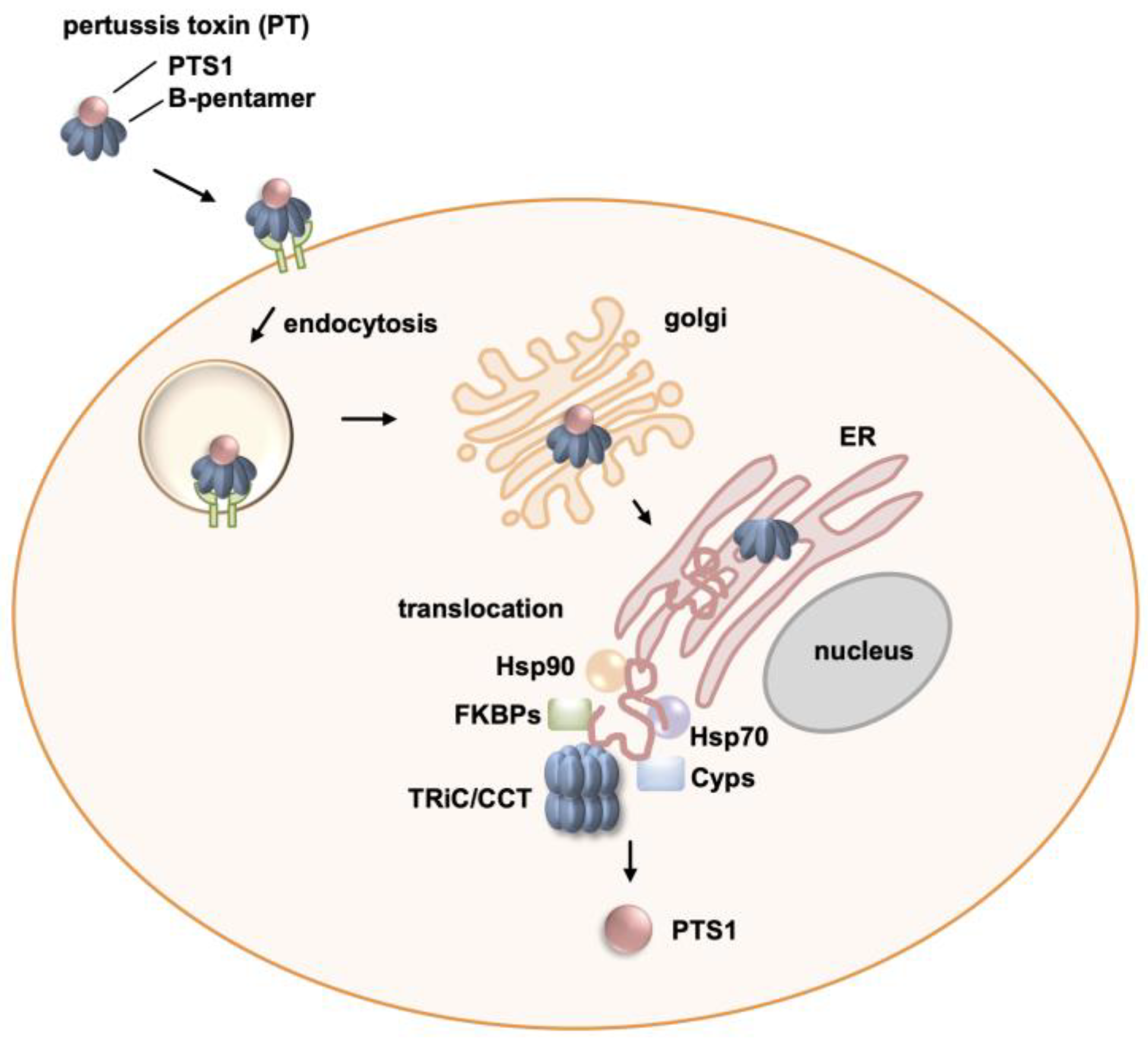
The Chaperonin TRiC/CCT Inhibitor HSF1A Protects Cells from Intoxication with Pertussis Toxin



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
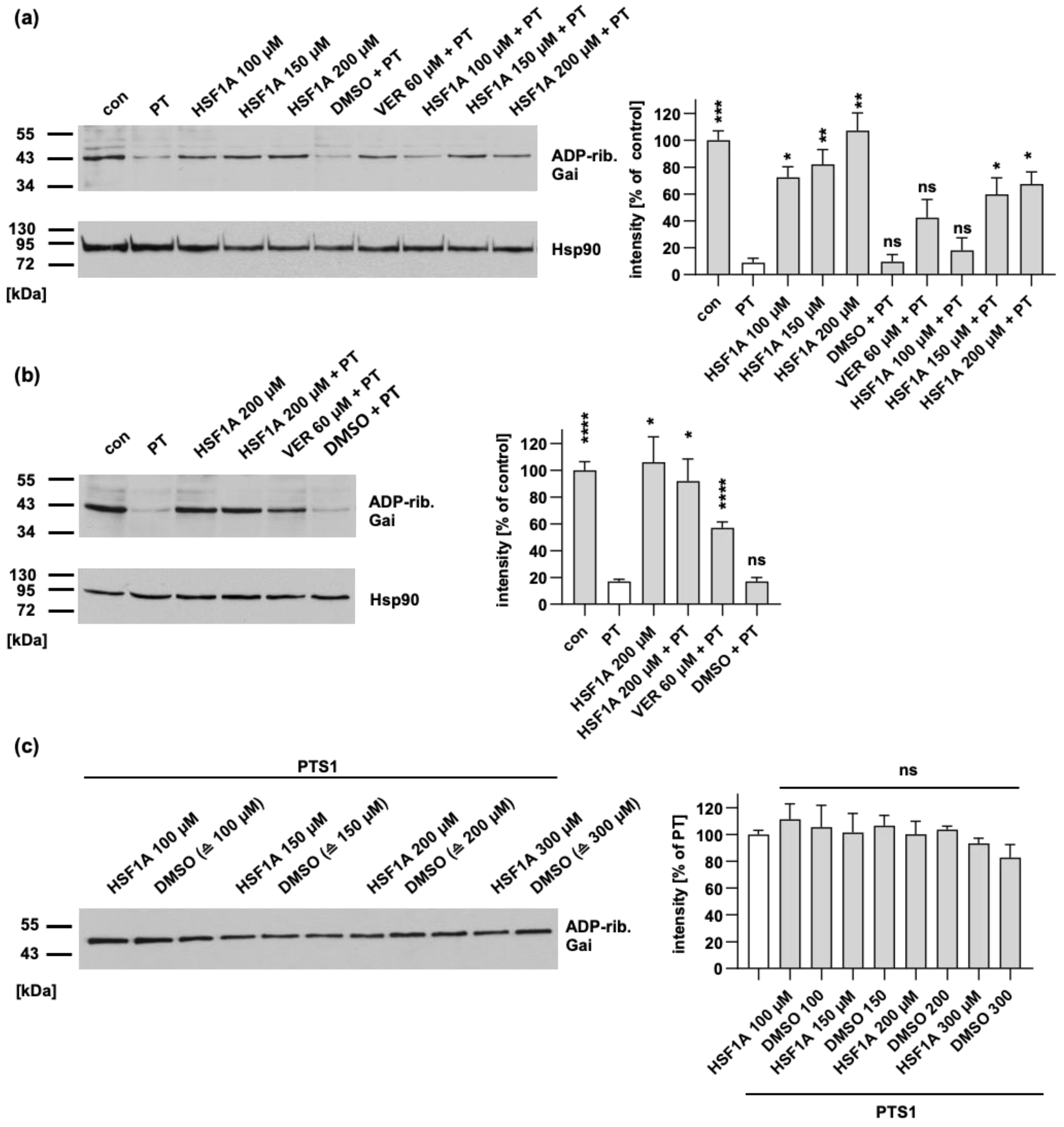
2.1. In PT-Treated Cells, HSF1A Reduces the Levels of ADP-Ribosylated Gαi without Causing Any Interference with the Enzyme Activity In Vitro or the Cellular Binding of PT
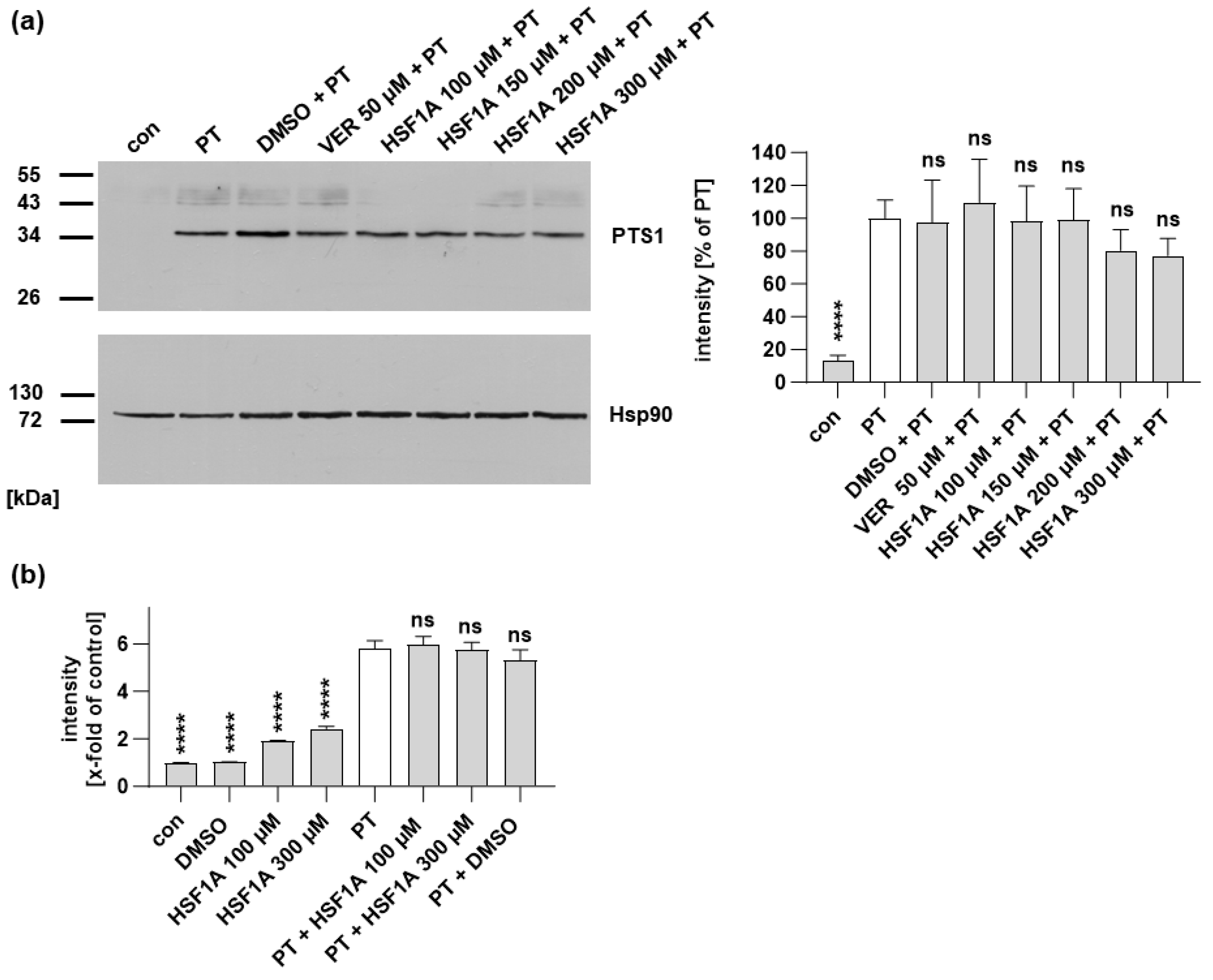
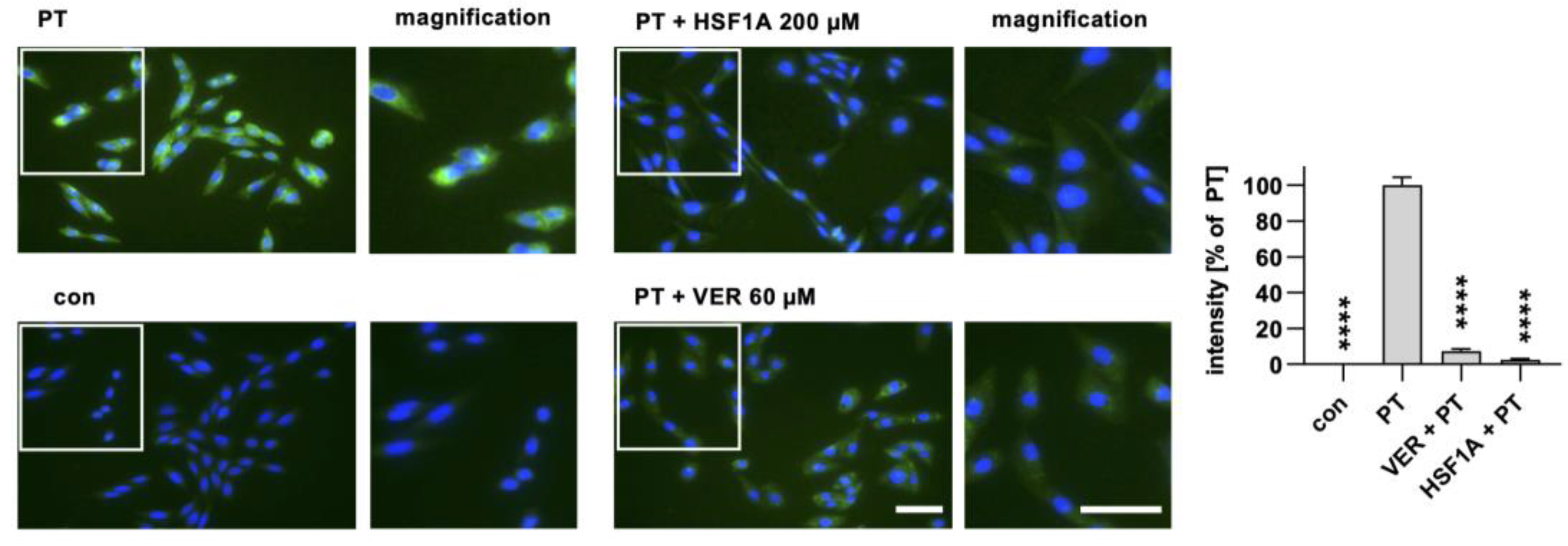
2.2. HSF1A Leads to Reduced Signal of PTS1 in Cells
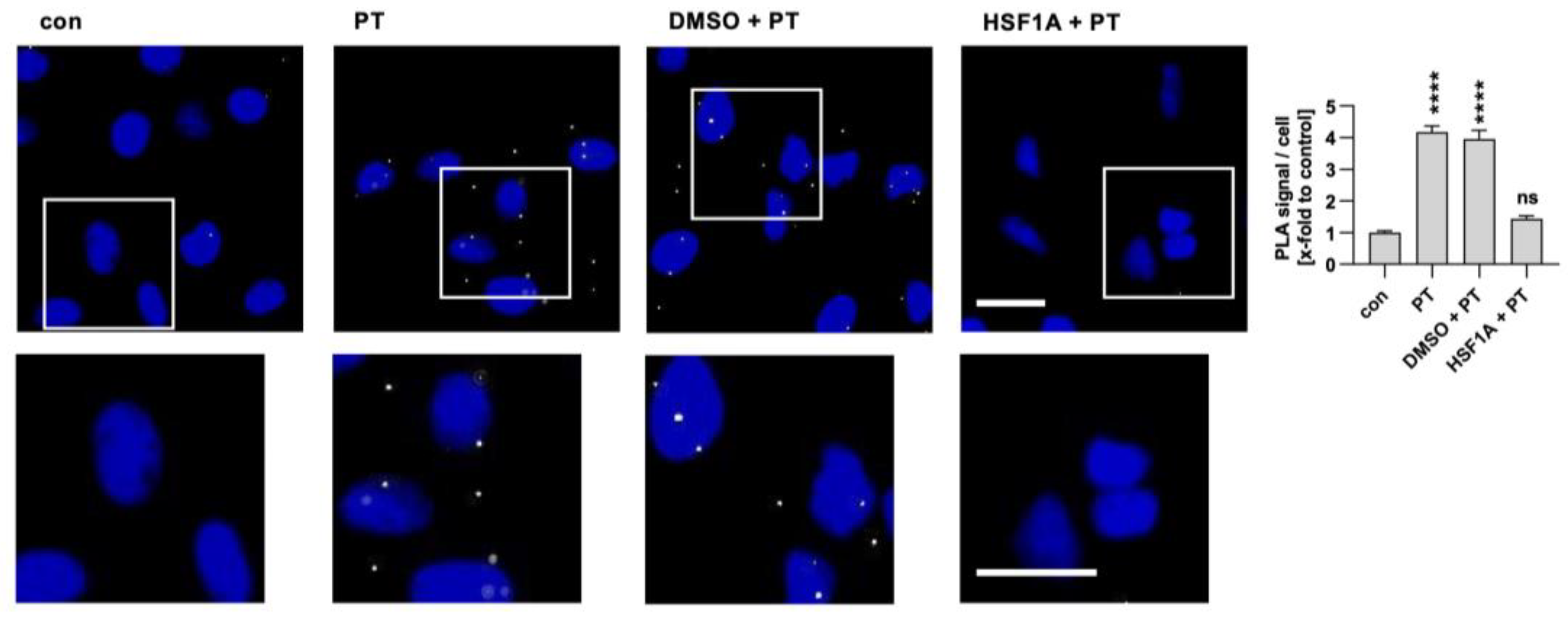
2.3. PTS1 Is Detected in Close Proximity with CCT5 in Cells
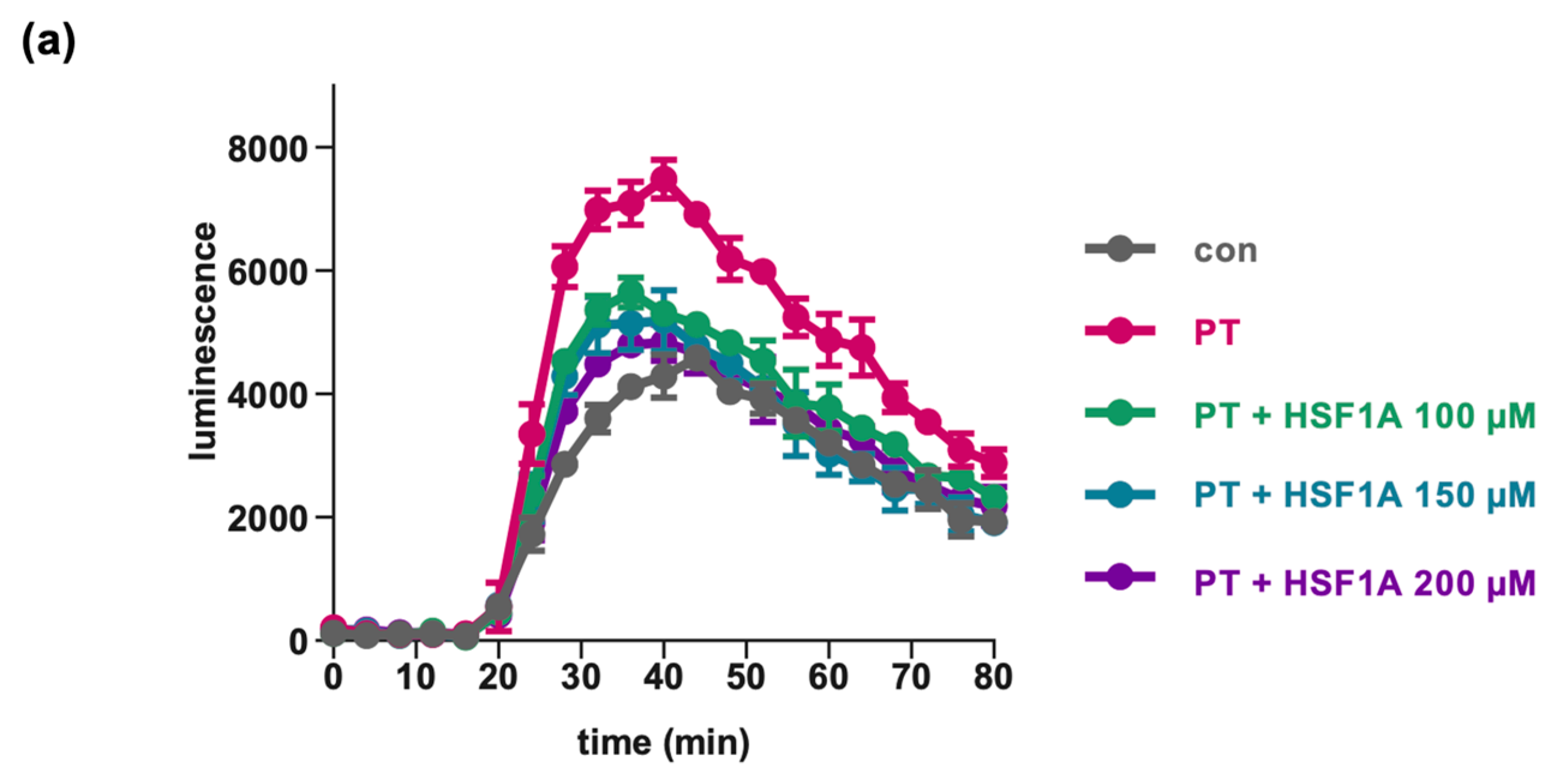
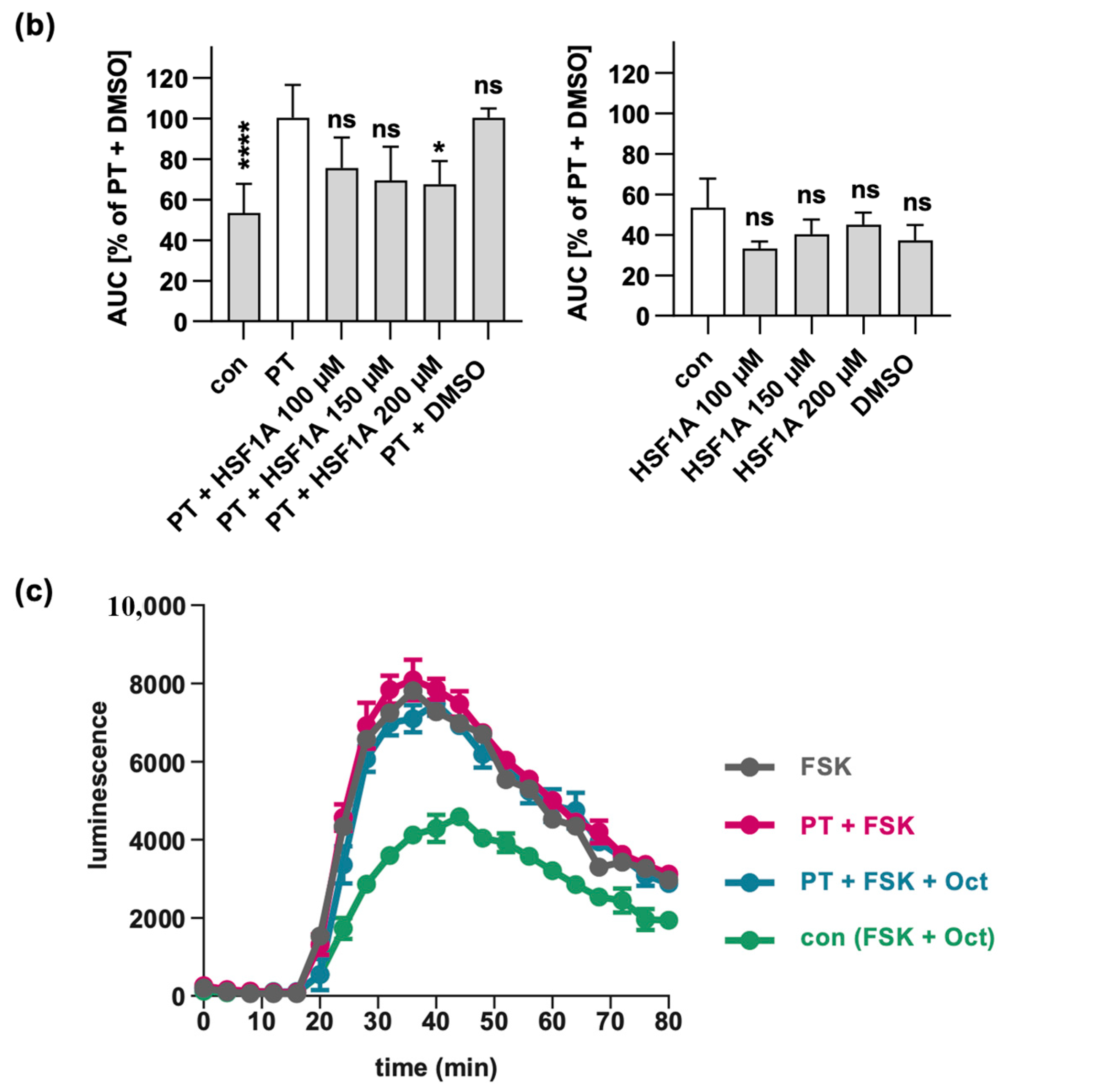
2.4. HSF1A Mitigates the Effect of PT on cAMP Signaling in Cells
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Cell Culture
4.3. Sequential ADP-Ribosylation of Gαi in Lysates from Toxin-treated Cells
4.4. In Vitro Enzyme Activity
4.5. Binding of PT to Cells by Western Blot Analysis and Flow Cytometry
4.6. Immunolabeling and Fluorescence Microscopy
4.7. Proximity Ligation Assay (PLA) for Detection of Protein Interaction in Cells
4.8. iGIST Bioassay
4.9. Reproducibility of Experiments and Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scanlon, K.; Skerry, C.; Carbonetti, N. Association of Pertussis Toxin with Severe Pertussis Disease. Toxins 2019, 11, 373. [Google Scholar] [CrossRef]
- Carbonetti, N.H. Contribution of Pertussis Toxin to the Pathogenesis of Pertussis Disease. Pathog. Dis. 2015, 73, ftv073. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, S.; Cherry, J.D. Molecular Pathogenesis, Epidemiology, and Clinical Manifestations of Respiratory Infections Due to Bordetella Pertussis and Other Bordetella Subspecies. Clin. Microbiol. Rev. 2005, 18, 326–382. [Google Scholar] [CrossRef]
- Paddock, C.D.; Sanden, G.N.; Cherry, J.D.; Gal, A.A.; Langston, C.; Tatti, K.M.; Wu, K.-H.; Goldsmith, C.S.; Greer, P.W.; Montague, J.L.; et al. Pathology and Pathogenesis of Fatal Bordetella Pertussis Infection in Infants. Clin. Infect. Dis. 2008, 47, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Surridge, J.; Segedin, E.R.; Grant, C.C. Pertussis Requiring Intensive Care. Arch. Dis. Child. 2007, 92, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, P.E.; Salim, A.M.; Zervos, M.J.; Schmitt, H.-J. Pertussis: Microbiology, Disease, Treatment, and Prevention. Clin. Microbiol. Rev. 2016, 29, 449–486. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, L.I.; Papin, J.F.; Warfel, J.; Wolf, R.F.; Kosanke, S.D.; Merkel, T.J. Histopathology of Bordetella Pertussis in the Baboon Model. Infect. Immun. 2018, 86, e00511-18. [Google Scholar] [CrossRef]
- WHO Immunization Coverage. Available online: https://www.who.int/news-room/fact-sheets/detail/immunization-coverage (accessed on 15 June 2023).
- Yeung, K.H.T.; Duclos, P.; Nelson, E.A.S.; Hutubessy, R.C.W. An Update of the Global Burden of Pertussis in Children Younger than 5 Years: A Modelling Study. Lancet Infect. Dis. 2017, 17, 974–980. [Google Scholar] [CrossRef]
- Locht, C.; Antoine, R. The History of Pertussis Toxin. Toxins 2021, 13, 623. [Google Scholar] [CrossRef]
- Domenech de Cellès, M.; Magpantay, F.M.G.; King, A.A.; Rohani, P. The Pertussis Enigma: Reconciling Epidemiology, Immunology and Evolution. Proc. Biol. Sci. 2016, 283, 20152309. [Google Scholar] [CrossRef]
- Althouse, B.M.; Scarpino, S.V. Asymptomatic Transmission and the Resurgence of Bordetella Pertussis. BMC Med. 2015, 13, 146. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Stefanelli, P.; Fry, N.K.; Fedele, G.; He, Q.; Paterson, P.; Tan, T.; Knuf, M.; Rodrigo, C.; Weil Olivier, C.; et al. Pertussis Prevention: Reasons for Resurgence, and Differences in the Current Acellular Pertussis Vaccines. Front. Immunol. 2019, 10, 1344. [Google Scholar] [CrossRef] [PubMed]
- Carbonetti, N.H. Pertussis Leukocytosis: Mechanisms, Clinical Relevance and Treatment. Pathog. Dis. 2016, 74, ftw087. [Google Scholar] [CrossRef]
- Cherry, J.D. Treatment of Pertussis-2017. J. Pediatr. Infect. Dis. Soc. 2017, 7, e123–e125. [Google Scholar] [CrossRef]
- Altunaiji, S.; Kukuruzovic, R.; Curtis, N.; Massie, J. Antibiotics for Whooping Cough (Pertussis). Cochrane Database Syst. Rev. 2007, 18, CD004404. [Google Scholar] [CrossRef]
- Connelly, C.E.; Sun, Y.; Carbonetti, N.H. Pertussis Toxin Exacerbates and Prolongs Airway Inflammatory Responses during Bordetella Pertussis Infection. Infect. Immun. 2012, 80, 4317–4332. [Google Scholar] [CrossRef] [PubMed]
- Pierce, C.; Klein, N.; Peters, M. Is Leukocytosis a Predictor of Mortality in Severe Pertussis Infection? Intensive Care Med. 2000, 26, 1512–1514. [Google Scholar] [CrossRef] [PubMed]
- Belcher, T.; Dubois, V.; Rivera-Millot, A.; Locht, C.; Jacob-Dubuisson, F. Pathogenicity and Virulence of Bordetella Pertussis and Its Adaptation to Its Strictly Human Host. Virulence 2021, 12, 2608–2632. [Google Scholar] [CrossRef]
- Ernst, K. Novel Strategies to Inhibit Pertussis Toxin. Toxins 2022, 14, 187. [Google Scholar] [CrossRef]
- Stein, P.E.; Boodhoo, A.; Armstrong, G.D.; Cockle, S.A.; Klein, M.H.; Read, R.J. The Crystal Structure of Pertussis Toxin. Structure 1994, 2, 45–57. [Google Scholar] [CrossRef]
- Tamura, M.; Nogimori, K.; Murai, S.; Yajima, M.; Ito, K.; Katada, T.; Ui, M.; Ishii, S. Subunit Structure of Islet-Activating Protein, Pertussis Toxin, in Conformity with the A-B Model. Biochemistry 1982, 21, 5516–5522. [Google Scholar] [CrossRef]
- Weiss, A.A.; Johnson, F.D.; Burns, D.L. Molecular Characterization of an Operon Required for Pertussis Toxin Secretion. Proc. Natl. Acad. Sci. USA 1993, 90, 2970–2974. [Google Scholar] [CrossRef] [PubMed]
- Pittman, M. The Concept of Pertussis as a Toxin-Mediated Disease. Pediatr. Infect. Dis. 1984, 3, 467–486. [Google Scholar] [CrossRef]
- Witvliet, M.H.; Burns, D.L.; Brennan, M.J.; Poolman, J.T.; Manclark, C.R. Binding of Pertussis Toxin to Eucaryotic Cells and Glycoproteins. Infect. Immun. 1989, 57, 3324–3330. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.D.; Howard, L.A.; Peppler, M.S. Use of Glycosyltransferases to Restore Pertussis Toxin Receptor Activity to Asialoagalactofetuin. J. Biol. Chem. 1988, 263, 8677–8684. [Google Scholar] [CrossRef] [PubMed]
- Hausman, S.Z.; Burns, D.L. Binding of Pertussis Toxin to Lipid Vesicles Containing Glycolipids. Infect. Immun. 1993, 61, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Plaut, R.D.; Carbonetti, N.H. Retrograde Transport of Pertussis Toxin in the Mammalian Cell. Cell. Microbiol. 2008, 10, 1130–1139. [Google Scholar] [CrossRef]
- el Bayâ, A.; Linnemann, R.; von Olleschik-Elbheim, L.; Robenek, H.; Schmidt, M.A. Endocytosis and Retrograde Transport of Pertussis Toxin to the Golgi Complex as a Prerequisite for Cellular Intoxication. Eur. J. Cell Biol. 1997, 73, 40–48. [Google Scholar]
- Burns, D.L.; Manclark, C.R. Adenine Nucleotides Promote Dissociation of Pertussis Toxin Subunits. J. Biol. Chem. 1986, 261, 4324–4327. [Google Scholar] [CrossRef]
- Hazes, B.; Boodhoo, A.; Cockle, S.A.; Read, R.J. Crystal Structure of the Pertussis Toxin-ATP Complex: A Molecular Sensor. J. Mol. Biol. 1996, 258, 661–671. [Google Scholar] [CrossRef]
- Plaut, R.D.; Scanlon, K.M.; Taylor, M.; Teter, K.; Carbonetti, N.H. Intracellular Disassembly and Activity of Pertussis Toxin Require Interaction with ATP. Pathog. Dis. 2016, 74, ftw065. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Cilenti, L.; Taylor, M.; Showman, A.; Tatulian, S.A.; Teter, K. Thermal Unfolding of the Pertussis Toxin S1 Subunit Facilitates Toxin Translocation to the Cytosol by the Mechanism of Endoplasmic Reticulum-Associated Degradation. Infect. Immun. 2016, 84, 3388–3398. [Google Scholar] [CrossRef]
- Pande, A.H.; Moe, D.; Jamnadas, M.; Tatulian, S.A.; Teter, K. The Pertussis Toxin S1 Subunit Is a Thermally Unstable Protein Susceptible to Degradation by the 20S Proteasome. Biochemistry 2006, 45, 13734–13740. [Google Scholar] [CrossRef]
- Worthington, Z.E.V.; Carbonetti, N.H. Evading the Proteasome: Absence of Lysine Residues Contributes to Pertussis Toxin Activity by Evasion of Proteasome Degradation. Infect. Immun. 2007, 75, 2946–2953. [Google Scholar] [CrossRef] [PubMed]
- Hazes, B.; Read, R.J. Accumulating Evidence Suggests That Several AB-Toxins Subvert the Endoplasmic Reticulum-Associated Protein Degradation Pathway to Enter Target Cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, G.M.; Katada, T.; Northup, J.K.; Hewlett, E.L.; Gilman, A.G. Identification of the Predominant Substrate for ADP-Ribosylation by Islet Activating Protein. J. Biol. Chem. 1983, 258, 2072–2075. [Google Scholar] [CrossRef]
- Katada, T.; Ui, M. Direct Modification of the Membrane Adenylate Cyclase System by Islet-Activating Protein Due to ADP-Ribosylation of a Membrane Protein. Proc. Natl. Acad. Sci. USA 1982, 79, 3129–3133. [Google Scholar] [CrossRef]
- Sakari, M.; Tran, M.T.; Rossjohn, J.; Pulliainen, A.T.; Beddoe, T.; Littler, D.R. Crystal Structures of Pertussis Toxin with NAD+ and Analogs Provide Structural Insights into the Mechanism of Its Cytosolic ADP-Ribosylation Activity. J. Biol. Chem. 2022, 298, 101892. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In Vivo Aspects of Protein Folding and Quality Control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Lopez, T.; Dalton, K.; Frydman, J. The Mechanism and Function of Group II Chaperonins. J. Mol. Biol. 2015, 427, 2919–2930. [Google Scholar] [CrossRef]
- Yam, A.Y.; Xia, Y.; Lin, H.-T.J.; Burlingame, A.; Gerstein, M.; Frydman, J. Defining the TRiC/CCT Interactome Links Chaperonin Function to Stabilization of Newly Made Proteins with Complex Topologies. Nat. Struct. Mol. Biol. 2008, 15, 1255–1262. [Google Scholar] [CrossRef]
- Leroux, M.R.; Hartl, F.U. Protein Folding: Versatility of the Cytosolic Chaperonin TRiC/CCT. Curr. Biol. 2000, 10, R260–R264. [Google Scholar] [CrossRef]
- Stemp, M.J.; Guha, S.; Hartl, F.U.; Barral, J.M. Efficient Production of Native Actin upon Translation in a Bacterial Lysate Supplemented with the Eukaryotic Chaperonin TRiC. Biol. Chem. 2005, 386, 753–757. [Google Scholar] [CrossRef]
- Chen, X.; Sullivan, D.S.; Huffaker, T.C. Two Yeast Genes with Similarity to TCP-1 Are Required for Microtubule and Actin Function in Vivo. Proc. Natl. Acad. Sci. USA 1994, 91, 9111–9115. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, M.B.; Farr, G.W.; Miklos, D.; Horwich, A.L.; Sternlicht, M.L.; Sternlicht, H. TCP1 Complex Is a Molecular Chaperone in Tubulin Biogenesis. Nature 1992, 358, 245–248. [Google Scholar] [CrossRef]
- Steinemann, M.; Schlosser, A.; Jank, T.; Aktories, K. The Chaperonin TRiC/CCT Is Essential for the Action of Bacterial Glycosylating Protein Toxins like Clostridium Difficile Toxins A and B. Proc. Natl. Acad. Sci. USA 2018, 115, 9580–9585. [Google Scholar] [CrossRef] [PubMed]
- Neef, D.W.; Jaeger, A.M.; Gomez-Pastor, R.; Willmund, F.; Frydman, J.; Thiele, D.J. A Direct Regulatory Interaction between Chaperonin TRiC and Stress-Responsive Transcription Factor HSF1. Cell Rep. 2014, 9, 955–966. [Google Scholar] [CrossRef]
- Neef, D.W.; Turski, M.L.; Thiele, D.J. Modulation of Heat Shock Transcription Factor 1 as a Therapeutic Target for Small Molecule Intervention in Neurodegenerative Disease. PLoS Biol. 2010, 8, e1000291. [Google Scholar] [CrossRef]
- Ernst, K. Requirement of Peptidyl-Prolyl Cis/Trans Isomerases and Chaperones for Cellular Uptake of Bacterial AB-Type Toxins. Front. Cell Infect. Microbiol. 2022, 12, 938015. [Google Scholar] [CrossRef]
- Ernst, K.; Eberhardt, N.; Mittler, A.-K.; Sonnabend, M.; Anastasia, A.; Freisinger, S.; Schiene-Fischer, C.; Malešević, M.; Barth, H. Pharmacological Cyclophilin Inhibitors Prevent Intoxication of Mammalian Cells with Bordetella Pertussis Toxin. Toxins 2018, 10, 181. [Google Scholar] [CrossRef] [PubMed]
- Ernst, K.; Mittler, A.-K.; Winkelmann, V.; Kling, C.; Eberhardt, N.; Anastasia, A.; Sonnabend, M.; Lochbaum, R.; Wirsching, J.; Sakari, M.; et al. Pharmacological Targeting of Host Chaperones Protects from Pertussis Toxin in Vitro and in Vivo. Sci. Rep. 2021, 11, 5429. [Google Scholar] [CrossRef]
- Kellner, A.; Taylor, M.; Banerjee, T.; Britt, C.B.T.; Teter, K. A Binding Motif for Hsp90 in the A Chains of ADP-Ribosylating Toxins That Move from the Endoplasmic Reticulum to the Cytosol. Cell. Microbiol. 2019, 21, e13074. [Google Scholar] [CrossRef] [PubMed]
- Kellner, A.; Cherubin, P.; Harper, J.K.; Teter, K. Proline Isomerization as a Key Determinant for Hsp90-Toxin Interactions. Front. Cell Infect. Microbiol. 2021, 11, 771653. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.C.; Guerrant, R.L.; Hewlett, E.L. The CHO Cell Clustering Response to Pertussis Toxin: History of Its Discovery and Recent Developments in Its Use. Toxins 2021, 13, 815. [Google Scholar] [CrossRef]
- Williamson, D.S.; Borgognoni, J.; Clay, A.; Daniels, Z.; Dokurno, P.; Drysdale, M.J.; Foloppe, N.; Francis, G.L.; Graham, C.J.; Howes, R.; et al. Novel Adenosine-Derived Inhibitors of 70 kDa Heat Shock Protein, Discovered through Structure-Based Design. J. Med. Chem. 2009, 52, 1510–1513. [Google Scholar] [CrossRef]
- Jia, J.; Braune-Yan, M.; Lietz, S.; Wahba, M.; Pulliainen, A.T.; Barth, H.; Ernst, K. Domperidone Inhibits Clostridium Botulinum C2 Toxin and Bordetella Pertussis Toxin. Toxins 2023, 15, 412. [Google Scholar] [CrossRef]
- Katada, T. The Inhibitory G Protein G(i) Identified as Pertussis Toxin-Catalyzed ADP-Ribosylation. Biol. Pharm. Bull. 2012, 35, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Paramonov, V.M.; Sahlgren, C.; Rivero-Müller, A.; Pulliainen, A.T. iGIST-A Kinetic Bioassay for Pertussis Toxin Based on Its Effect on Inhibitory GPCR Signaling. ACS Sens. 2020, 5, 3438–3448. [Google Scholar] [CrossRef]
- Teter, K. Intracellular Trafficking and Translocation of Pertussis Toxin. Toxins 2019, 11, 437. [Google Scholar] [CrossRef]
- Aktories, K.; Schwan, C.; Jank, T. Clostridium Difficile Toxin Biology. Annu. Rev. Microbiol. 2017, 71, 281–307. [Google Scholar] [CrossRef]
- Concilli, M.; Petruzzelli, R.; Parisi, S.; Catalano, F.; Sirci, F.; Napolitano, F.; Renda, M.; Galietta, L.J.V.; Di Bernardo, D.; Polishchuk, R.S. Pharmacoproteomics Pinpoints HSP70 Interaction for Correction of the Most Frequent Wilson Disease-Causing Mutant of ATP7B. Proc. Natl. Acad. Sci. USA 2020, 117, 32453–32463. [Google Scholar] [CrossRef] [PubMed]
- Burress, H.; Taylor, M.; Banerjee, T.; Tatulian, S.A.; Teter, K. Co- and Post-Translocation Roles for HSP90 in Cholera Intoxication. J. Biol. Chem. 2014, 289, 33644–33654. [Google Scholar] [CrossRef] [PubMed]
- Burress, H.; Kellner, A.; Guyette, J.; Tatulian, S.A.; Teter, K. HSC70 and HSP90 Chaperones Perform Complementary Roles in Translocation of the Cholera Toxin A1 Subunit from the Endoplasmic Reticulum to the Cytosol. J. Biol. Chem. 2019, 294, 12122–12131. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Navarro-Garcia, F.; Huerta, J.; Burress, H.; Massey, S.; Ireton, K.; Teter, K. Hsp90 Is Required for Transfer of the Cholera Toxin A1 Subunit from the Endoplasmic Reticulum to the Cytosol. J. Biol. Chem. 2010, 285, 31261–31267. [Google Scholar] [CrossRef]
- Haug, G.; Leemhuis, J.; Tiemann, D.; Meyer, D.K.; Aktories, K.; Barth, H. The Host Cell Chaperone Hsp90 Is Essential for Translocation of the Binary Clostridium Botulinum C2 Toxin into the Cytosol. J. Biol. Chem. 2003, 278, 32266–32274. [Google Scholar] [CrossRef]
- Kaiser, E.; Pust, S.; Kroll, C.; Barth, H. Cyclophilin A Facilitates Translocation of the Clostridium Botulinum C2 Toxin across Membranes of Acidified Endosomes into the Cytosol of Mammalian Cells. Cell. Microbiol. 2009, 11, 780–795. [Google Scholar] [CrossRef]
- Kaiser, E.; Böhm, N.; Ernst, K.; Langer, S.; Schwan, C.; Aktories, K.; Popoff, M.; Fischer, G.; Barth, H. FK506-Binding Protein 51 Interacts with Clostridium Botulinum C2 Toxin and FK506 Inhibits Membrane Translocation of the Toxin in Mammalian Cells. Cell. Microbiol. 2012, 14, 1193–1205. [Google Scholar] [CrossRef]
- Ernst, K.; Langer, S.; Kaiser, E.; Osseforth, C.; Michaelis, J.; Popoff, M.R.; Schwan, C.; Aktories, K.; Kahlert, V.; Malesevic, M.; et al. Cyclophilin-Facilitated Membrane Translocation as Pharmacological Target to Prevent Intoxication of Mammalian Cells by Binary Clostridial Actin ADP-Ribosylated Toxins. J. Mol. Biol. 2015, 427, 1224–1238. [Google Scholar] [CrossRef]
- Kaiser, E.; Kroll, C.; Ernst, K.; Schwan, C.; Popoff, M.; Fischer, G.; Buchner, J.; Aktories, K.; Barth, H. Membrane Translocation of Binary Actin-ADP-Ribosylating Toxins from Clostridium Difficile and Clostridium Perfringens Is Facilitated by Cyclophilin A and Hsp90. Infect. Immun. 2011, 79, 3913–3921. [Google Scholar] [CrossRef]
- Haug, G.; Wilde, C.; Leemhuis, J.; Meyer, D.K.; Aktories, K.; Barth, H. Cellular Uptake of Clostridium Botulinum C2 Toxin: Membrane Translocation of a Fusion Toxin Requires Unfolding of Its Dihydrofolate Reductase Domain. Biochemistry 2003, 42, 15284–15291. [Google Scholar] [CrossRef]
- Dmochewitz, L.; Lillich, M.; Kaiser, E.; Jennings, L.D.; Lang, A.E.; Buchner, J.; Fischer, G.; Aktories, K.; Collier, R.J.; Barth, H. Role of CypA and Hsp90 in Membrane Translocation Mediated by Anthrax Protective Antigen. Cell. Microbiol. 2011, 13, 359–373. [Google Scholar] [CrossRef]
- Biebl, M.M.; Buchner, J. Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb. Perspect. Biol. 2019, 11, a034017. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Toft, D.O. Steroid Receptor Interactions with Heat Shock Protein and Immunophilin Chaperones. Endocr. Rev. 1997, 18, 306–360. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Toft, D.O. Regulation of Signaling Protein Function and Trafficking by the Hsp90/Hsp70-Based Chaperone Machinery. Exp. Biol. Med. 2003, 228, 111–133. [Google Scholar] [CrossRef] [PubMed]
- Ernst, K.; Sailer, J.; Braune, M.; Barth, H. Intoxication of Mammalian Cells with Binary Clostridial Enterotoxins Is Inhibited by the Combination of Pharmacological Chaperone Inhibitors. Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 941–954. [Google Scholar] [CrossRef]
- Ernst, K.; Kling, C.; Landenberger, M.; Barth, H. Combined Pharmacological Inhibition of Cyclophilins, FK506-Binding Proteins, Hsp90, and Hsp70 Protects Cells From Clostridium Botulinum C2 Toxin. Front. Pharmacol. 2018, 9, 1287. [Google Scholar] [CrossRef]
- Azarnia Tehran, D.; Pirazzini, M.; Leka, O.; Mattarei, A.; Lista, F.; Binz, T.; Rossetto, O.; Montecucco, C. Hsp90 Is Involved in the Entry of Clostridial Neurotoxins into the Cytosol of Nerve Terminals. Cell. Microbiol. 2016, 19, e12647. [Google Scholar] [CrossRef]
- Pirazzini, M.; Azarnia Tehran, D.; Zanetti, G.; Rossetto, O.; Montecucco, C. Hsp90 and Thioredoxin-Thioredoxin Reductase Enable the Catalytic Activity of Clostridial Neurotoxins inside Nerve Terminals. Toxicon 2018, 147, 32–37. [Google Scholar] [CrossRef]
- Ashok, Y.; Miettinen, M.; de Oliveira, D.K.H.; Tamirat, M.Z.; Näreoja, K.; Tiwari, A.; Hottiger, M.O.; Johnson, M.S.; Lehtiö, L.; Pulliainen, A.T. Discovery of Compounds Inhibiting the ADP-Ribosyltransferase Activity of Pertussis Toxin. ACS Infect. Dis. 2020, 6, 588–602. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, J.; Zoeschg, M.; Barth, H.; Pulliainen, A.T.; Ernst, K. The Chaperonin TRiC/CCT Inhibitor HSF1A Protects Cells from Intoxication with Pertussis Toxin. Toxins 2024, 16, 36. https://doi.org/10.3390/toxins16010036
Jia J, Zoeschg M, Barth H, Pulliainen AT, Ernst K. The Chaperonin TRiC/CCT Inhibitor HSF1A Protects Cells from Intoxication with Pertussis Toxin. Toxins. 2024; 16(1):36. https://doi.org/10.3390/toxins16010036
Chicago/Turabian StyleJia, Jinfang, Manuel Zoeschg, Holger Barth, Arto T. Pulliainen, and Katharina Ernst. 2024. "The Chaperonin TRiC/CCT Inhibitor HSF1A Protects Cells from Intoxication with Pertussis Toxin" Toxins 16, no. 1: 36. https://doi.org/10.3390/toxins16010036
APA StyleJia, J., Zoeschg, M., Barth, H., Pulliainen, A. T., & Ernst, K. (2024). The Chaperonin TRiC/CCT Inhibitor HSF1A Protects Cells from Intoxication with Pertussis Toxin. Toxins, 16(1), 36. https://doi.org/10.3390/toxins16010036