Simplified Synthesis and Stability Assessment of Aflatoxin B1-Lysine and Aflatoxin G1-Lysine


Abstract
:
1. Introduction
2. Results
2.1. Conversion of AFB1 to AFB2a and AFG1 to AFG2a
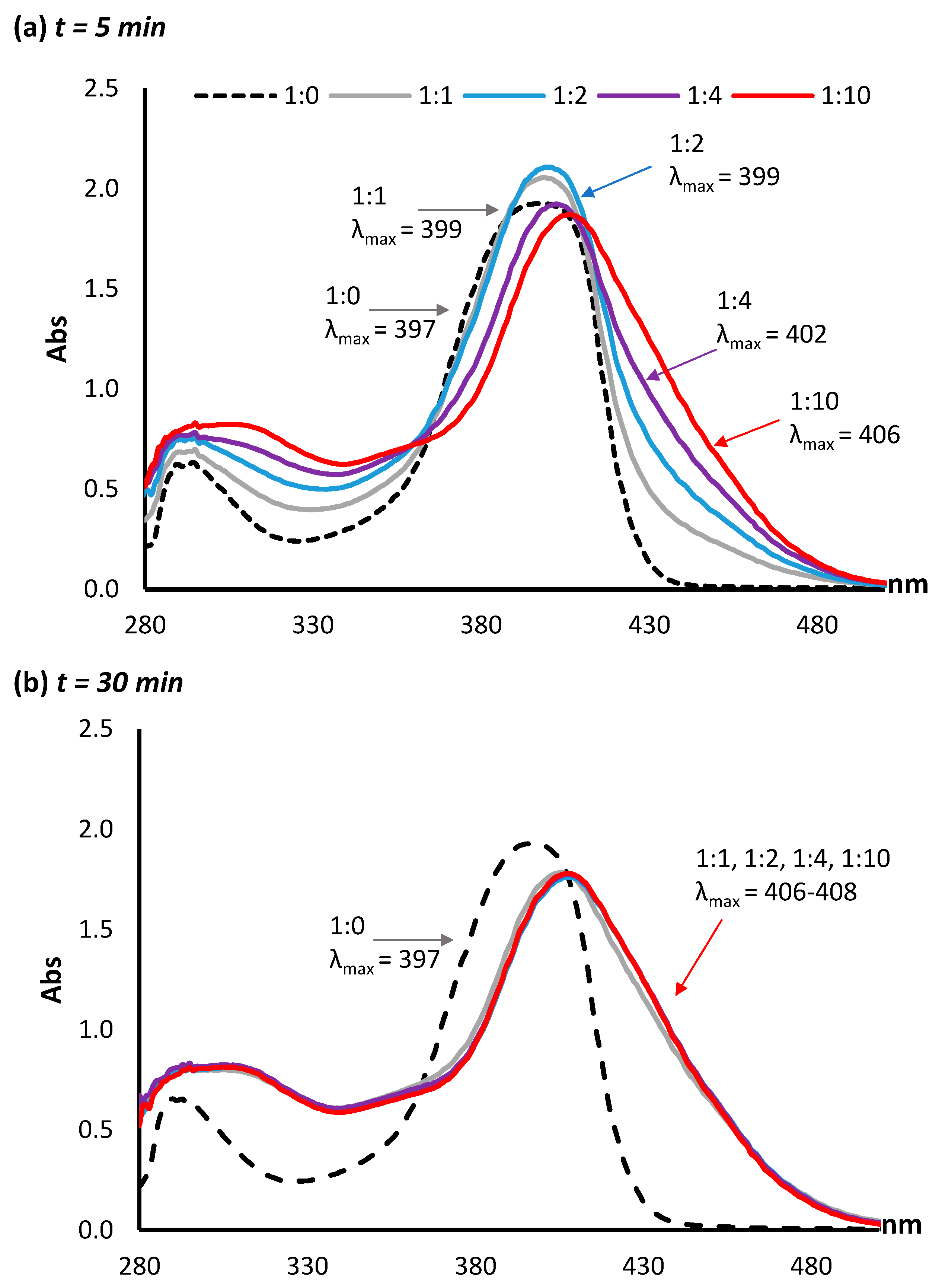
2.2. Coupling of AFB2a or AFG2a to Lysine
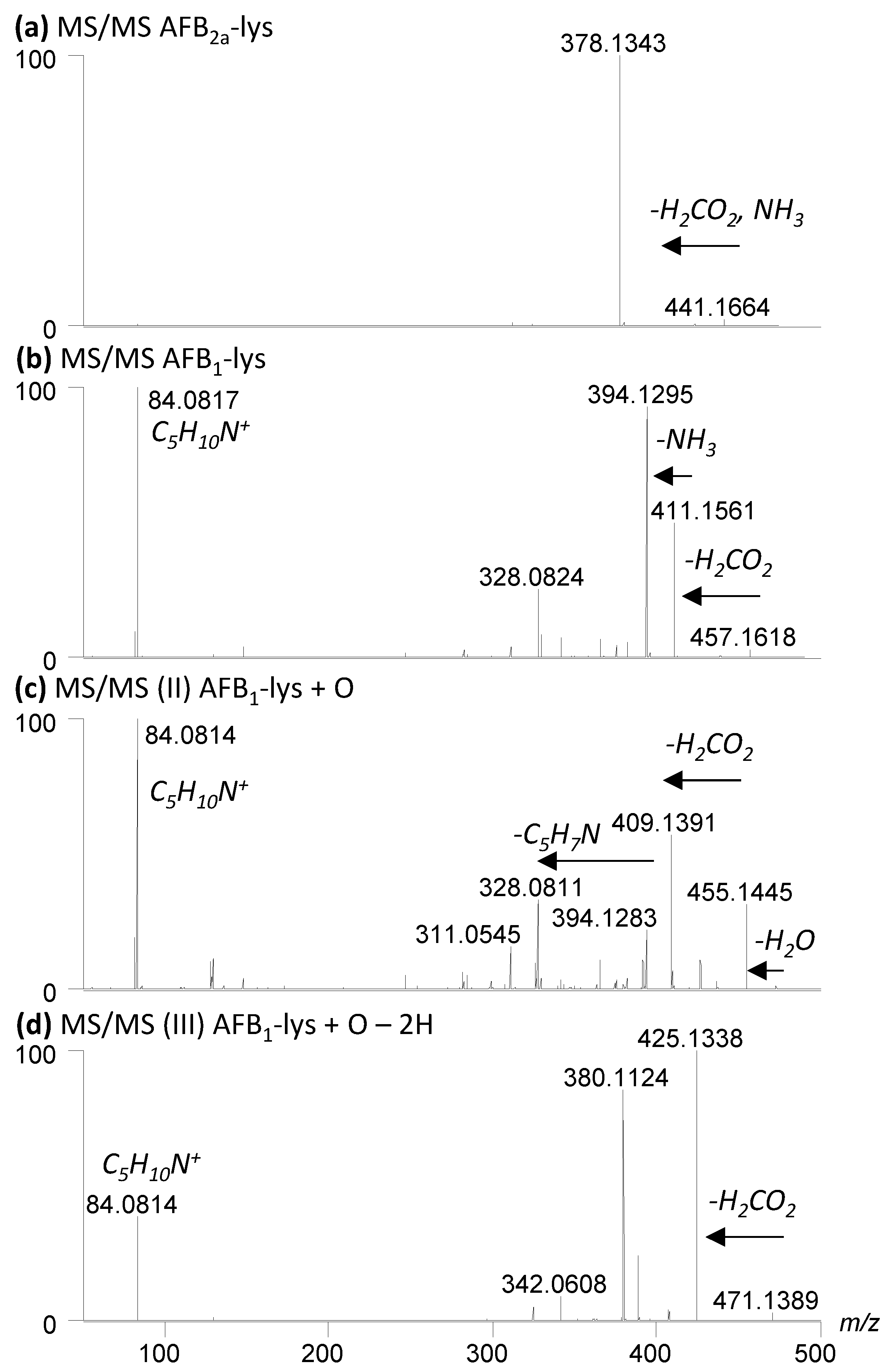
2.3. Oxidation of AFB2a-Lys to AFB1-Lys
2.4. Synthesis of Isotopically Labelled Standards
2.5. Quantitative NMR
2.6. Stability
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Conversion of AFB1/AFG1 to AFB2a/AFG2a
4.3. Coupling of Lysine to AFB2a
4.4. Oxidation of AFB2a-Lysine to AFB1-Lysine
4.5. Purification by HPLC
4.6. Quantitative NMR
4.7. Stability Assay
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evaluation of Certain Contaminants in Food: Eighty-Third Report of the Joint Fao/Who Expert Committee on Food Additives; Joint Food and Agriculture Organization/World Health Organization: Geneva, Switzerland, 2017.
- Pitt, J.I.; Wild, C.P.; Baan, R.A.; Gelderblom, W.C.; Miller, J.; Riley, R.; Wu, F. Improving Public Health through Mycotoxin Control; International Agency for Research on Cancer: Lyon, France, 2012. [Google Scholar]
- Wild, C.P.; Miller, J.D.; Groopman, J.D. Mycotoxin Control in Low-and Middle-Income Countries; International Agency for Research on Cancer: Lyon, France, 2015. [Google Scholar]
- Wojnowski, L.; Turner, P.C.; Pedersen, B.; Hustert, E.; Brockmöller, J.; Mendy, M.; Whittle, H.C.; Kirk, G.; Wild, C.P. Increased levels of aflatoxin-albumin adducts are associated with CYP3A5 polymorphisms in The Gambia, West Africa. Pharmacogenet. Genom. 2004, 14, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.; Stone, M.; Gopalakrishnan, S.; Baertschi, S.; Raney, K.; Byrd, S. Aflatoxin B1 epoxide, the ultimate carcinogenic form of aflatoxin B1: Synthesis and reaction with DNA. J. Toxicol. Toxin Rev. 1989, 8, 111–120. [Google Scholar] [CrossRef]
- Martin, C.N.; Garner, R.C. Aflatoxin B-oxide generated by chemical or enzymic oxidation of aflatoxin B1 causes guanine substitution in nucleic acids. Nature 1977, 267, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Hsieh, D. Mutagenicity of aflatoxins related to their metabolism and carcinogenic potential. Proc. Natl. Acad. Sci. USA 1976, 73, 2241–2244. [Google Scholar] [CrossRef] [Green Version]
- Theumer, M.G.; Henneb, Y.; Khoury, L.; Snini, S.; Tadrist, S.; Canlet, C.; Puel, O.; Oswald, I.P.; Audebert, M. Genotoxicity of aflatoxins and their precursors in human cells. Toxicol. Lett. 2018, 287, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.W.; Harris, T.M.; Guengerich, F.P. Kinetics and mechanism of hydrolysis of aflatoxin B1 exo-8, 9-epoxide and rearrangement of the dihydrodiol. J. Am. Chem. Soc. 1996, 118, 8213–8220. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Voehler, M.; Williams, K.M.; Deng, Z.; Harris, T.M. Structure of the aflatoxin B1 dialdehyde adduct formed from reaction with methylamine. Chem. Res. Toxicol. 2002, 15, 793–798. [Google Scholar] [CrossRef]
- Sabbioni, G.; Skipper, P.L.; Büchi, G.; Tannenbaum, S.R. Isolation and characterization of the major serum albumin adduct formed by aflatoxin B1 in vivo in rats. Carcinogenesis 1987, 8, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.; Megalla, S.; Abd El-Fattah, H.; Hafez, A.; El-Deap, T. Binding of aflatoxin b1, g1 and m to plasma albumin. Mycopathologia 1982, 79, 35–38. [Google Scholar] [CrossRef]
- Gan, L.-S.; Skipper, P.L.; Peng, X.; Groopman, J.D.; Chen, J.-S.; Wogan, G.N.; Tannenbaum, S.R. Serum albumin adducts in the molecular epidemiology of aflatoxin carcinogenesis: Correlation with aflatoxin B1 intake and urinary excretion of aflatoxin m 1. Carcinogenesis 1988, 9, 1323–1325. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.K.; Yu, M.; Henderson, B.; Yuan, J.-M.; Qian, G.-S.; Tu, J.-T.; Gao, Y.-T.; Wogan, G.; Groopman, J. Urinary aflatoxin biomarkers and risk of hepatocellular carcinoma. Lancet 1992, 339, 943–946. [Google Scholar] [CrossRef]
- Groopman, J.D.; Wild, C.P.; Hasler, J.; Junshi, C.; Wogan, G.N.; Kensler, T.W. Molecular epidemiology of aflatoxin exposures: Validation of aflatoxin-N7-guanine levels in urine as a biomarker in experimental rat models and humans. Environ. Health Perspect. 1993, 99, 107–113. [Google Scholar] [CrossRef]
- Wild, C.; Turner, P. The toxicology of aflatoxins as a basis for public health decisions. Mutagenesis 2002, 17, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, J.; Trabjerg, E.; Grevys, A.; Azevedo, C.; Brennan, S.O.; Stensland, M.; Wilson, J.; Sand, K.M.K.; Bern, M.; Dalhus, B. An intact C-terminal end of albumin is required for its long half-life in humans. Commun. Biol. 2020, 3, 1–11. [Google Scholar] [CrossRef]
- Wild, C.P.; Jiang, Y.-Z.; Sabbioni, G.; Chapot, B.; Montesano, R. Evaluation of methods for quantitation of aflatoxin-albumin adducts and their application to human exposure assessment. Cancer Res. 1990, 50, 245–251. [Google Scholar] [PubMed]
- Chapot, B.; Wild, C. ELISA for quantification of aflatoxin-albumin adducts and their application to human exposure assessment. Tech. Diagn. Pathol. 1991, 2, 135–155. [Google Scholar]
- Wild, C.P.; Hudson, G.J.; Sabbioni, G.; Chapot, B.; Hall, A.J.; Wogan, G.N.; Whittle, H.; Montesano, R.; Groopman, J.D. Dietary intake of aflatoxins and the level of albumin-bound aflatoxin in peripheral blood in The Gambia, West Africa. Cancer Epidemiol. Prev. Biomark. 1992, 1, 229–234. [Google Scholar]
- Xue, K.S.; Cai, W.; Tang, L.; Wang, J.-S. Aflatoxin B1-lysine adduct in dried blood spot samples of animals and humans. Food Chem. Toxicol. 2016, 98, 210–219. [Google Scholar] [CrossRef]
- McCoy, L.F.; Scholl, P.F.; Schleicher, R.L.; Groopman, J.D.; Powers, C.D.; Pfeiffer, C.M. Analysis of aflatoxin B1-lysine adduct in serum using isotope-dilution liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2203–2210. [Google Scholar] [CrossRef]
- Scholl, P.F.; Turner, P.C.; Sutcliffe, A.E.; Sylla, A.; Diallo, M.S.; Friesen, M.D.; Groopman, J.D.; Wild, C.P. Quantitative comparison of aflatoxin B1 serum albumin adducts in humans by isotope dilution mass spectrometry and elisa. Cancer Epidemiol. Biomark. Prev. 2006, 15, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Baertschi, S.W.; Raney, K.D.; Shimada, T.; Harris, T.M.; Guengerich, F.P. Comparison of rates of enzymic oxidation of aflatoxin b1, aflatoxin g1, and sterigmatocystin and activities of the epoxides in forming guanyl-N7 adducts and inducing different genetic responses. Chem. Res. Toxicol. 1989, 2, 114–122. [Google Scholar] [CrossRef]
- Sabbioni, G.; Wild, C.P. Identification of an aflatoxin G1–serum albumin adduct and its relevance to the measurement of human exposure to aflatoxins. Carcinogenesis 1991, 12, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Purchase, I.; Steyn, M. The metabolism of aflatoxin B1 in rats. Br. J. Cancer 1969, 23, 800–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruprich, J. Preparation and identification of aflatoxin B2a under laboratory conditions. Vet. Med. 1983, 28, 361–366. [Google Scholar]
- Ciegler, A.; Peterson, R. Aflatoxin detoxification: Hydroxydihydro-aflatoxin B. Appl. Microbiol. 1968, 16, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Bean, T.; Yourtee, D.; Akande, B.; Ogunlewe, J. Aflatoxin metabolites in the urine of Nigerians comparison of chromatographic methods. J. Toxicol. Toxin Rev. 1989, 8, 43–52. [Google Scholar] [CrossRef]
- Ashoor, S.H.; Chu, F.S. Reduction of aflatoxin B2a with sodium borohydride. J. Agric. Food Chem. 1975, 23, 445–447. [Google Scholar] [CrossRef]
- Rushing, B.R.; Selim, M.I. Structure and oxidation of pyrrole adducts formed between aflatoxin B2a and biological amines. Chem. Res. Toxicol. 2017, 30, 1275–1285. [Google Scholar] [CrossRef]
- Rushing, B.R.; Selim, M.I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food Chem. Toxicol. 2019, 124, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Cristina Sass, D.; Vincenzi Jager, A.; Gustavo Tonin, F.; Naira Zambelli Ramalho, L.; Silva Ramalho, F.; Gomes Constantino, M.; Augusto Fernandes Oliveira, C. Methods for chemical preparation of aflatoxin B1 adducts, AFB1-N7-guanine and AFB1-lysine. Toxin Rev. 2013, 32, 68–74. [Google Scholar] [CrossRef]
- McMillan, A.; Renaud, J.B.; Burgess, K.M.; Orimadegun, A.E.; Akinyinka, O.O.; Allen, S.J.; Miller, J.D.; Reid, G.; Sumarah, M.W. Aflatoxin exposure in nigerian children with severe acute malnutrition. Food Chem. Toxicol. 2018, 111, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.S.; Harris, T.M. Preparation of aflatoxin B1 8, 9-epoxide using m-chloroperbenzoic acid. Chem. Res. Toxicol. 1993, 6, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Sass, D.C.; Jager, A.V.; Tonin, F.G.; Rosim, R.E.; Constantino, M.G.; Oliveira, C.A.F. Synthesis and purification of the aflatoxin B1-lysine adduct. Toxin Rev. 2015, 34, 53–59. [Google Scholar] [CrossRef]
- Sabbioni, G. Chemical and physical properties of the major serum albumin adduct of aflatoxin B1 and their implications for the quantification in biological samples. Chem.-Biol. Interact. 1990, 75, 1–15. [Google Scholar] [CrossRef]
- Scholl, P.F.; Groopman, J.D. Synthesis of 5, 5, 6, 6-D4-l-lysine-aflatoxin Bl for use as a mass spectrometric internal standard. J. Label. Compd. Radiopharm. 2004, 47, 807–815. [Google Scholar] [CrossRef]
- Patterson, D.; Roberts, B. Aflatoxin metabolism in duck-liver homogenates: The relative importance of reversible cyclopentenone reduction and hemiacetal formation. Food Cosmet. Toxicol. 1972, 10, 501–512. [Google Scholar] [CrossRef]
- Howard, J.K.; Rihak, K.J.; Bissember, A.C.; Smith, J.A. The oxidation of pyrrole. Chem.-Asian J. 2016, 11, 155–167. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ε (pH 7.4) | Concentration by UV–VIS (mM) | Concentration by qNMR (mM) |
|---|---|---|---|
| AFB1-Lys | 30,866 | 1.15 | 1.01 |
| AFG1-Lys | 27,783 | 0.634 | 1.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renaud, J.B.; Walsh, J.P.; Sumarah, M.W. Simplified Synthesis and Stability Assessment of Aflatoxin B1-Lysine and Aflatoxin G1-Lysine. Toxins 2022, 14, 56. https://doi.org/10.3390/toxins14010056
Renaud JB, Walsh JP, Sumarah MW. Simplified Synthesis and Stability Assessment of Aflatoxin B1-Lysine and Aflatoxin G1-Lysine. Toxins. 2022; 14(1):56. https://doi.org/10.3390/toxins14010056
Chicago/Turabian StyleRenaud, Justin B., Jacob P. Walsh, and Mark W. Sumarah. 2022. "Simplified Synthesis and Stability Assessment of Aflatoxin B1-Lysine and Aflatoxin G1-Lysine" Toxins 14, no. 1: 56. https://doi.org/10.3390/toxins14010056
APA StyleRenaud, J. B., Walsh, J. P., & Sumarah, M. W. (2022). Simplified Synthesis and Stability Assessment of Aflatoxin B1-Lysine and Aflatoxin G1-Lysine. Toxins, 14(1), 56. https://doi.org/10.3390/toxins14010056