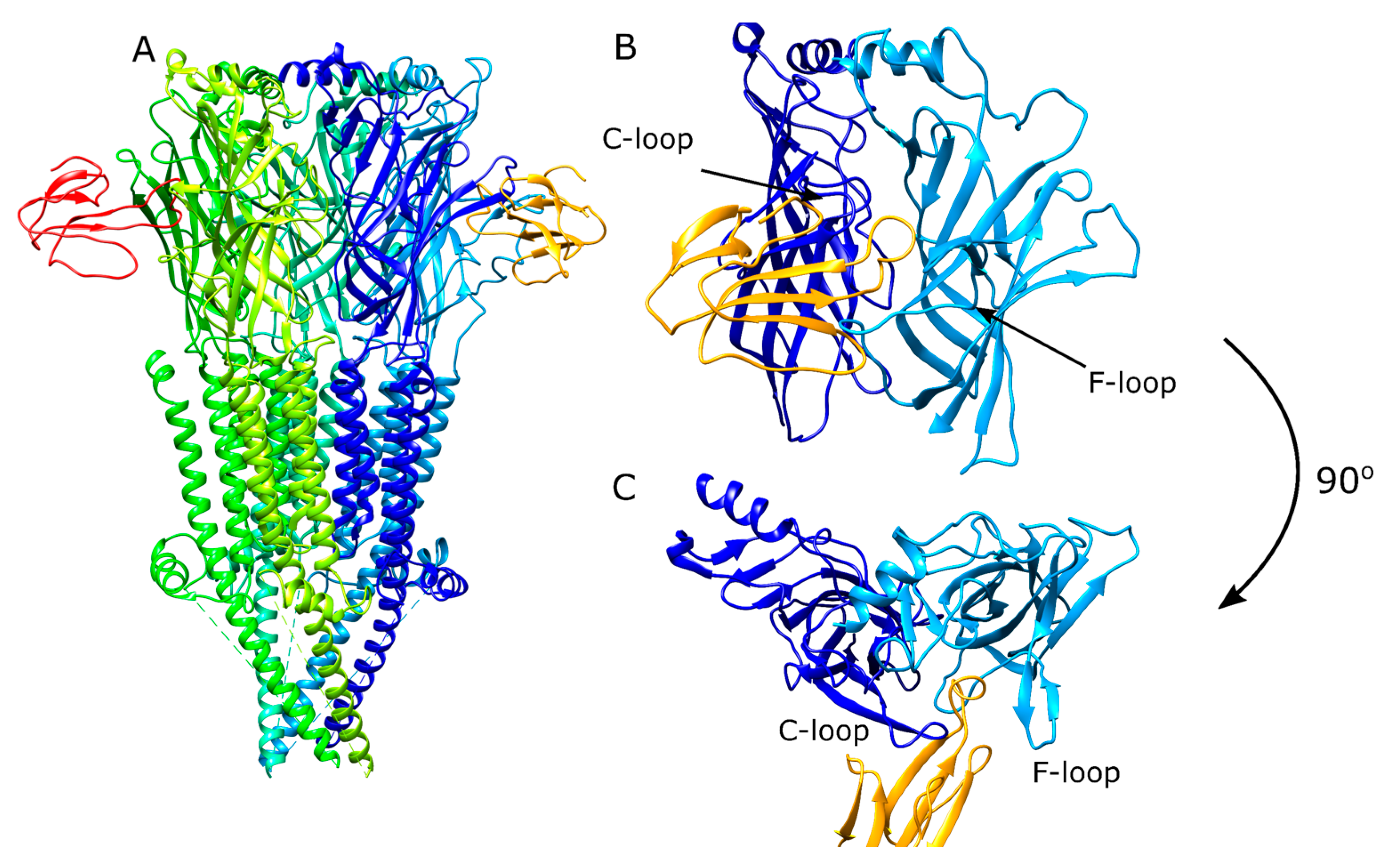
1.4. Structural Information from AChBP Can Be Used to Model Peptide–nAChR Interactions
1.5. Rosetta Peptide Docking Can Be Used to Identify 3FTX–nAChR Interactions
2. Results
2.1. 3FTX and α-Conotoxin Structures Were Considered for the Re-Docking and Cross-Docking Calculations
2.2. The Docking Protocol Consists of Peptide–Protein Complex Relaxing and Protein–Protein Docking Steps
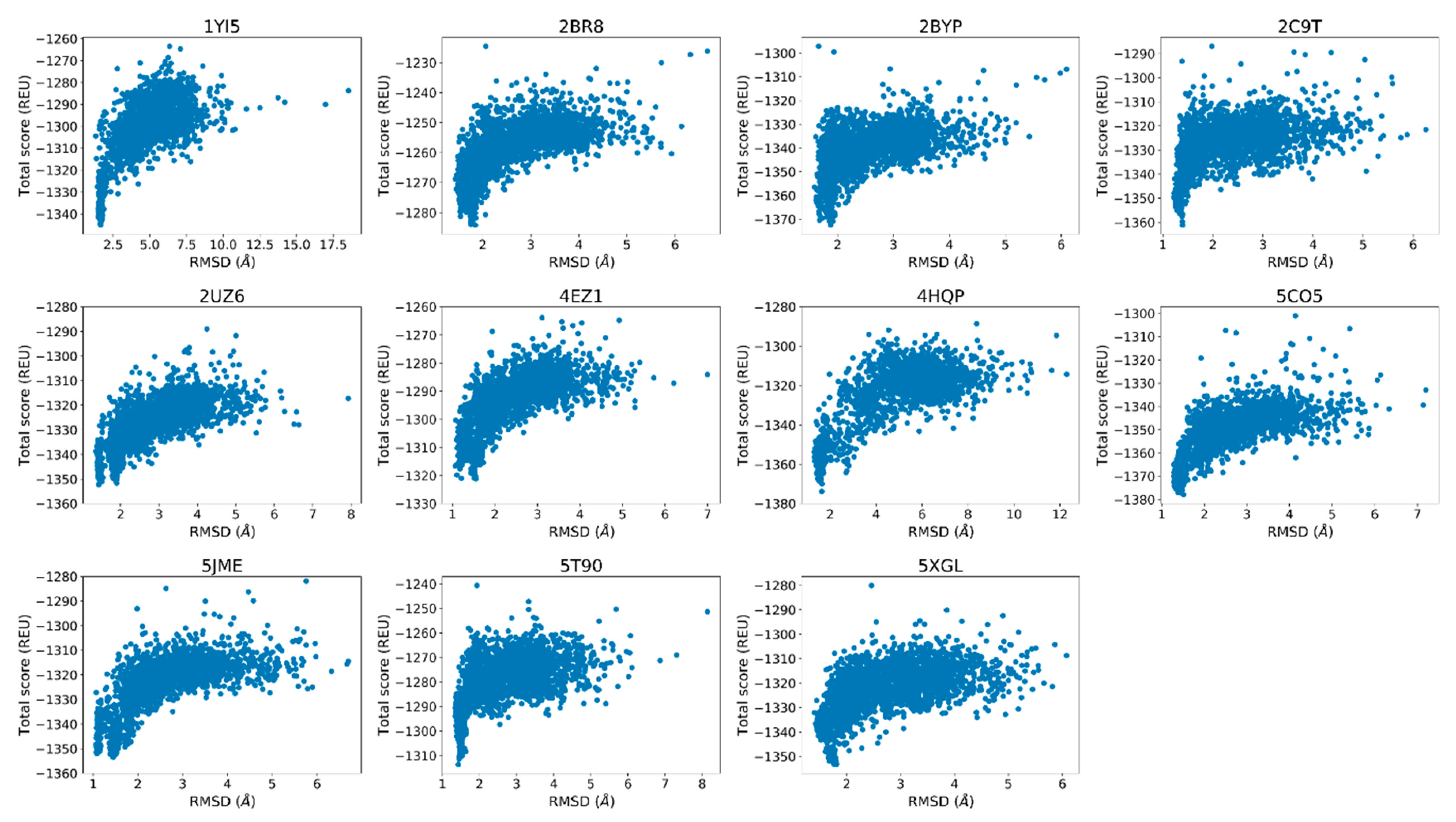
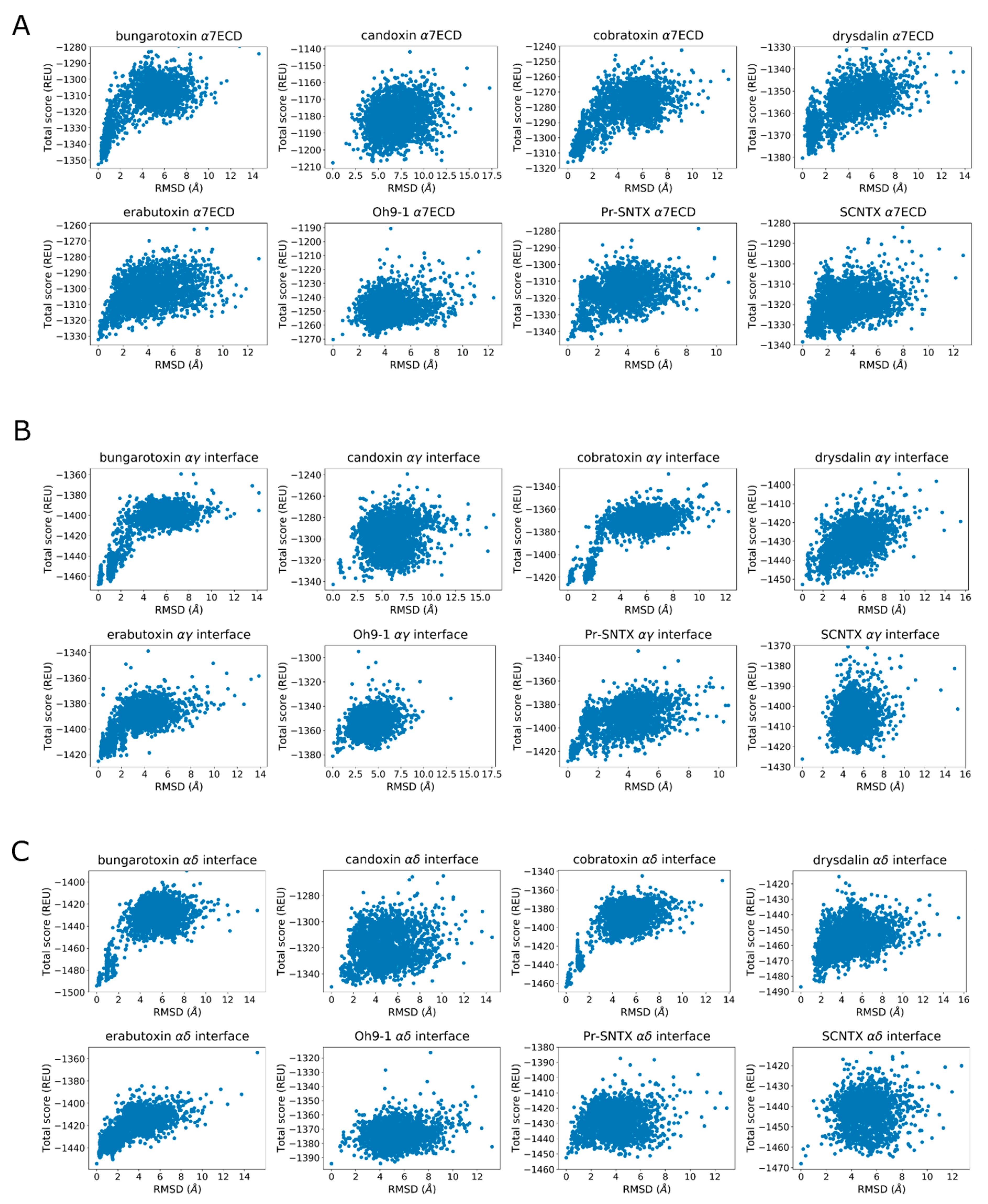
2.3. Rosetta Re-Docking Protocol Predicts Binding Conformation of the Peptides with RMSD Values Less than 2 Å
2.4. Fraction of Native Contacts Conserved Indicate Rosetta’s Success in Predicting Side-Chain Conformations
2.5. Cross-Docking Calculations Yield Slightly Diminished Accuracy Compared to the Native Re-Docking Calculations
2.6. Experimental AChBP Binding Data Was Used to Test the Success of the Docking Protocol
2.7. The Docking Protocol Accurately Predicts the Binding Properties of Long-Chain 3FTX to AChBP
2.8. Most Non-Binders Interact Only with the Outer Face of the C-Loop and have No Significant Interactions with the Aromatic Cage Residues
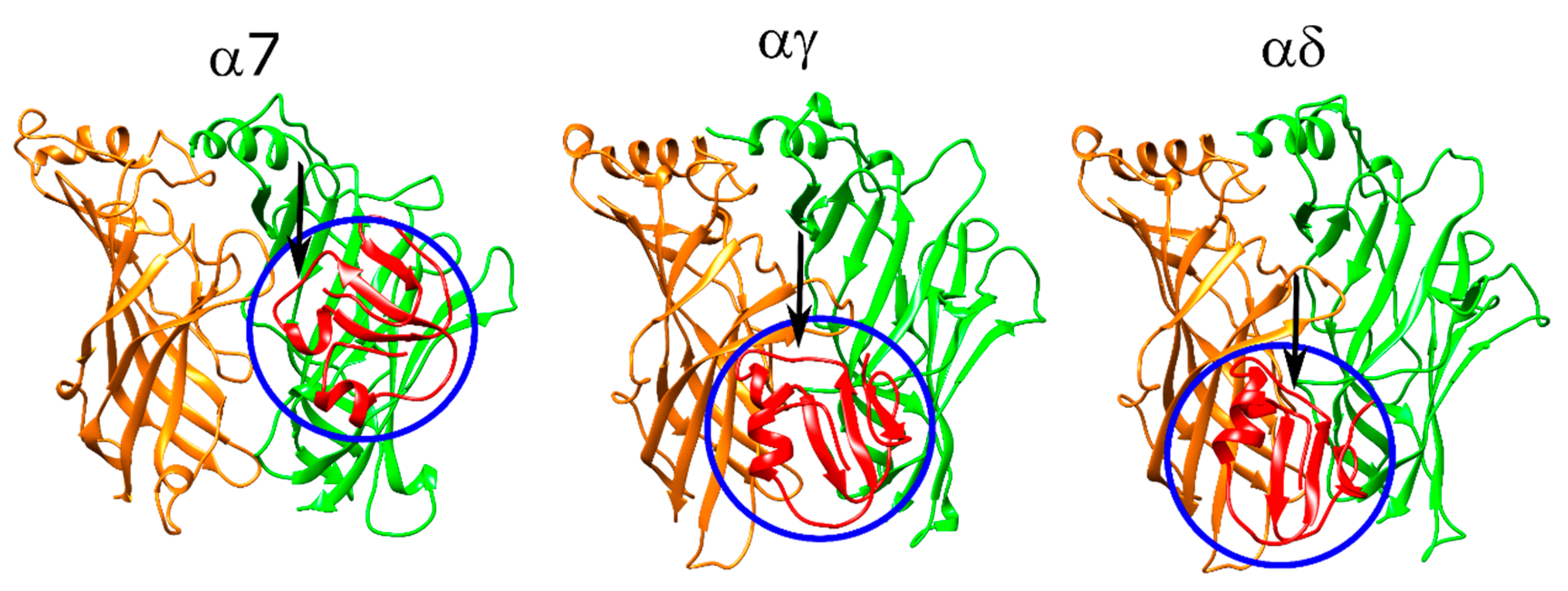
2.9. Peptides with Diverse Properties Were Docked to α7 and Muscle-Type nAChR to Identify Interactions Associated with Binding
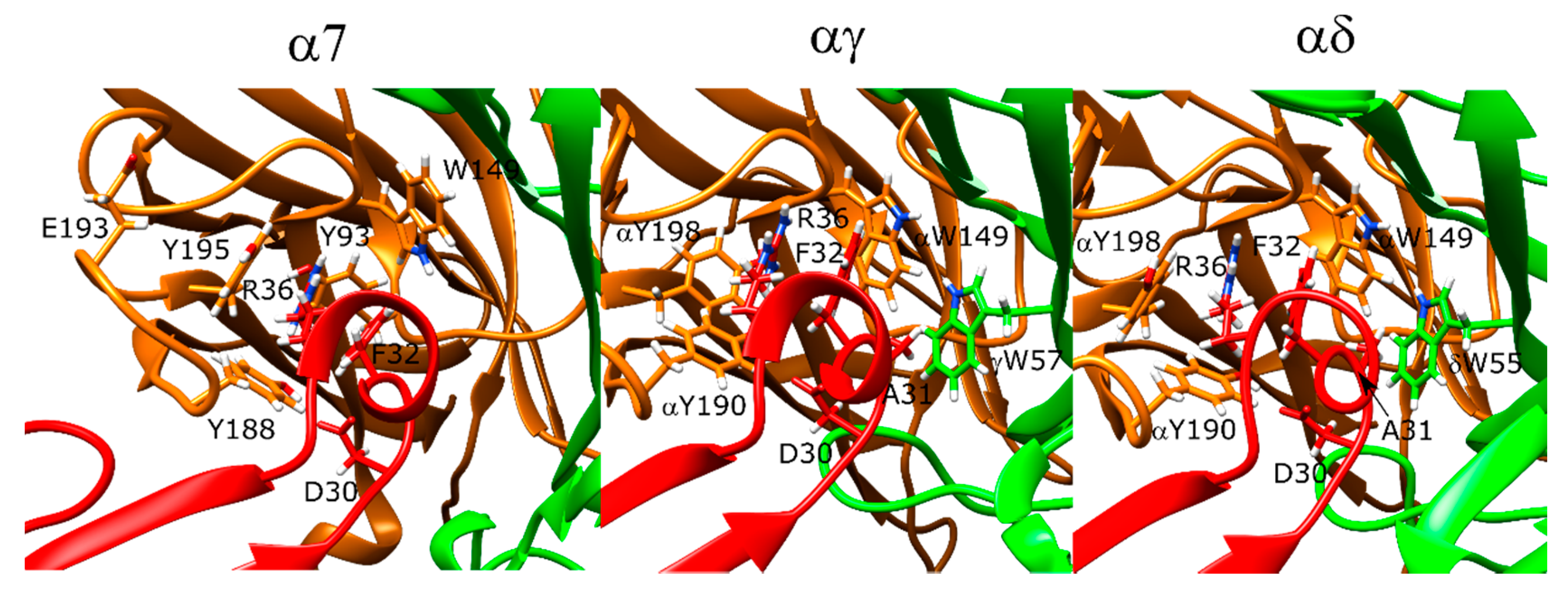
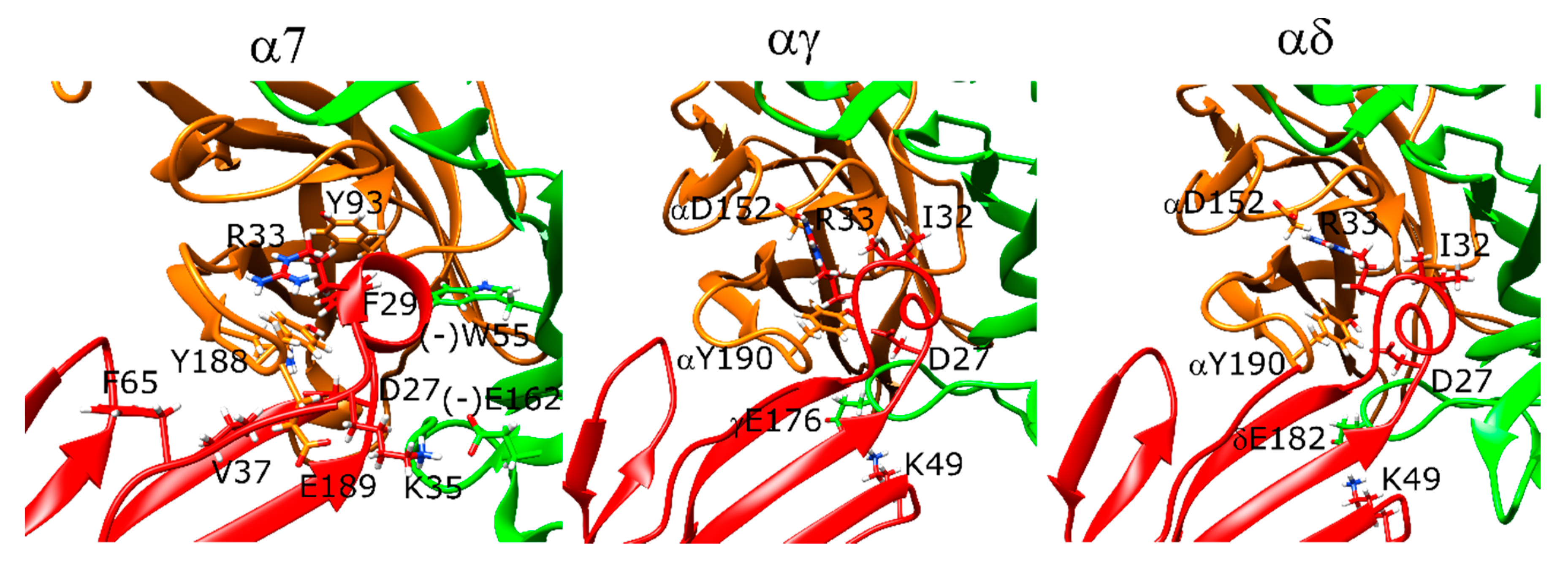
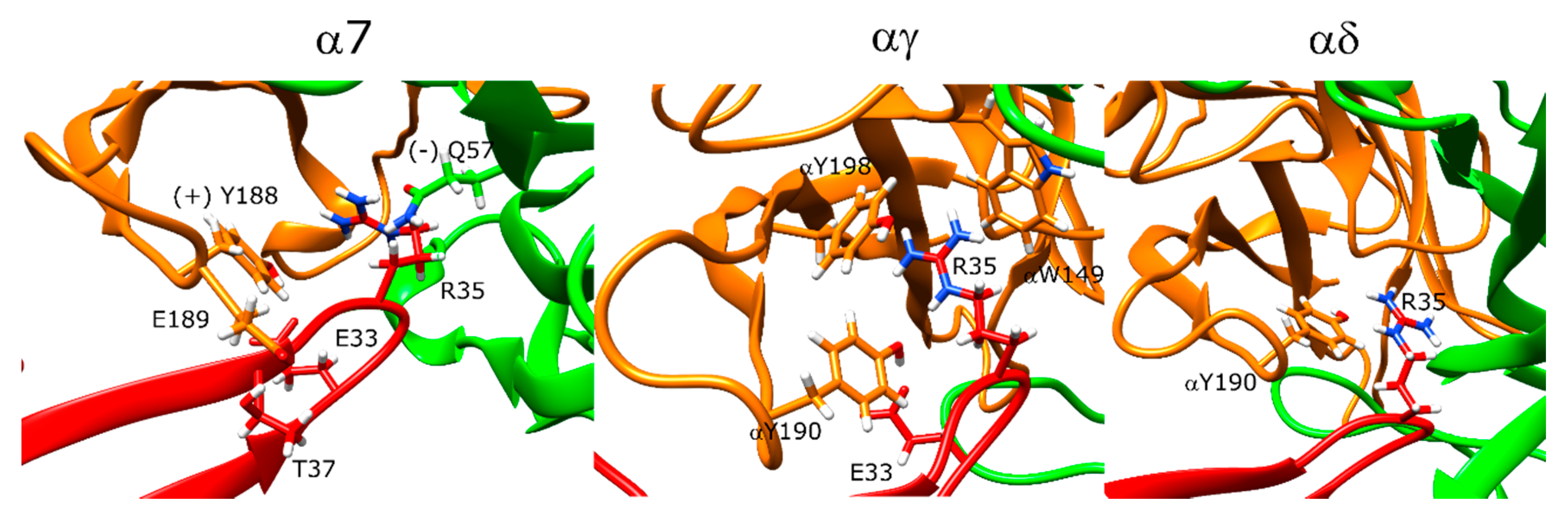
2.10. α-Bungarotoxin and α-Cobratoxin Poses Are Consistent with Homologous Crystal Structures and Mutagenesis Experiments with Slight Differences
2.11. Drysdalin Behaves Differently than α-Cobratoxin and α-Bungarotoxin Due to Conformational Differences
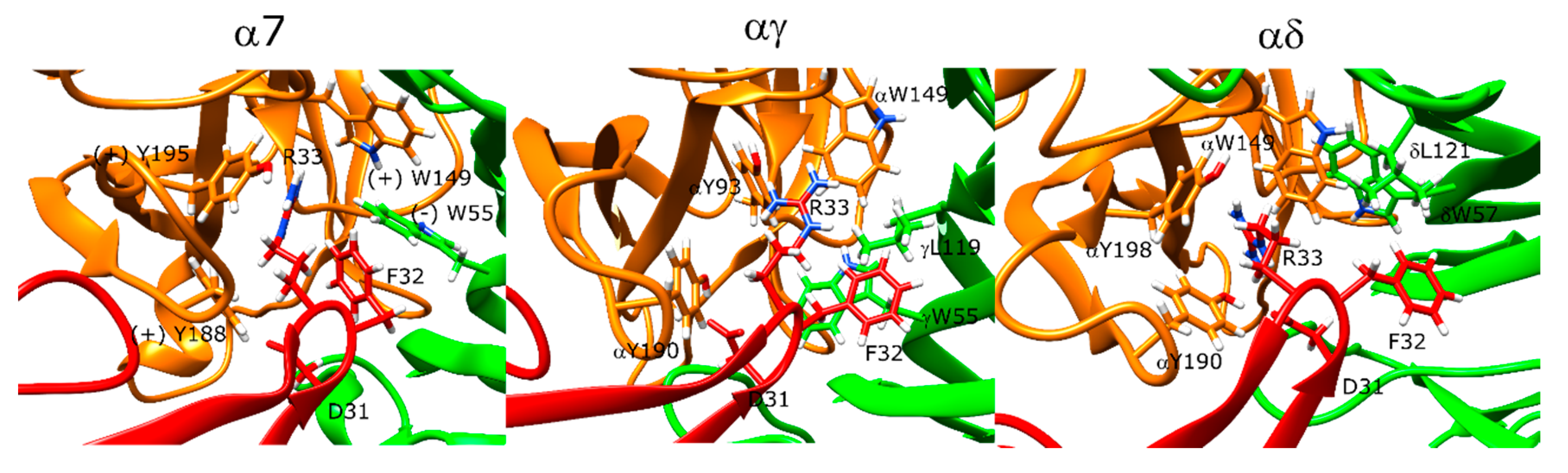
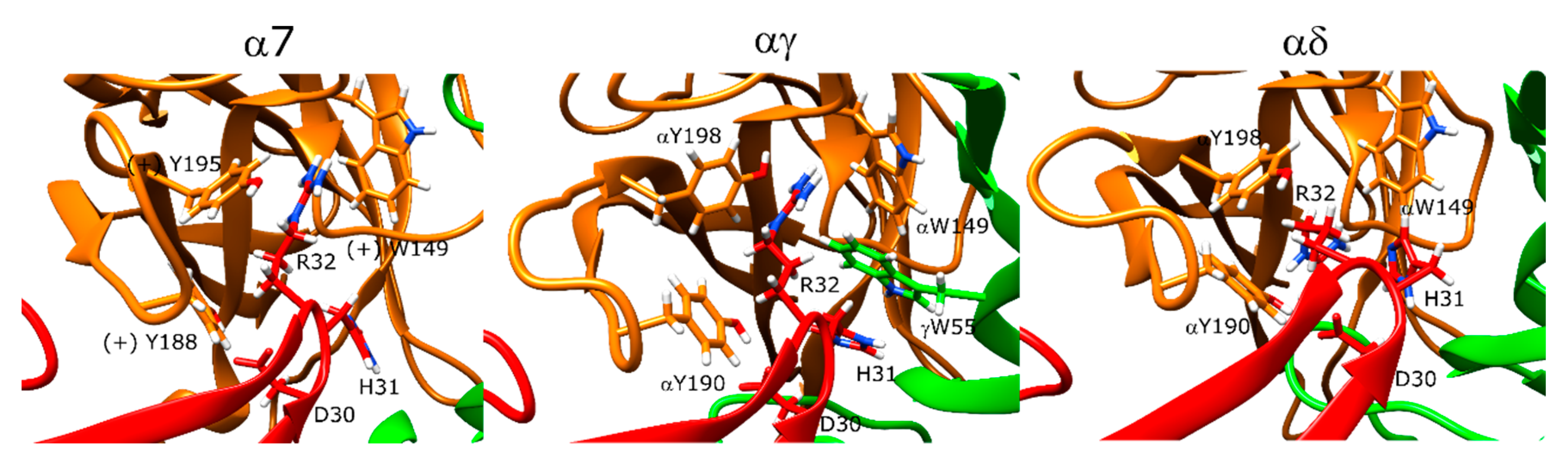
2.12. Candoxin Binds α7 and Muscle-Type nAChR Differently
2.13. Erabutoxin-a Interacts with α7 and Muscle-Type nAChR
2.14. SCNTX Bound to α7 nAChR but Not to Muscle-Type nAChR
2.15. Oh9-1 Binding to nAChR Was Tested with Two Different Configurations
2.16. Rotation of the Oh9-1 Starting Poses Affects the Results of the Docking Calculations
2.17. Pr-SNTX May Bind to α7 nAChR with Weak Affinity
3. Discussion
3.1. Rosetta Protein–Protein Docking Can Accurately Model Peptide–Protein Interactions
3.2. Long-Chain 3FTX Have Docking Poses Consistent with Experimental Data
3.3. Erabutoxin-a and Pr-SNTX Stably Bind to α7 and Muscle-Type nAChR
3.4. Candoxin Binding Was Inconsistent with Previous Experimental and Computational Data
3.5. SCNTX Binding to Muscle-Type nAChR Was Not Predicted by the Docking Protocol
3.6. ω-Bungarotoxin Oh9-1 May Bind to nAChR Configurations Different than Observed for Long and Short-Chain 3FTX
3.7. Limitations of the Protocol
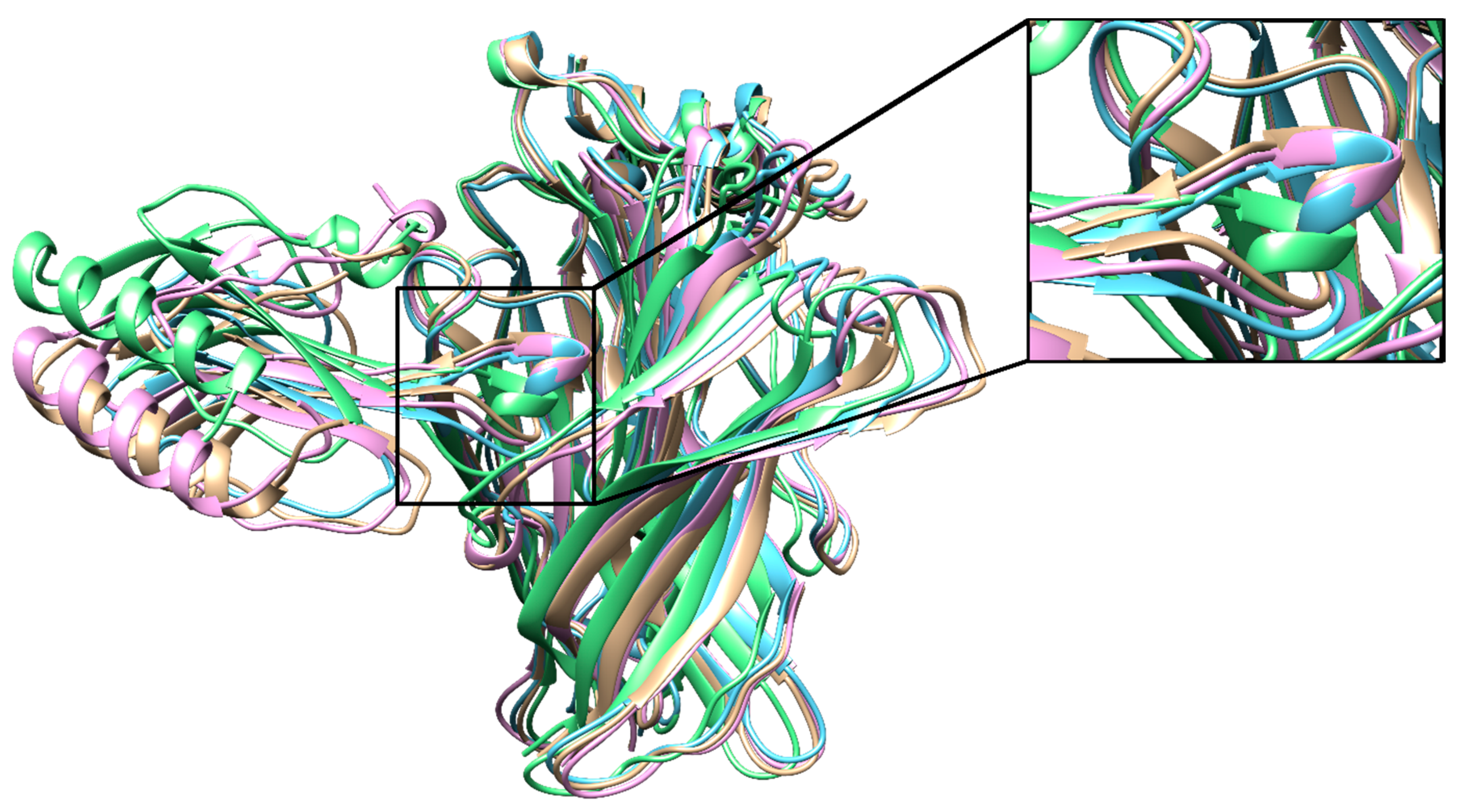
3.8. Homology Modeling Yielded Models with Structural Variability
3.9. Mobility of the C- and F-Loops May Drastically Change nAChR Conformations
3.10. Lack of Partial Covalent Interactions in the Rosetta Score Function May Affect Correct Assignment of Side Chain Conformations
4. Conclusions
5. Materials and Methods
5.1. Selection of the Crystal Structures for the Re-Docking and Cross-Docking Calculations
5.2. Homology Modeling of the Venom Peptides for AChBP Binding Calculations
5.3. Modeling of nAChR
5.4. Relax with the Bound Peptides
5.5. Protein–Protein Docking Calculations
5.6. Analysis of the Docked Structures
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hogg, R.C.; Raggenbass, M.; Bertrand, D. Nicotinic acetylcholine receptors: From structure to brain function. Rev. Physiol. Biochem. Pharmacol. 2003, 147, 1–46. [Google Scholar]
- Zhong, W.; Gallivan, J.P.; Zhang, Y.; Li, L.; Lester, H.A.; Dougherty, D.A. From ab initio quantum mechanics to molecular neurobiology: A cation–π binding site in the nicotinic receptor. Proc. Natl. Acad. Sci. USA 1998, 95, 12088–12093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puskar, N.L.; Xiu, X.; Lester, H.A.; Dougherty, D.A. Two Neuronal Nicotinic Acetylcholine Receptors, α4β4 and α7, Show Differential Agonist Binding Modes. J. Biol. Chem. 2011, 286, 14618–14627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luetje, C.W.; Patrick, J. Both α- and β-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J. Neurosci. 1991, 11, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Palma, E.; Bertrand, S.; Binzoni, T.; Bertrand, D. Neuronal nicotinic α7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllycaconitine. J. Physiol. 1996, 491, 151–161. [Google Scholar] [CrossRef]
- Kessler, P.; Marchot, P.; Silva, M.; Servent, D. The three-finger toxin fold: A multifunctional structural scaffold able to modulate cholinergic functions. J. Neurochem. 2017, 142, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Teixeira-Clerc, F.; Ménez, A.; Kessler, P. How do short neurotoxins bind to a muscular-type nicotinic acetylcholine receptor? J. Biol. Chem. 2002, 277, 25741–25747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirthanan, S.; Gwee, M.C.E. Three-Finger α-Neurotoxins and the Nicotinic Acetylcholine Receptor, Forty Years on. J. Pharmacol. Sci. 2004, 94, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirthanan, S.; Charpantier, E.; Gopalakrishnakone, P.; Gwee, M.C.E.; Khoo, H.E.; Cheah, L.S.; Bertrand, D.; Manjunatha Kini, R. Candoxin, a novel toxin from Bungarus candidus, is a reversible antagonist of muscle (αβγδ) but a poorly reversible antagonist of neuronal α7 nicotinic acetylcholine receptors. J. Biol. Chem. 2002, 277, 17811–17820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, C.; Li, B.; Hu, L.; Wei, X.; Feng, L.; Fu, W.; Lu, W. Micelle-Based Brain-Targeted Drug Delivery Enabled by a Nicotine Acetylcholine Receptor Ligand. Angew. Chemie 2011, 123, 5596–5599. [Google Scholar] [CrossRef]
- Roy, A.; Zhou, X.; Chong, M.Z.; D’Hoedt, D.; Foo, C.S.; Rajagopalan, N.; Nirthanan, S.; Bertrand, D.; Sivaraman, J.; Manjunatha Kini, R. Structural and functional characterization of a novel homodimeric three-finger neurotoxin from the venom of Ophiophagus hannah (King cobra). J. Biol. Chem. 2010, 285, 8302–8315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foo, C.S.; Jobichen, C.; Hassan-Puttaswamy, V.; Dekan, Z.; Tae, H.S.; Bertrand, D.; Adams, D.J.; Alewood, P.F.; Sivaraman, J.; Nirthanan, S.; et al. Fulditoxin, representing a new class of dimeric snake toxins, defines novel pharmacology at nicotinic ACh receptors. Br. J. Pharmacol. 2020, 177, 1822–1840. [Google Scholar] [CrossRef] [PubMed]
- Servent, D.; Winckler-Dietrich, V.; Hu, H.Y.; Kessler, P.; Drevet, P.; Bertrand, D.; Ménez, A. Only snake curaremimetic toxins with a fifth disulfide bond have high affinity for the neuronal α7 nicotinic receptor. J. Biol. Chem. 1997, 272, 24279–24286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahsavar, A.; Gajhede, M.; Kastrup, J.S.; Balle, T. Structural Studies of Nicotinic Acetylcholine Receptors: Using Acetylcholine-Binding Protein as a Structural Surrogate. Basic Clin. Pharmacol. Toxicol. 2015, 118, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, A.B.; Syed, N.I.; Schaap, D.; van Minnen, J.; Klumperman, J.; Kits, K.S.; Lodder, H.; van der Schors, R.C.; van Elk, R.; Sorgedrager, B.; et al. A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature 2001, 411, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Sixma, T.K.; Smit, A.B. Acetylcholine binding protein (AChBP): A secreted glial protein that provides a high-resolution model for the extracellular domain of pentameric ligand-gated ion channels. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 311–334. [Google Scholar] [CrossRef]
- Brejc, K.; Van Dijk, W.J.; Klaassen, R.V.; Schuurmans, M.; Van Der Oost, J.; Smit, A.B.; Sixma, T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411, 269–276. [Google Scholar] [CrossRef]
- Rahman, M.; Teng, J.; Worrell, B.T.; Karlin, A.; Stowell, M.H.B.; Hibbs, R.E.; Rahman, M.; Teng, J.; Worrell, B.T.; Noviello, C.M.; et al. Structure of the Native Muscle-type Nicotinic Receptor and Inhibition by Snake Venom Toxins Article Structure of the Native Muscle-type Nicotinic Receptor and Inhibition by Snake Venom Toxins. Neuron 2020, 106, 952–962. [Google Scholar] [CrossRef]
- Huang, S.; Li, S.-X.; Bren, N.; Cheng, K.; Gomoto, R.; Chen, L.; Sine, S.M. Complex between α-bungarotoxin and an α7 nicotinic receptor ligand-binding domain chimaera. Biochem. J. 2013, 454, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Dellisanti, C.D.; Yao, Y.; Stroud, J.C.; Wang, Z.-Z.; Chen, L. Crystal structure of the extracellular domain of nAChR α1 bound to α-bungarotoxin at 1.94 Å resolution. Nat. Neurosci. 2007, 10, 953–962. [Google Scholar] [CrossRef]
- Zouridakis, M.; Giastas, P.; Zarkadas, E.; Chroni-Tzartou, D.; Bregestovski, P.; Tzartos, S.J. Crystal structures of free and antagonist-bound states of human ± 9 nicotinic receptor extracellular domain. Nat. Struct. Mol. Biol. 2014, 21, 976–980. [Google Scholar] [CrossRef]
- Bourne, Y.; Talley, T.T.; Hansen, S.B.; Taylor, P.; Marchot, P. Crystal structure of a Cbtx–AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J. 2005, 24, 1512–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Mordvintsev, D.Y.; Polyak, Y.L.; Levtsova, O.V.; Tourleigh, Y.V.; Kasheverov, I.E.; Shaitan, K.V.; Utkin, Y.N.; Tsetlin, V.I. A model for short α-neurotoxin bound to nicotinic acetylcholine receptor from Torpedo californica: Comparison with long-chain α-neurotoxins and α-conotoxins. Comput. Biol. Chem. 2005, 29, 398–411. [Google Scholar] [CrossRef]
- Mordvitsev, D.Y.; Polyak, Y.L.; Kuzmin, D.A.; Levtsova, O.V.; Tourleigh, Y.V.; Utkin, Y.N.; Shaitan, K.V.; Tsetlin, V.I. Computer modeling of binding of diverse weak toxins to nicotinic acetylcholine receptors. Comput. Biol. Chem. 2007, 31, 72–81. [Google Scholar] [CrossRef]
- Lyukmanova, E.N.; Shulepko, M.A.; Shenkarev, Z.O.; Kasheverov, I.E.; Chugunov, A.O.; Kulbatskii, D.S.; Myshkin, M.Y.; Utkin, Y.N.; Efremov, R.G.; Tsetlin, V.I.; et al. Central loop of non-conventional toxin WTX from Naja kaouthia is important for interaction with nicotinic acetylcholine receptors. Toxicon 2016, 119, 274–279. [Google Scholar] [CrossRef]
- Leffler, A.E.; Kuryatov, A.; Zebroski, H.A.; Powell, S.R.; Filipenko, P.; Hussein, A.K.; Gorson, J.; Heizmann, A.; Lyskov, S.; Tsien, R.W.; et al. Discovery of peptide ligands through docking and virtual screening at nicotinic acetylcholine receptor homology models. Proc. Natl. Acad. Sci. USA 2017, 114, E8100–E8109. [Google Scholar] [CrossRef] [Green Version]
- Chaudhury, S.; Berrondo, M.; Weitzner, B.D.; Muthu, P.; Bergman, H.; Gray, J.J. Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS ONE 2011, 6, e22477. [Google Scholar] [CrossRef] [Green Version]
- Celie, P.H.N.; Kasheverov, I.E.; Mordvintsev, D.Y.; Hogg, R.C.; van Nierop, P.; van Elk, R.; van Rossum-Fikkert, S.E.; Zhmak, M.N.; Bertrand, D.; Tsetlin, V.; et al. Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an alpha-conotoxin PnIA variant. Nat. Struct. Mol. Biol. 2005, 12, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.B.; Sulzenbacher, G.; Huxford, T.; Marchot, P.; Taylor, P.; Bourne, Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005, 24, 3635–3646. [Google Scholar] [CrossRef]
- Ulens, C.; Hogg, R.C.; Celie, P.H.; Bertrand, D.; Tsetlin, V.; Smit, A.B.; Sixma, T.K. Structural determinants of selective α-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc. Natl. Acad. Sci. USA 2006, 103, 3615–3620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, S.; Ulens, C.; Büttner, R.; Fish, A.; van Elk, R.; Kendel, Y.; Hopping, G.; Alewood, P.F.; Schroeder, C.; Nicke, A.; et al. AChBP-targeted α-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J. 2007, 26, 3858–3867. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.; Luo, S.; Xiang, S.-H.; Zhangsun, D.; Wu, Y.; Xu, M.; Lin, B.; Tsetlin, V.I.; Hu, Y.; et al. From crystal structure of α-conotoxin GIC in complex with Ac-AChBP to molecular determinants of its high selectivity for α3β2 nAChR. Sci. Rep. 2016, 6, 22349. [Google Scholar]
- Abraham, N.; Healy, M.; Ragnarsson, L.; Brust, A.; Alewood, P.F.; Lewis, R.J. Structural mechanisms for α-conotoxin activity at the human α3β4 nicotinic acetylcholine receptor. Sci. Rep. 2017, 7, 45466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Zhu, X.; Yu, J.; Yu, J.; Luo, S.; Wang, X. The crystal structure of Ac-AChBP in complex with α-conotoxin LvIA reveals the mechanism of its selectivity towards different nAChR subtypes. Protein Cell 2017, 8, 675–685. [Google Scholar] [CrossRef]
- Bhardwaj, G.; Mulligan, V.K.; Bahl, C.D.; Gilmore, J.M.; Harvey, P.J.; Cheneval, O.; Buchko, G.W.; Pulavarti, S.V.S.R.K.; Kaas, Q.; Eletsky, A.; et al. Accurate de novo design of hyperstable constrained peptides. Nature 2016, 538, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Albulescu, L.O.; Kazandjian, T.; Slagboom, J.; Bruyneel, B.; Ainsworth, S.; Alsolaiss, J.; Wagstaff, S.C.; Whiteley, G.; Harrison, R.A.; Ulens, C.; et al. A decoy-receptor approach using nicotinic acetylcholine receptor mimics reveals their potential as novel therapeutics against neurotoxic snakebite. Front. Pharmacol. 2019, 10, 848. [Google Scholar] [CrossRef] [Green Version]
- Sine, S.M.; Huang, S.; Li, S.X.; Dacosta, C.J.B.; Chen, L. Inter-residue coupling contributes to high-affinity subtype-selective binding of α-bungarotoxin to nicotinic receptors. Biochem. J. 2013, 454, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Chandna, R.; Tae, H.-S.; Seymour, V.A.L.; Chathrath, S.; Adams, D.J.; Kini, R.M. Drysdalin, an antagonist of nicotinic acetylcholine receptors highlights the importance of functional rather than structural conservation of amino acid residues. FASEB BioAdvances 2019, 1, 115–131. [Google Scholar] [CrossRef] [Green Version]
- de la Rosa, G.; Corrales-García, L.L.; Rodriguez-Ruiz, X.; López-Vera, E.; Corzo, G. Short-chain consensus alpha-neurotoxin: A synthetic 60-mer peptide with generic traits and enhanced immunogenic properties. Amino Acids 2018, 50, 885–895. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Kimoto, H.; Yang, X.; Filkin, S.; Utkin, Y.; Kubo, T.; Inagaki, H. Pr-SNTX, a short-chain three-finger toxin from Papuan pigmy mulga snake, is an antagonist of muscle-type nicotinic acetylcholine receptor (α2βδε). Biosci. Biotechnol. Biochem. 2016, 80, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Hassan-Puttaswamy, V.; Adams, D.J.; Kini, R.M. A Distinct Functional Site in ω-Neurotoxins: Novel Antagonists of Nicotinic Acetylcholine Receptors from Snake Venom. ACS Chem. Biol. 2015, 10, 2805–2815. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.O.; Scherf, T.; Eisenstein, M.; Chill, J.H.; Anglister, J. The mechanism for acetylcholine receptor inhibition by α-neurotoxins and species-specific resistance to α-bungarotoxin revealed by NMR. Neuron 2002, 35, 319–332. [Google Scholar] [CrossRef] [Green Version]
- McLane, K.E.; Wu, X.; Conti-Tronconi, B.M. An α-Bungarotoxin-Binding Sequence on the Torpedo Nicotinic Acetylcholine Receptor α-Subunit: Conservative Amino Acid Substitutions Reveal Side-Chain Specific Interactions. Biochemistry 1994, 33, 2576–2585. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, V.; Donnelly-Roberts, D.L.; Lentz, T.L. Effects of Mutations of Torpedo Acetylcholine Receptor α1 Subunit Residues 184–200 on α-Bungarotoxin Binding in a Recombinant Fusion Protein. Biochemistry 1993, 32, 9570–9576. [Google Scholar] [CrossRef] [PubMed]
- Sine, S.M. Identification of equivalent residues in the γ, δ, and ε subunits of the nicotinic receptor that contribute to α-bungarotoxin binding. J. Biol. Chem. 1997, 272, 23521–23527. [Google Scholar] [CrossRef] [Green Version]
- Servent, D.; Antil-Delbeke, S.; Gaillard, C.; Corringer, P.J.; Changeux, J.P.; Ménez, A. Molecular characterization of the specificity of interactions of various neurotoxins on two distinct nicotinic acetylcholine receptors. Eur. J. Pharmacol. 2000, 393, 197–204. [Google Scholar] [CrossRef]
- Ackermann, E.J.; Ang, E.T.H.; Kanter, J.R.; Tsigelny, I.; Taylor, P. Identification of pairwise interactions in the α-neurotoxin-nicotinic acetylcholine receptor complex through double mutant cycles. J. Biol. Chem. 1998, 273, 10958–10964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antil-Delbeke, S.; Gaillard, C.; Tamiya, T.; Corringer, P.J.; Changeux, J.P.; Servent, D.; Ménez, A. Molecular determinants by which a long chain toxin from snake venom interacts with the neuronal α7-nicotinic acetylcholine receptor. J. Biol. Chem. 2000, 275, 29594–29601. [Google Scholar] [CrossRef] [Green Version]
- Pillet, L.; Tremeau, O.; Ducancel, F.; Drevet, P.; Zinn-justin, S.; Pinkasfeld, S.; Boulain, J. Genetic Engineering of Snake Toxins. J. Biol. Chem. 1993, 268, 909–916. [Google Scholar]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Dimaio, F.; Wang, R.Y.R.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Tyka, M.D.; Keedy, D.A.; André, I.; Dimaio, F.; Song, Y.; Richardson, D.C.; Richardson, J.S.; Baker, D. Alternate states of proteins revealed by detailed energy landscape mapping. J. Mol. Biol. 2011, 405, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Protein | PDB ID |
|---|---|---|
| α-bungarotoxin | α7/Ls-AChBP | 4HQP [19] |
| α-cobratoxin | Ls-AChBP | 1YI5 [22] |
| α-conotoxin PnIA | Ac-AChBP | 2BR8 [29] |
| α-conotoxin ImI | Ac-AChBP | 2BYP, 2C9T [30,31] |
| α-conotoxin TxIA (A10L) | Ac-AChBP | 2UZ6 [32] |
| α-conotoxin BuIA | Ac-AChBP | 4EZ1 |
| α-conotoxin GIC | Ac-AChBP | 5CO5 [33] |
| α-conotoxin PeIA | Ac-AChBP | 5JME |
| α-conotoxin LsIA | Ls-AChBP | 5T90 [34] |
| α-conotoxin LvIA | Ac-AChBP | 5XGL [35] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulsevin, A.; Meiler, J. An Investigation of Three-Finger Toxin—nAChR Interactions through Rosetta Protein Docking. Toxins 2020, 12, 598. https://doi.org/10.3390/toxins12090598
Gulsevin A, Meiler J. An Investigation of Three-Finger Toxin—nAChR Interactions through Rosetta Protein Docking. Toxins. 2020; 12(9):598. https://doi.org/10.3390/toxins12090598
Chicago/Turabian StyleGulsevin, Alican, and Jens Meiler. 2020. "An Investigation of Three-Finger Toxin—nAChR Interactions through Rosetta Protein Docking" Toxins 12, no. 9: 598. https://doi.org/10.3390/toxins12090598
APA StyleGulsevin, A., & Meiler, J. (2020). An Investigation of Three-Finger Toxin—nAChR Interactions through Rosetta Protein Docking. Toxins, 12(9), 598. https://doi.org/10.3390/toxins12090598