Advances in Deoxynivalenol Toxicity Mechanisms: The Brain as a Target

{kind=link}
{kind=link}
Abstract
:1. Introduction
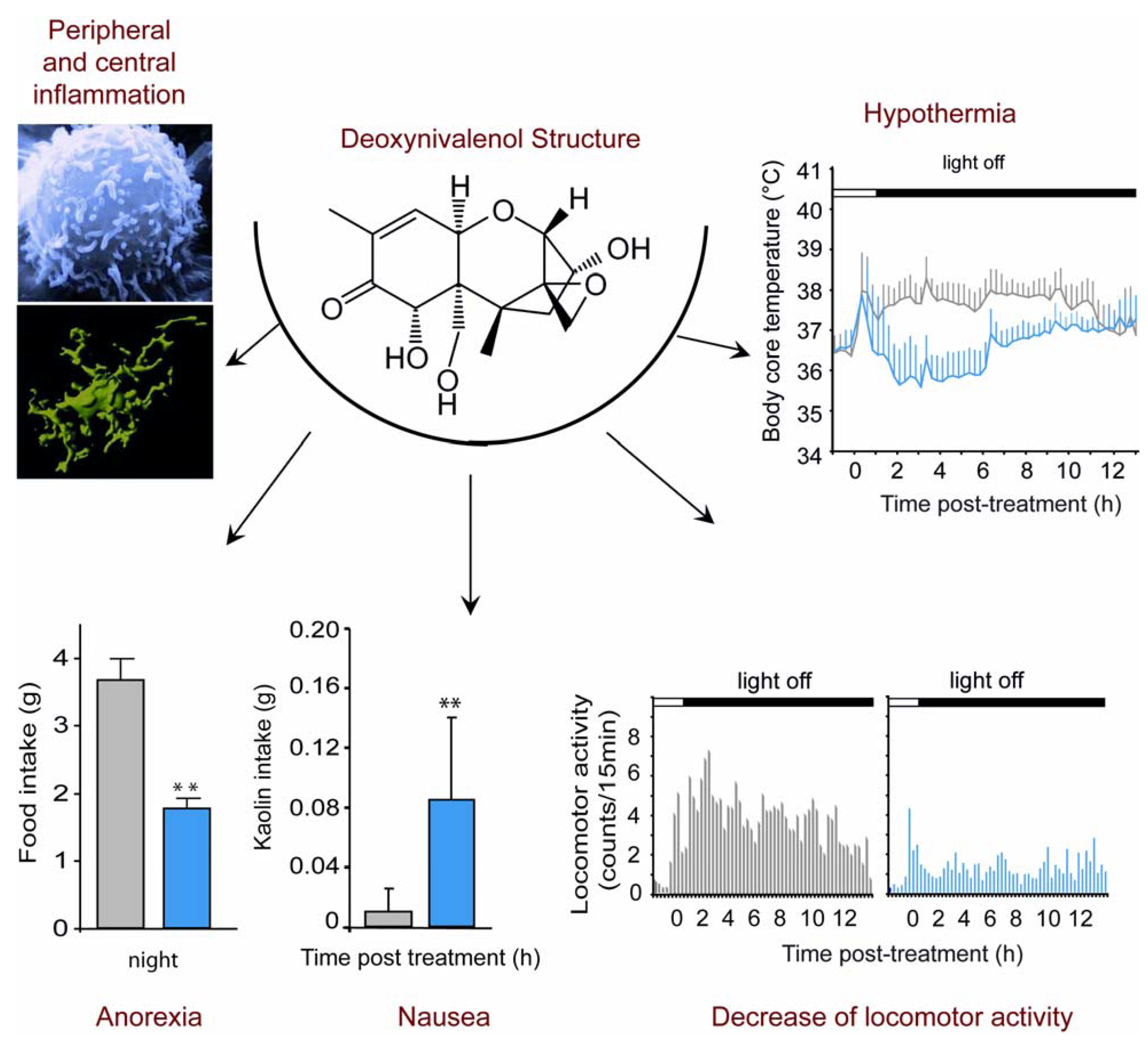
2. DON, Food Refusal, Emesis and Anorexia

3. DON-Induced Anorexia in Mice Models
4. Sickness Behavior and Central Cytokines Expression
5. DON, Cytokines Expression and Anorexia

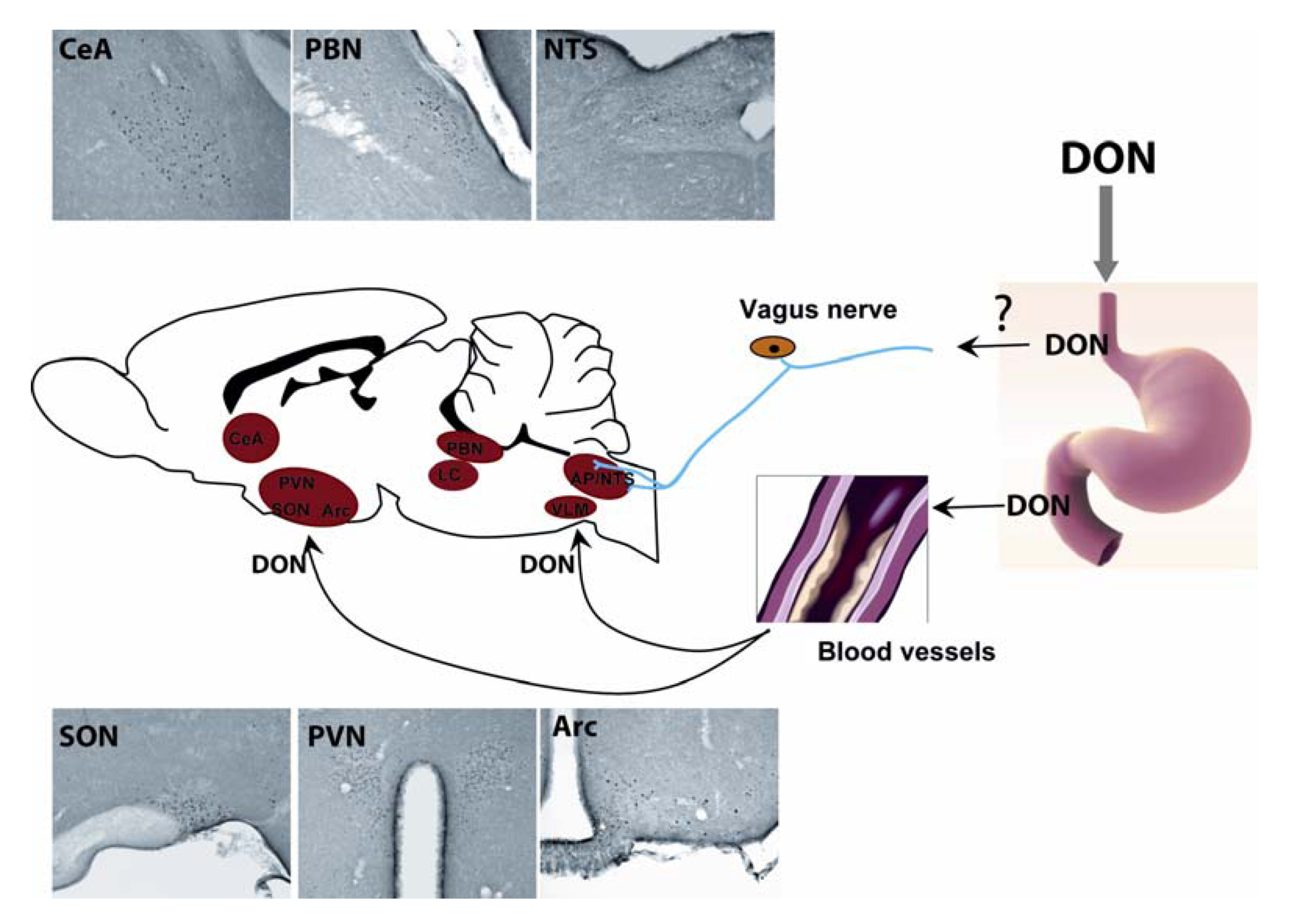
6. Brain Structures Activated during Acute DON Intoxication
7. Brain Neurochemistry and DON
8. Feeding Circuits and DON
9. DON May Target the Brain
10. Perspectives
11. Conclusions
Acknowledgment
Conflict of Interest
References
- Ueno, Y. Mode of action of trichotecenes. Ann. Nutr. Aliment. 1977, 31, 885–900. [Google Scholar]
- Lombaert, G.A.; Pellaers, P.; Roscoe, V.; Mankotia, M.; Neil, R.; Scott, P.M. Mycotoxins in infant cereal foods from the Canadian retail market. Food Addit. Contam. 2003, 20, 494–504. [Google Scholar] [CrossRef]
- Schothorst, R.C.; van Egmond, H.P. Report from SCOOP task 3.2.10 “collection of occurrence data of Fusarium toxins in food and assessment of dietary intake by the population of EU member states”. Subtask: Trichothecenes. Toxicol. Lett. 2004, 153, 133–143. [Google Scholar] [CrossRef]
- Turner, P.C.; White, K.L.; Burley, V.J.; Hopton, R.P.; Rajendram, A.; Fisher, J.; Cade, J.E.; Wild, C.P. A comparison of deoxynivalenol intake and urinary deoxynivalenol in UK adults. Biomarkers 2010, 15, 553–562. [Google Scholar] [CrossRef]
- Turner, P.C.; Rothwell, J.A.; White, K.L.; Gong, Y.; Cade, J.E.; Wild, C.P. Urinary deoxynivalenol is correlated with cereal intake in individuals from the United kingdom. Environ. Health Perspect. 2008, 116, 21–25. [Google Scholar]
- Vesonder, R.F.; Ciegler, A.; Jensen, A.H. Isolation of the emetic principle from Fusarium-infected corn. Appl. Microbiol. 1973, 26, 1008–1010. [Google Scholar]
- Pestka, J.J. Deoxynivalenol: Mechanisms of action, human exposure, and toxicological relevance. Arch. Toxicol. 2010, 84, 663–679. [Google Scholar] [CrossRef]
- Pestka, J.J.; Smolinski, A.T. Deoxynivalenol: Toxicology and potential effects on humans. J. Toxicol. Environ. Health B 2005, 8, 39–69. [Google Scholar]
- Forsyth, D.M.; Yoshizawa, T.; Morooka, N.; Tuite, J. Emetic and refusal activity of deoxynivalenol to swine. Appl. Environ. Microbiol. 1977, 34, 547–552. [Google Scholar]
- Pestka, J.J.; Lin, W.S.; Miller, E.R. Emetic activity of the trichothecene 15-acetyldeoxynivalenol in swine. Food Chem. Toxicol. 1987, 25, 855–858. [Google Scholar]
- Prelusky, D.B.; Trenholm, H.L. The efficacy of various classes of anti-emetics in preventing deoxynivalenol-induced vomiting in swine. Nat. Toxins 1993, 1, 296–302. [Google Scholar] [CrossRef]
- Young, L.G.; McGirr, L.; Valli, V.E.; Lumsden, J.H.; Lun, A. Vomitoxin in corn fed to young pigs. J. Anim. Sci. 1983, 57, 655–664. [Google Scholar]
- Abbas, H.K.; Mirocha, C.J.; Tuite, J. Natural occurrence of deoxynivalenol, 15-acetyl-deoxynivalenol, and zearalenone in refusal factor corn stored since 1972. Appl. Environ. Microbiol. 1986, 51, 841–843. [Google Scholar]
- Trenholm, H.L.; Hamilton, R.M.; Friend, D.W.; Thompson, B.K.; Hartin, K.E. Feeding trials with vomitoxin (deoxynivalenol)-contaminated wheat, effects on swine, poultry, and dairy cattle. J. Am. Vet. Med. Assoc. 1984, 185, 527–531. [Google Scholar]
- Forsell, J.H.; Witt, M.F.; Tai, J.H.; Jensen, R.; Pestka, J.J. Effects of 8-week exposure of the B6C3F1 mouse to dietary deoxynivalenol (vomitoxin) and zearalenone. Food Chem. Toxicol. 1986, 24, 213–219. [Google Scholar]
- Robbana-Barnat, S.; Loridon-Rosa, B.; Cohen, H.; Lafarge-Frayssinet, C.; Neish, G.A.; Frayssinet, C. Protein synthesis inhibition and cardiac lesions associated with deoxynivalenol ingestion in mice. Food Addit. Contam. 1987, 4, 49–56. [Google Scholar] [CrossRef]
- Arnold, D.L.; Karpinski, K.F.; McGuire, P.F.; Nera, E.A.; Zawidzka, Z.Z.; Lok, E.; Campbell, J.S.; Tryphonas, L.; Scott, P.M. A short-term feeding study with deoxynivalenol (vomitoxin) using rats. Fundam. Appl. Toxicol. 1986, 6, 691–696. [Google Scholar] [CrossRef]
- Hughes, D.M.; Gahl, M.J.; Graham, C.H.; Grieb, S.L. Overt signs of toxicity to dogs and cats of dietary deoxynivalenol. J. Anim. Sci. 1999, 77, 693–700. [Google Scholar]
- Amuzie, C.J.; Flannery, B.M.; Ulrich, A.M.; Pestka, J.J. Effects of deoxynivalenol consumption on body weight and adiposity in the diet-induced obese mouse. J. Toxicol. Environ. Health Part A 2011, 74, 658–667. [Google Scholar] [CrossRef]
- Flannery, B.M.; Wu, W.; Pestka, J.J. Characterization of deoxynivalenol-induced anorexia using mouse bioassay. Food Chem. Toxicol. 2011, 49, 1863–1869. [Google Scholar]
- Girardet, C.; Bonnet, M.S.; Jdir, R.; Sadoud, M.; Thirion, S.; Tardivel, C.; Roux, J.; Lebrun, B.; Wanaverbecq, N.; Mounien, L.; et al. The food-contaminant deoxynivalenol modifies eating by targeting anorexigenic neurocircuitry. PLoS One 2011, 6, e26134. [Google Scholar]
- Girardet, C.; Bonnet, M.S.; Jdir, R.; Sadoud, M.; Thirion, S.; Tardivel, C.; Roux, J.; Lebrun, B.; Mounien, L.; Trouslard, J.; et al. Central inflammation and sickness-like behavior induced by the food contaminant deoxynivalenol: A PGE2-independent mechanism. Toxicol. Sci. 2011, 124, 179–191. [Google Scholar] [CrossRef]
- Kobayashi-Hattori, K.; Amuzie, C.J.; Flannery, B.M.; Pestka, J.J. Body composition and hormonal effects following exposure to mycotoxin deoxynivalenol in the high-fat diet-induced obese mouse. Mol. Nutr. Food Res. 2011, 55, 1070–1078. [Google Scholar]
- Porter, M.H.; Arnold, M.; Langhans, W. Lipopolysaccharide-induced anorexia following hepatic portal vein and vena cava administration. Physiol. Behav. 1998, 64, 581–584. [Google Scholar] [CrossRef]
- West, D.B.; Greenwood, M.R.; Marshall, K.A.; Woods, S.C. Lithium chloride, cholecystokinin and meal patterns: Evidence that cholecystokinin suppresses meal size in rats without causing malaise. Appetite 1987, 8, 221–227. [Google Scholar]
- Yamamoto, K.; Matsunaga, S.; Matsui, M.; Takeda, N.; Yamatodani, A. Pica in mice as a new model for the study of emesis. Methods Find. Exp. Clin. Pharmacol. 2002, 24, 135–138. [Google Scholar] [CrossRef]
- Flynn, M.C.; Scott, T.R.; Pritchard, T.C.; Plata-Salamán, C.R. Mode of action of OB protein (leptin) on feeding. Am. J. Physiol. 1998, 275, R174–R179. [Google Scholar]
- Hsiao, S.; Wang, C.H. Continuous infusion of cholecystokinin and meal pattern in the rat. Peptides 1983, 4, 15–17. [Google Scholar] [CrossRef]
- Le Roux, C.W.; Batterham, R.L.; Aylwin, S.J.; Patterson, M.; Borg, C.M.; Wynne, K.J.; Kent, A.; Vincent, R.P.; Gardiner, J.; Ghatei, M.A.; Bloom, S.R. Attenuated peptide YY release in obese subjects is associated with reduced satiety. Endocrinology 2006, 147, 3–8. [Google Scholar] [CrossRef]
- VanderWeele, D.A. Insulin is a prandial satiety hormone. Physiol. Behav. 1994, 56, 619–622. [Google Scholar] [CrossRef]
- Hart, B.L. Biological basis of the behavior of sick animals. Neurosci. Biobehav. Rev. 1988, 12, 123–137. [Google Scholar]
- Langhans, W. Anorexia of infection: Current prospects. Nutrition 2000, 16, 996–1005. [Google Scholar] [CrossRef]
- Goehler, L.E.; Relton, J.K.; Dripps, D.; Kiechle, R.; Tartaglia, N.; Maier, S.F.; Watkins, L.R. Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: A possible mechanism for immune-to-brain communication. Brain Res. Bull. 1997, 43, 357–364. [Google Scholar] [CrossRef]
- Konsman, J.P.; Parnet, P.; Dantzer, R. Cytokine-induced sickness behaviour: Mechanisms and implications. Trends Neurosci. 2002, 25, 154–159. [Google Scholar] [CrossRef]
- Chakravarty, S.; Herkenham, M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J. Neurosci. 2005, 25, 1788–1796. [Google Scholar]
- Wisse, B.E.; Ogimoto, K.; Morton, G.J.; Williams, D.L.; Schwartz, M.W. Central interleukin-1 (IL1) signaling is required for pharmacological, but not physiological, effects of leptin on energy balance. Brain Res. 2007, 1144, 101–106. [Google Scholar] [CrossRef]
- Azcona-Olivera, J.I.; Ouyang, Y.; Murtha, J.; Chu, F.S.; Pestka, J.J. Induction of cytokine mRNAs in mice after oral exposure to the trichothecene vomitoxin (deoxynivalenol): Relationship to toxin distribution and protein synthesis inhibition. Toxicol. Appl. Pharmacol. 1995, 133, 109–120. [Google Scholar]
- Amuzie, C.J.; Harkema, J.R.; Pestka, J.J. Tissue distribution and proinflammatory cytokine induction by the trichothecene deoxynivalenol in the mouse: Comparison of nasal vs. oral exposure. Toxicology 2008, 248, 39–44. [Google Scholar] [CrossRef]
- Mikami, O.; Yamaguchi, H.; Murata, H.; Nakajima, Y.; Miyazaki, S. Induction of apoptotic lesions in liver and lymphoid tissues and modulation of cytokine mRNA expression by acute exposure to deoxynivalenol in piglets. J. Vet. Sci. 2010, 11, 107–113. [Google Scholar]
- Pestka, J.J.; Amuzie, C.J. Tissue distribution and proinflammatory cytokine gene expression following acute oral exposure to deoxynivalenol: Comparison of weanling and adult mice. Food Chem. Toxicol. 2008, 46, 2826–2831. [Google Scholar] [CrossRef]
- Zhou, H.R.; Yan, D.; Pestka, J.J. Induction of cytokine gene expression in mice after repeated and subchronic oral exposure to vomitoxin (Deoxynivalenol): Differential toxin-induced hyporesponsiveness and recovery. Toxicol. Appl. Pharmacol. 1998, 151, 347–358. [Google Scholar]
- Zhou, H.R.; Yan, D.; Pestka, J.J. Differential cytokine mRNA expression in mice after oral exposure to the trichothecene vomitoxin (deoxynivalenol), dose response and time course. Toxicol. Appl. Pharmacol. 1997, 144, 294–305. [Google Scholar] [CrossRef]
- Sugita-Konishi, Y.; Pestka, J.J. Differential upregulation of TNF-alpha, IL-6, and IL-8 production by deoxynivalenol (vomitoxin) and other 8-ketotrichothecenes in a human macrophage model. J. Toxicol. Environ. Health A 2001, 64, 619–636. [Google Scholar] [CrossRef]
- Pestka, J.J.; Uzarski, R.L.; Islam, Z. Induction of apoptosis and cytokine production in the Jurkat human T cells by deoxynivalenol: Role of mitogen-activated protein kinases and comparison to other 8-ketotrichothecenes. Toxicology 2005, 206, 207–219. [Google Scholar]
- Gray, J.S.; Pestka, J.J. Transcriptional regulation of deoxynivalenol-induced IL-8 expression in human monocytes. Toxicol. Sci. 2007, 99, 502–511. [Google Scholar] [CrossRef]
- Döll, S.; Schrickx, J.A.; Dänicke, S.; Fink-Gremmels, J. Interactions of deoxynivalenol and lipopolysaccharides on cytokine excretion and mRNA expression in porcine hepatocytes and Kupffer cell enriched hepatocyte cultures. Toxicol. Lett. 2009, 190, 96–105. [Google Scholar]
- Wong, S.S.; Zhou, H.R.; Marin-Martinez, M.L.; Brooks, K.; Pestka, J.J. Modulation of IL-1beta, IL-6 and TNF-alpha secretion and mRNA expression by the trichothecene vomitoxin in the RAW 264.7 murine macrophage cell line. Food Chem. Toxicol. 1998, 36, 409–419. [Google Scholar] [CrossRef]
- Zhou, H.R.; Harkema, J.R.; Yan, D.; Pestka, J.J. Amplified proinflammatory cytokine expression and toxicity in mice coexposed to lipopolysaccharide and the trichothecene vomitoxin (deoxynivalenol). J. Toxicol. Environ. Health A 1999, 57, 115–136. [Google Scholar]
- Pestka, J.J. Mechanisms of deoxynivalenol-induced gene expression and apoptosis. Food Addit. Contam. Part A 2008, 25, 1128–1140. [Google Scholar]
- Pestka, J.J.; Zhou, H.R. Effects of tumor necrosis factor type 1 and 2 receptor deficiencies on anorexia, growth and IgA dysregulation in mice exposed to the trichothecene vomitoxin. Food Chem. Toxicol. 2002, 40, 1623–1631. [Google Scholar] [CrossRef]
- Pestka, J.J.; Zhou, H.R. Interleukin-6-deficient mice refractory to IgA dysregulation but not anorexia induction by vomitoxin (deoxynivalenol) ingestion. Food Chem. Toxicol. 2000, 38, 565–575. [Google Scholar]
- Matsumura, K.; Kobayashi, S. Signaling the brain in inflammation: The role of endothelial cells. Front Biosci. 2004, 9, 2819–2826. [Google Scholar] [CrossRef]
- Trebino, C.E.; Eskra, J.D.; Wachtmann, T.S.; Perez, J.R.; Carty, T.J.; Audoly, L.P. Redirection of eicosanoid metabolism in mPGES-1-deficient macrophages. J. Biol. Chem. 2005, 280, 16579–16585. [Google Scholar]
- Levine, A.S.; Morley, J.E. The effect of prostaglandins (PGE2 and PGF2 alpha) on food intake in rats. Pharmacol. Biochem. Behav. 1981, 15, 735–738. [Google Scholar] [CrossRef]
- Jakobsson, P.J.; Thorén, S.; Morgenstern, R.; Samuelsson, B. Identification of human prostaglandin E synthase: A microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. USA 1999, 96, 7220–7225. [Google Scholar]
- Dieter, P.; Scheibe, R.; Jakobsson, P.J.; Watanabe, K.; Kolada, A.; Kamionka, S. Functional coupling of cyclooxygenase 1 and 2 to discrete prostanoid synthases in liver macrophages. Biochem. Biophys. Res. Commun. 2000, 276, 488–492. [Google Scholar]
- Masuko-Hongo, K.; Berenbaum, F.; Humbert, L.; Salvat, C.; Goldring, M.B.; Thirion, S. Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: Critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 2004, 50, 2829–2838. [Google Scholar] [CrossRef]
- Soler, M.; Camacho, M.; Escudero, J.R.; Iñiguez, M.A.; Vila, L. Human vascular smooth muscle cells but not endothelial cells express prostaglandin E synthase. Circ. Res. 2000, 87, 504–507. [Google Scholar] [CrossRef]
- Pecchi, E.; Dallaporta, M.; Thirion, S.; Salvat, C.; Berenbaum, F.; Jean, A.; Troadec, J.D. Involvement of central microsomal prostaglandin E synthase-1 in IL-1beta-induced anorexia. Physiol. Genomics 2006, 25, 485–492. [Google Scholar] [CrossRef]
- Jia, Q.; Pestka, J.J. Role of cyclooxygenase-2 in deoxynivalenol-induced immunoglobulin a nephropathy. Food. Chem. Toxicol. 2005, 43, 721–728. [Google Scholar]
- Dragunow, M.; Faull, R. The use of c-fos as a metabolic marker in neuronal pathway tracing. J. Neurosci. Methods. 1989, 29, 261–265. [Google Scholar] [CrossRef]
- Morgan, J.I.; Curran, T. Proto-oncogene transcription factors and epilepsy. Trends Pharmacol. Sci. 1991, 12, 343–349. [Google Scholar]
- Elmquist, J.K.; Saper, C.B. Activation of neurons projecting to the paraventricular hypothalamic nucleus by intravenous lipopolysaccharide. J. Comp. Neurol. 1996, 374, 315–331. [Google Scholar] [CrossRef]
- Lacroix, S.; Rivest, S. Functional circuitry in the brain of immune-challenged rats: Partial involvement of prostaglandins. J. Comp. Neurol. 1997, 387, 307–324. [Google Scholar]
- Lacroix, S.; Rivest, S. Role of cyclo-oxygenase pathways in the stimulatory. Influence of immune challenge on the transcription of a specific CRF receptor. Subtype in the rat brain. J. Chem. Neuroanat. 1996, 10, 53–71. [Google Scholar] [CrossRef]
- Marvel, F.A.; Chen, C.C.; Badr, N.; Gaykema, R.P.; Goehler, L.E. Reversible inactivation of the dorsal vagal complex blocks lipopolysaccharide-induced social withdrawal and c-Fos expression in central autonomic nuclei. Brain Behav. Immun. 2004, 18, 123–134. [Google Scholar]
- Ossenkopp, K.P.; Hirst, M.; Rapley, W.A. Deoxynivalenol (vomitoxin)-induced conditioned taste aversions in rats are mediated by the chemosensitive area postrema. Pharmacol. Biochem. Behav. 1994, 47, 363–367. [Google Scholar] [CrossRef]
- Boulant, J.A. Role of the preoptic-anterior hypothalamus in thermoregulation and fever. Clin. Infect. Dis. 2000, 31, S157–S161. [Google Scholar] [CrossRef]
- Takahashi, A.; Ishimaru, H.; Ikarashi, Y.; Kishi, E.; Maruyama, Y. Opposite regulation of body temperature by cholinergic input to the paraventricular nucleus and supraoptic nucleus in rats. Brain Res. 2001, 909, 102–111. [Google Scholar]
- Girish, C.K.; MacDonald, E.J.; Scheinin, M.; Smith, T.K. Effects of feedborne fusarium mycotoxins on brain regional neurochemistry of turkeys. Poult. Sci. 2008, 87, 1295–1302. [Google Scholar] [CrossRef]
- Lam, D.D.; Garfield, A.S.; Marston, O.J.; Shaw, J.; Heisler, L.K. Brain serotonin system in the coordination of food intake and body weight. Pharmacol. Biochem. Behav. 2010, 97, 84–91. [Google Scholar] [CrossRef]
- Prelusky, D.B. The effect of low-level deoxynivalenol on neurotransmitter levels measured in pig cerebral spinal fluid. J. Environ. Sci. Health B 1993, 28, 731–761. [Google Scholar]
- Prelusky, D.B.; Yeung, J.M.; Thompson, B.K.; Trenholm, H.L. Effect of deoxynivalenol on neurotransmitters in discrete regions of swine brain. Arch. Environ. Contam. Toxicol. 1992, 22, 36–40. [Google Scholar]
- Swamy, H.V.; Smith, T.K.; Karrow, N.A.; Boermans, H.J. Effects of feeding blends of grains naturally contaminated with Fusarium mycotoxins on growth and immunological parameters of broiler chickens. Poult. Sci. 2004, 83, 533–543. [Google Scholar]
- Swamy, H.V.; Smith, T.K.; MacDonald, E.J.; Boermans, H.J.; Squires, E.J. Effects of feeding a blend of grains naturally contaminated with Fusarium mycotoxins on swine performance, brain regional neurochemistry, and serum chemistry and the efficacy of a polymeric glucomannan mycotoxin adsorbent. J. Anim. Sci. 2002, 80, 3257–3267. [Google Scholar]
- Fitzpatrick, D.W.; Boyd, K.E.; Wilson, L.M.; Wilson, J.R. Effect of the trichothecene deoxynivalenol on brain biogenic monoamines concentrations in rats and chickens. J. Environ. Sci. Health B 1988, 23, 159–170. [Google Scholar]
- Prelusky, D.B. The effect of deoxynivalenol on serotoninergic neurotransmitter levels in pig blood. J. Environ. Sci. Health B 1994, 29, 1203–1218. [Google Scholar] [CrossRef]
- Prelusky, D.B. A study on the effect of deoxynivalenol on serotonin receptor binding in pig brain membranes. J. Environ. Sci. Health B 1996, 31, 1103–1117. [Google Scholar]
- Garfield, A.S.; Lam, D.D.; Marston, O.J.; Przydzial, M.J.; Heisler, L.K. Role of central melanocortin pathways in energy homeostasis. Trends Endocrinol. Metab. 2009, 20, 203–215. [Google Scholar]
- Oh-I, S.; Shimizu, H.; Satoh, T.; Okada, S.; Adachi, S.; Inoue, K.; Eguchi, H.; Yamamoto, M.; Imaki, T.; Hashimoto, K.; et al. Identification of nesfatin-1 as a satiety molecule in the hypothalamus. Nature 2006, 443, 709–712. [Google Scholar]
- Shimizu, H.; Oh-I, S.; Hashimoto, K.; Nakata, M.; Yamamoto, S.; Yoshida, N.; Eguchi, H.; Kato, I.; Inoue, K.; Satoh, T.; et al. Peripheral administration of Nesfatin-1 reduces food intake in mice: The leptin-independent mechanism. Endocrinology 2009, 150, 662–671. [Google Scholar]
- Zhang, A.Q.; Li, X.L.; Jiang, C.Y.; Lin, L.; Shi, R.H.; Chen, J.D.; Oomura, Y. Expression of nesfatin-1/NUCB2 in rodent digestive system. World J. Gastroenterol. 20 2010, 16, 1735–1741. [Google Scholar] [CrossRef]
- Foo, K.S.; Brismar, H.; Broberger, C. Distribution and neuropeptide coexistence of nucleobindin-2 mRNA/nesfatin-like immunoreactivity in the rat CNS. Neuroscience 2008, 156, 563–579. [Google Scholar]
- Kohno, D.; Nakata, M.; Maejima, Y.; Shimizu, H.; Sedbazar, U.; Yoshida, N.; Dezaki, K.; Onaka, T.; Mori, M.; Yada, T. Nesfatin-1 neurons in paraventricular and supraoptic nuclei of the rat hypothalamus coexpress oxytocin and vasopressin and are activated by refeeding. Endocrinology 2008, 149, 1295–1301. [Google Scholar]
- Noetzel, S.; Stengel, A.; Inhoff, T.; Goebel, M.; Wisser, A.S.; Bannert, N.; Wiedenmann, B.; Klapp, B.F.; Taché, Y.; Mönnikes, H.; Kobelt, P. CCK-8S activates c-Fos in a dose-dependent manner in nesfatin-1 immunoreactive neurons in the paraventricular nucleus of the hypothalamus and in the nucleus of the solitary tract of the brainstem. Regul. Pept. 2009, 157, 84–91. [Google Scholar] [CrossRef]
- Prelusky, D.B.; Hartin, K.E.; Trenholm, H.L. Distribution of deoxynivalenol in cerebral spinal fluid following administration to swine and sheep. J. Environ. Sci. Health B 1990, 25, 395–413. [Google Scholar]
- Prelusky, D.B.; Hamilton, R.M.; Trenholm, H.L.; Miller, J.D. Tissue distribution and excretion of radioactivity following administration of 14C-labeled deoxynivalenol to White Leghorn hens. Fundam. Appl. Toxicol. 1986, 7, 635–645. [Google Scholar] [CrossRef]
- Dantzer, R.; Bluthé, R.M.; Layé, S.; Bret-Dibat, J.L.; Parnet, P.; Kelley, K.W. Cytokines and sickness behavior. Ann. N. Y. Acad. Sci. 1998, 840, 586–590. [Google Scholar]
- Bluthé, R.M.; Dantzer, R.; Kelley, K.W. Effects of interleukin-1 receptor antagonist on the behavioral effects of lipopolysaccharide in rat. Brain Res. 1992, 573, 318–320. [Google Scholar]
- Kozak, W.; Zheng, H.; Conn, C.A.; Soszynski, D.; van der Ploeg, L.H.; Kluger, M.J. Thermal and behavioral effects of lipopolysaccharide and influenza in interleukin-1 beta-deficient mice. Am. J. Physiol. 1995, 269, R969–R977. [Google Scholar]
- Luheshi, G.; Miller, A.J.; Brouwer, S.; Dascombe, M.J.; Rothwell, N.J.; Hopkins, S.J. Interleukin-1 receptor antagonist inhibits endotoxin fever and systemic interleukin-6 induction in the rat. Am. J. Physiol. 1996, 270, E91–E95. [Google Scholar]
- Boraschi, D.; Tagliabue, A. The interleukin-1 receptor family. Vitam. Horm. 2006, 74, 229–254. [Google Scholar] [CrossRef]
- Moon, Y.; Pestka, J.J. Vomitoxin-induced cyclooxygenase-2 gene expression in macrophages mediated by activation of ERK and p38 but not JNK mitogen-activated protein kinases. Toxicol. Sci. 2002, 69, 373–382. [Google Scholar]
- Yang, G.H.; Jarvis, B.B.; Chung, Y.J.; Pestka, J.J. Apoptosis induction by the satratoxins and other trichothecene mycotoxins: Relationship to ERK, p38 MAPK, and SAPK/JNK activation. Toxicol. Appl. Pharmacol. 2000, 164, 149–160. [Google Scholar] [CrossRef]
- Zhou, H.R.; Islam, Z.; Pestka, J.J. Rapid, sequential activation of mitogen-activated protein kinases and transcription factors precedes proinflammatory cytokine mRNA expression in spleens of mice exposed to the trichothecene vomitoxin. Toxicol. Sci. 2003, 72, 130–142. [Google Scholar] [CrossRef]
- Bae, H.; Gray, J.S.; Li, M.; Vines, L.; Kim, J.; Pestka, J.J. Hematopoietic cell kinase associates with the 40S ribosomal subunit and mediates the ribotoxic stress response to deoxynivalenol in mononuclear phagocytes. Toxicol. Sci. 2010, 115, 444–452. [Google Scholar]
- He, K.; Zhou, H.R.; Pestka, J.J. Targets and intracellular signaling mechanisms for deoxynivalenol-induced ribosomal RNA cleavage. Toxicol. Sci. 2012, 127, 382–390. [Google Scholar] [CrossRef]
- Zhou, H.R.; Lau, A.S.; Pestka, J.J. Role of double-stranded RNA-activated protein kinase R (PKR) in deoxynivalenol-induced ribotoxic stress response. Toxicol. Sci. 2003, 74, 335–344. [Google Scholar]
- Bunner, D.L.; Morris, E.R. Alteration of multiple cell membrane functions in L-6 myoblasts by T-2 toxin: An important mechanism of action. Toxicol. Appl. Pharmacol. 1988, 92, 113–121. [Google Scholar] [CrossRef]
- Yoshino, N.; Takizawa, M.; Akiba, H.; Okumura, H.; Tashiro, F.; Honda, M.; Ueno, Y. Transient elevation of intracellular calcium ion levels as an early event in T-2 toxin-induced apoptosis in human promyelotic cell line HL-60. Nat. Toxins 1996, 4, 234–241. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bonnet, M.S.; Roux, J.; Mounien, L.; Dallaporta, M.; Troadec, J.-D. Advances in Deoxynivalenol Toxicity Mechanisms: The Brain as a Target. Toxins 2012, 4, 1120-1138. https://doi.org/10.3390/toxins4111120
Bonnet MS, Roux J, Mounien L, Dallaporta M, Troadec J-D. Advances in Deoxynivalenol Toxicity Mechanisms: The Brain as a Target. Toxins. 2012; 4(11):1120-1138. https://doi.org/10.3390/toxins4111120
Chicago/Turabian StyleBonnet, Marion S., Julien Roux, Lourdes Mounien, Michel Dallaporta, and Jean-Denis Troadec. 2012. "Advances in Deoxynivalenol Toxicity Mechanisms: The Brain as a Target" Toxins 4, no. 11: 1120-1138. https://doi.org/10.3390/toxins4111120
APA StyleBonnet, M. S., Roux, J., Mounien, L., Dallaporta, M., & Troadec, J. -D. (2012). Advances in Deoxynivalenol Toxicity Mechanisms: The Brain as a Target. Toxins, 4(11), 1120-1138. https://doi.org/10.3390/toxins4111120