Immune Dysfunction in Uremia—An Update

Abstract
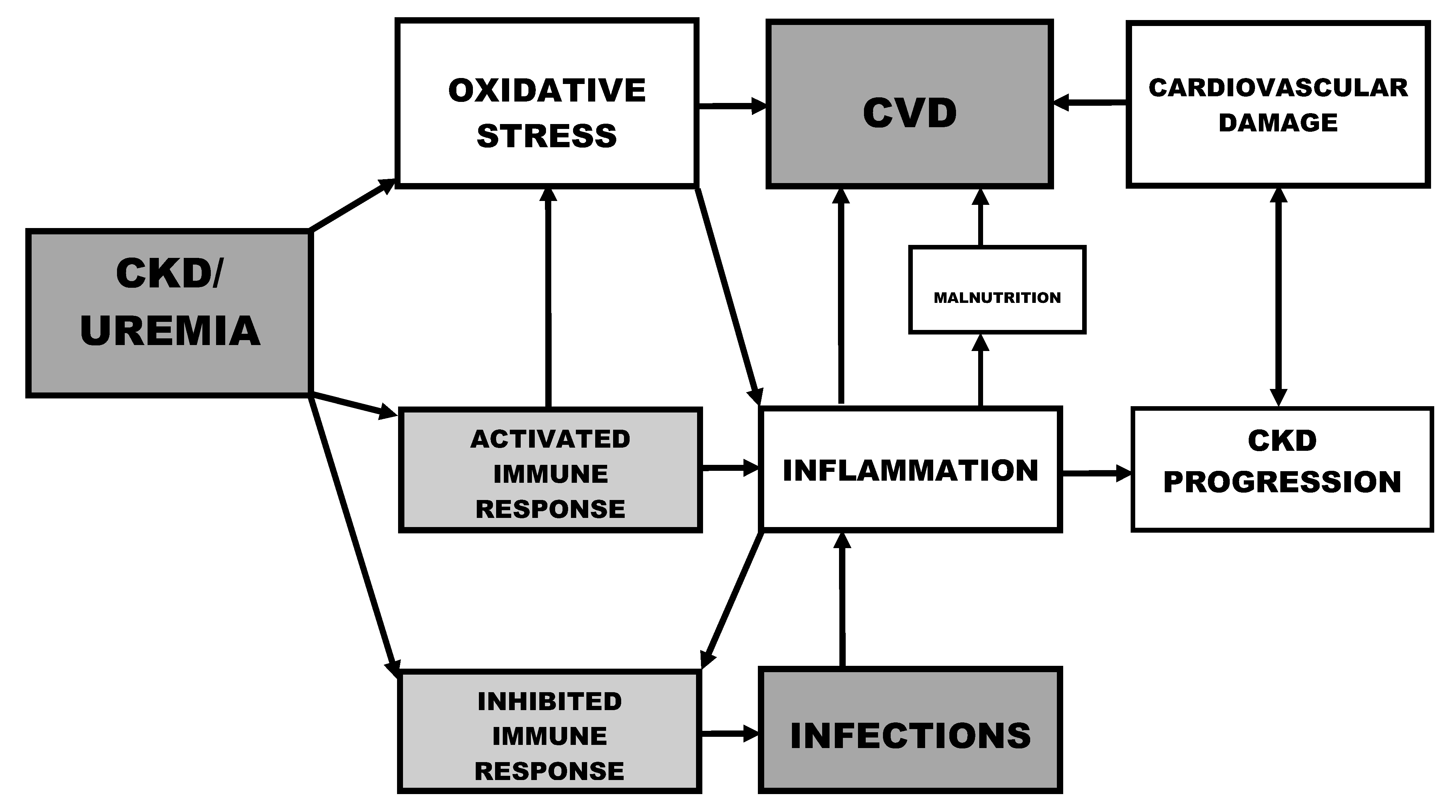
:1. Magnitude of the Problem: Mortality in Uremia

2. Oxidative Stress and Inflammation
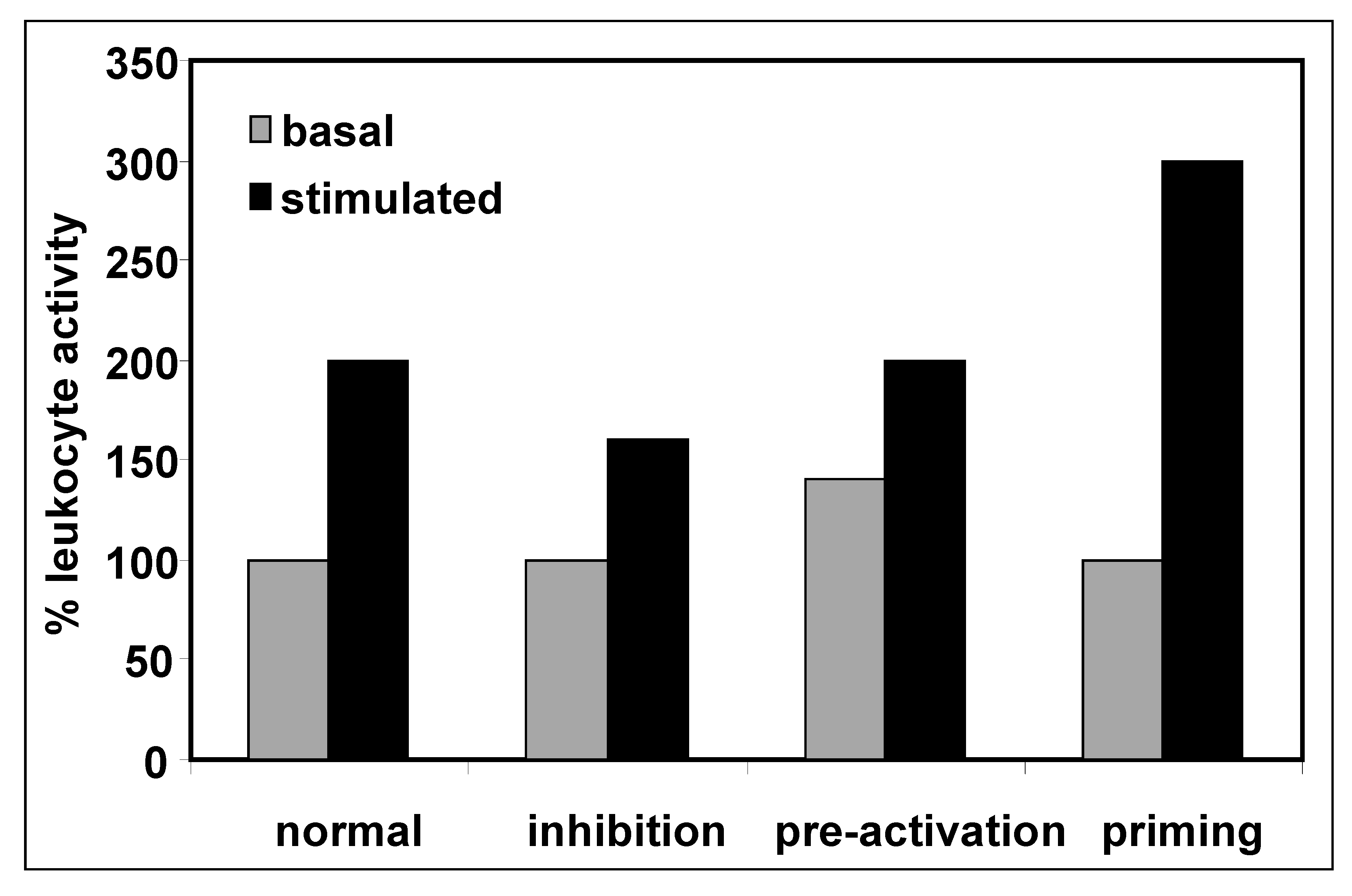
3. Priming of Immune Cells

4. Apoptosis
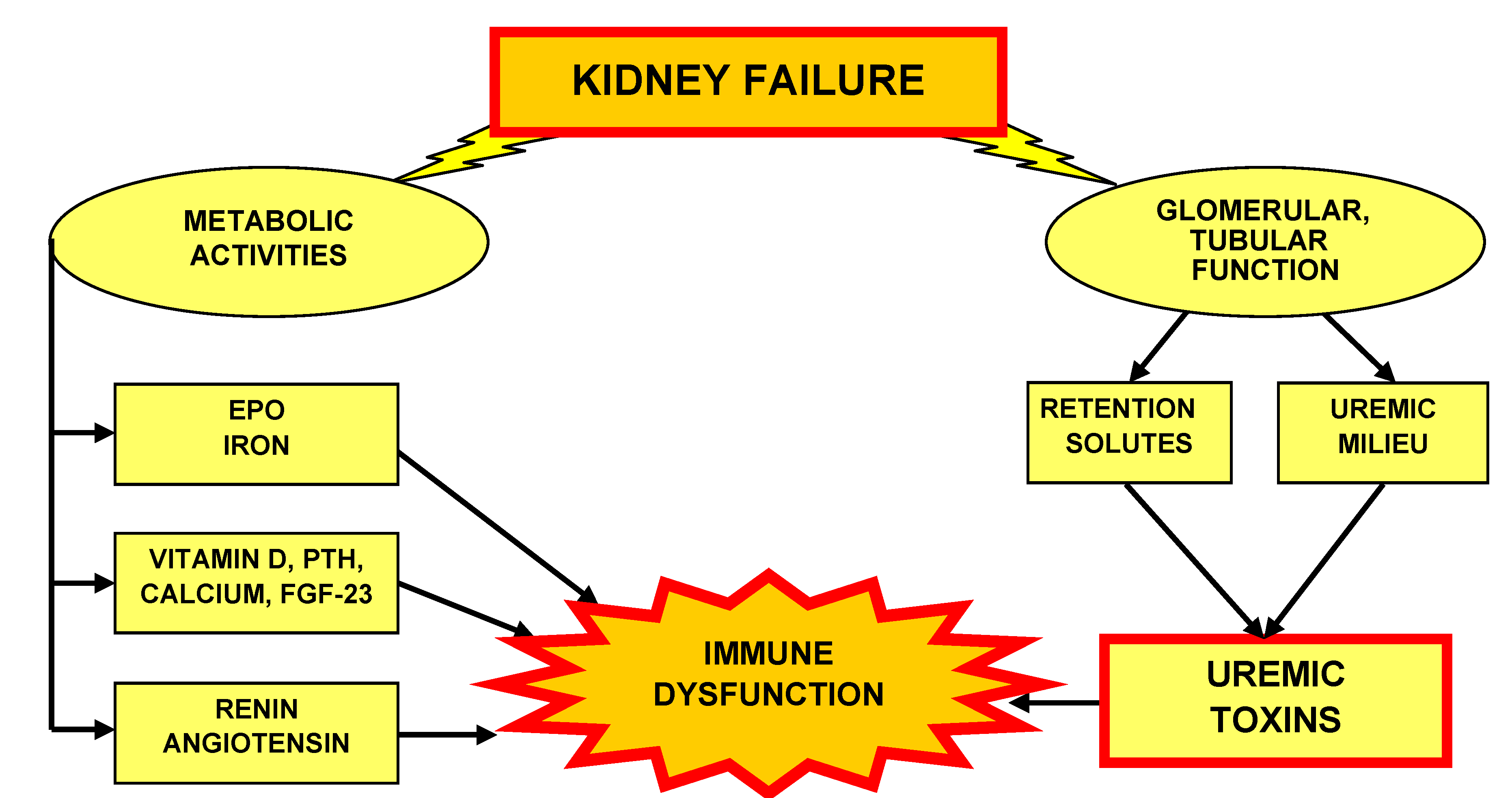
5. Metabolic Kidney Activities

5.1. Erythropoietin and Iron
5.2. Vitamin D, Calcium, Parathyroid Hormone and Fibroblast Growth Factor 23
5.3. Renin, Angiotensin
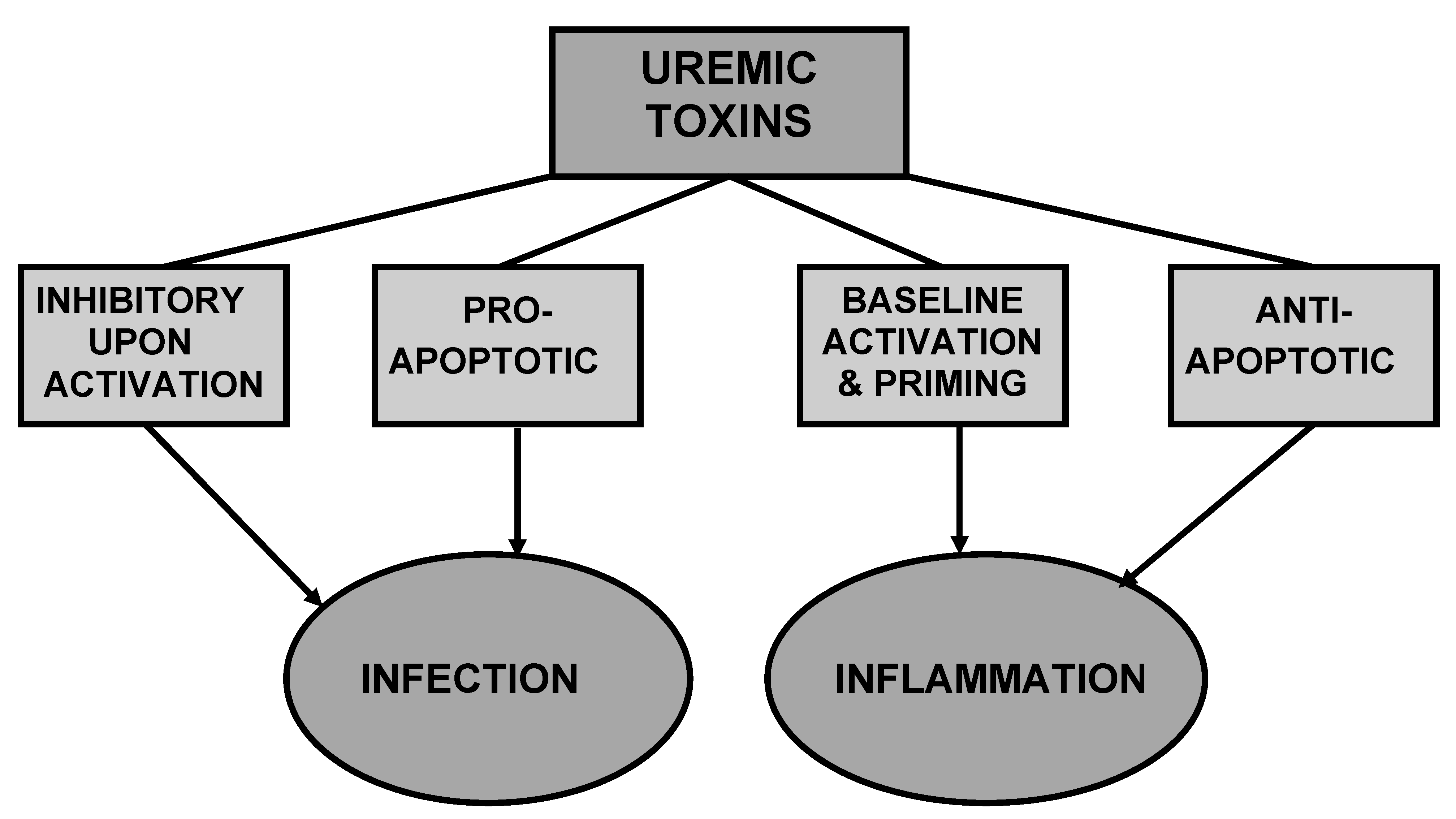
6. Uremic Toxins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uremic toxin | Functional disturbance |
|---|---|
| LMW Solutes | |
| Phenylacetic acid (PAA) | Macrophages: inducible nitric oxide synthase ↓ [106]; |
| PMNLs: oxidative burst, phagocytosis and integrin expression ↑; apoptosis ↓ [49] | |
| Dinucleoside polyphosphates | Leukocytes: oxidative burst ↑ [107]. |
| Guanidino compounds | Monocytes/macrophages: pro- and anti-inflammatory [108,109,110] |
| Indoxyl sulfate | Endothel: E-selectin ↑ [111] |
| P-cresyl sulfate | Leukocytes: basal oxidative burst ↑ [112] |
| Homocysteine (Hcy) | ICAM-1 ↑ [113]; damage of DNA [114] and proteins [115] |
| Methylglyoxal (MGO) | PMNLs: apoptosis ↑ [116], oxidative burst ↑ [117]; |
| Monocytes: apoptosis ↑ [118] | |
| Middle Molecules, Proteins | |
| Immunoglobulin light chains (IgLCs) | PMNLs: chemotaxis ↓, glucose uptake stimulation ↓, glucose uptake basal ↑ [119]; apoptosis ↓ [47] |
| Retinol binding protein (RBP) | PMNLs: chemotaxis ↓, oxidative burst ↓, apoptosis ↓ [120] |
| Leptin | PMNLs: chemotaxis ↓, oxidative burst ↓ [121] |
| Resistin | PMNLs: chemotaxis ↓, oxidative burst ↓ [122] |
| Tamm-Horsfall protein (THP) | PMNLs: (high concentrations) apoptosis ↓, chemotaxis ↓, phagocytosis ↑; (low concentrations) chemotaxis ↑ [123] |
| High-density lipoprotein (HDL) | Loss of anti-inflammatory properties in uremia [124,125] |
| Protein Modifications | |
| Glucose-modified proteins | PMNLs: chemotaxis ↑, glucose uptake ↑, apoptosis ↑ [48] |
| AGE-modified albumin | Leukocytes: activating, pro- atherogenic [126] |
| AGEs | Macrophages: TNF and IL-1 secretion ↑ [127] |
| Monocytes: Chemotaxis ↑ [128] | |
| Glycated collagen | PMNLs: Adhesion ↑ [129] |
| Advanced oxidation protein products (AOPPs) | PMNLs and monocytes: oxidative burst ↑ [130] |
| Oxidized low-density lipoproteins (oxLDLs) | Macrophage activation [131]; |
| PMNLs and eosinophils: chemotaxis ↑, degranulation ↑ [132]; | |
| Regulatory T cells: proteasome activity ↓ → cell cycle arrest and apoptosis [133] | |
| Homocysteinylated albumin | Monocytes: adhesion ↑[134] |
6.1. LMW Solutes
6.2. Middle Molecules, Proteins
6.3. Protein Modifications
6.3.1. Advanced Glycation End-Products
6.3.2. Oxidative Modifications
6.3.3. Carbamoylation, Carbonylation and Homocysteinylation
7. Further Aspects of Immune Dysfunction in Uremia
7.1. Antigen-Presenting Cells
7.2. Epigenetics
7.3. Antineutrophil Cytoplasmic Autoantibodies
8. Conclusions
Acknowledgments
Conflict of Interest
References
- Wen, C.P.; Cheng, T.Y.; Tsai, M.K.; Chang, Y.C.; Chan, H.T.; Tsai, S.P.; Chiang, P.H.; Hsu, C.C.; Sung, P.K.; Hsu, Y.H.; et al. All-cause mortality attributable to chronic kidney disease: A prospective cohort study based on 462 293 adults in Taiwan. Lancet 2008, 371, 2173–2182. [Google Scholar] [CrossRef]
- Drey, N.; Roderick, P.; Mullee, M.; Rogerson, M. A population-based study of the incidence and outcomes of diagnosed chronic kidney disease. Am. J. Kidney Dis. 2003, 42, 677–684. [Google Scholar] [CrossRef]
- Kato, S.; Chmielewski, M.; Honda, H.; Pecoits-Filho, R.; Matsuo, S.; Yuzawa, Y.; Tranaeus, A.; Stenvinkel, P.; Lindholm, B. Aspects of immune dysfunction in end-stage renal disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1526–1533. [Google Scholar]
- Tonelli, M.; Wiebe, N.; Culleton, B.; House, A.; Rabbat, C.; Fok, M.; McAlister, F.; Garg, A.X. Chronic kidney disease and mortality risk: a systematic review. J. Am. Soc. Nephrol. 2006, 17, 2034–2047. [Google Scholar] [CrossRef]
- Meyer, T.W.; Hostetter, T.H. Uremia. N. Engl. J. Med. 2007, 357, 1316–1325. [Google Scholar]
- Haag-Weber, M.; Hörl, W.H. Dysfunction of polymorphonuclear leukocytes in uremia. Semin. Nephrol. 1996, 16, 192–201. [Google Scholar]
- Chonchol, M. Neutrophil dysfunction and infection risk in end-stage renal disease. Semin. Dial. 2006, 19, 291–296. [Google Scholar] [CrossRef]
- James, M.T.; Laupland, K.B.; Tonelli, M.; Manns, B.J.; Culleton, B.F.; Hemmelgarn, B.R. Risk of bloodstream infection in patients with chronic kidney disease not treated with dialysis. Arch. Intern. Med. 2008, 168, 2333–2339. [Google Scholar]
- Sarnak, M.J.; Jaber, B.L. Mortality caused by sepsis in patients with end-stage renal disease compared with the general population. Kidney Int. 2000, 58, 1758–1764. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Antoniadi, G.; Liakopoulos, V.; Kartsios, C.; Stefanidis, I. Disturbances of acquired immunity in hemodialysis patients. Semin. Dial. 2007, 20, 440–451. [Google Scholar]
- Vanholder, R.; Massy, Z.; Argiles, A.; Spasovski, G.; Verbeke, F.; Lameire, N. Chronic kidney disease as cause of cardiovascular morbidity and mortality. Nephrol. Dial. Transplant. 2005, 20, 1048–1056. [Google Scholar] [CrossRef]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Epidemiology of cardiovascular disease in chronic renal disease. J. Am. Soc. Nephrol. 1998, 9, S16–S23. [Google Scholar]
- Coresh, J.; Longenecker, J.C.; Miller, E.R., III; Young, H.J.; Klag, M.J. Epidemiology of cardiovascular risk factors in chronic renal disease. J. Am. Soc. Nephrol. 1998, 9, S24–S30. [Google Scholar]
- Fried, L.F.; Katz, R.; Cushman, M.; Sarnak, M.; Shlipak, M.G.; Kuller, L.; Newman, A.B. Change in cardiovascular risk factors with progression of kidney disease. Am. J. Nephrol. 2008, 29, 334–341. [Google Scholar]
- Ryan, T.P.; Fisher, S.G.; Elder, J.L.; Winters, P.C.; Beckett, W.; Tacci, J.; Sloand, J.A. Increased cardiovascular risk associated with reduced kidney function. Am. J. Nephrol. 2009, 29, 620–625. [Google Scholar] [CrossRef]
- Libetta, C.; Sepe, V.; Esposito, P.; Galli, F.; Dal Canton, A. Oxidative stress and inflammation: Implications in uremia and hemodialysis. Clin. Biochem. 2011, 44, 1189–1198. [Google Scholar]
- Dounousi, E.; Papavasiliou, E.; Makedou, A.; Ioannou, K.; Katopodis, K.P.; Tselepis, A.; Siamopoulos, K.C.; Tsakiris, D. Oxidative stress is progressively enhanced with advancing stages of CKD. Am. J. Kidney Dis. 2006, 48, 752–760. [Google Scholar] [CrossRef]
- Morena, M.; Cristol, J.P.; Senecal, L.; Leray-Moragues, H.; Krieter, D.; Canaud, B. Oxidative stress in hemodialysis patients: is NADPH oxidase complex the culprit? Kidney Int. 2002, 61, S109–S114. [Google Scholar] [CrossRef]
- Rodriguez-Ayala, E.; Anderstam, B.; Suliman, M.E.; Seeberger, A.; Heimburger, O.; Lindholm, B.; Stenvinkel, P. Enhanced RAGE-mediated NFkappaB stimulation in inflamed hemodialysis patients. Atherosclerosis 2005, 180, 333–340. [Google Scholar] [CrossRef]
- Stinghen, A.E.; Bucharles, S.; Riella, M.C.; Pecoits-Filho, R. Immune mechanisms involved in cardiovascular complications of chronic kidney disease. Blood Purif. 2010, 29, 114–120. [Google Scholar]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef]
- Gollapudi, P.; Yoon, J.W.; Gollapudi, S.; Pahl, M.V.; Vaziri, N.D. Leukocyte toll-like receptor expression in end-stage kidney disease. Am. J. Nephrol. 2010, 31, 247–254. [Google Scholar] [CrossRef]
- Okamura, D.M.; Pennathur, S.; Pasichnyk, K.; Lopez-Guisa, J.M.; Collins, S.; Febbraio, M.; Heinecke, J.; Eddy, A.A. CD36 regulates oxidative stress and inflammation in hypercholesterolemic CKD. J. Am. Soc. Nephrol. 2009, 20, 495–505. [Google Scholar]
- Rutkowski, P.; Malgorzewicz, S.; Slominska, E.; Renke, M.; Lysiak-Szydlowska, W.; Swierczynski, J.; Rutkowski, B. Interrelationship between uremic toxicity and oxidative stress. J. Ren. Nutr. 2006, 16, 190–193. [Google Scholar] [CrossRef]
- Galli, F. Protein damage and inflammation in uraemia and dialysis patients. Nephrol. Dial. Transplant. 2007, 22 Suppl. 5, V20–V36. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Carrero, J.J.; Axelsson, J.; Lindholm, B.; Stenvinkel, P. Low-grade inflammation in chronic kidney disease patients before the start of renal replacement therapy: Sources and consequences. Clin. Nephrol. 2007, 68, 1–9. [Google Scholar]
- Zoccali, C. Traditional and emerging cardiovascular and renal risk factors: An epidemiologic perspective. Kidney Int. 2006, 70, 26–33. [Google Scholar] [CrossRef]
- Carrero, J.J.; Stenvinkel, P. Persistent inflammation as a catalyst for other risk factors in chronic kidney disease: A hypothesis proposal. Clin. J. Am. Soc. Nephrol. 2009, 4 Suppl. 1, S49–S55. [Google Scholar] [CrossRef]
- Miyamoto, T.; Carrero, J.J.; Stenvinkel, P. Inflammation as a risk factor and target for therapy in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2011, 20, 662–668. [Google Scholar]
- Cirillo, P.; Sautin, Y.Y.; Kanellis, J.; Kang, D.H.; Gesualdo, L.; Nakagawa, T.; Johnson, R.J. Systemic inflammation, metabolic syndrome and progressive renal disease. Nephrol. Dial. Transplant. 2009, 24, 1384–1387. [Google Scholar] [CrossRef]
- Yao, Q.; Axelsson, J.; Stenvinkel, P.; Lindholm, B. Chronic systemic inflammation in dialysis patients: an update on causes and consequences. ASAIO J. 2004, 50, lii–lvii. [Google Scholar]
- Helal, I.; Smaoui, W.; Hamida, F.B.; Ouniss, M.; Aderrahim, E.; Hedri, H.; Elyounsi, F.; Maiz, H.B.; Abdallah, T.B.; Kheder, A. Cardiovascular risk factors in hemodialysis and peritoneal dialysis patients. Saudi J. Kidney Dis. Transpl. 2010, 21, 59–62. [Google Scholar]
- Jofre, R.; Rodriguez-Benitez, P.; Lopez-Gomez, J.M.; Perez-Garcia, R. Inflammatory syndrome in patients on hemodialysis. J. Am. Soc. Nephrol. 2006, 17, S274–S280. [Google Scholar] [CrossRef]
- Cazzavillan, S.; Ratanarat, R.; Segala, C.; Corradi, V.; de Cal, M.; Cruz, D.; Ocampo, C.; Polanco, N.; Rassu, M.; Levin, N.; et al. Inflammation and subclinical infection in chronic kidney disease: A molecular approach. Blood Purif. 2007, 25, 69–76. [Google Scholar] [CrossRef]
- Wann, J.G.; Hsu, Y.H.; Yang, C.C.; Lin, C.S.; Tai, D.W.; Chen, J.S.; Hsiao, C.W.; Chen, C.F. Neutrophils in acidotic haemodialysed patients have lower intracellular pH and inflamed state. Nephrol. Dial. Transplant. 2007, 22, 2613–2622. [Google Scholar] [CrossRef]
- Swain, S.D.; Rohn, T.T.; Quinn, M.T. Neutrophil priming in host defense: role of oxidants as priming agents. Antioxid. Redox. Signal. 2002, 4, 69–83. [Google Scholar]
- Koenderman, L.; Yazdanbakhsh, M.; Roos, D.; Verhoeven, A.J. Dual mechanisms in priming of the chemoattractant-induced respiratory burst in human granulocytes. A Ca2+-dependent and a Ca2+-independent route. J. Immunol. 1989, 142, 623–628. [Google Scholar]
- Chilvers, E.R.; Cadwallader, K.A.; Reed, B.J.; White, J.F.; Condliffe, A.M. The function and fate of neutrophils at the inflamed site: prospects for therapeutic intervention. J. R. Coll. Physicians. Lond. 2000, 34, 68–74. [Google Scholar]
- Klein, J.B.; McLeish, K.R.; Ward, R.A. Transplantation, not dialysis, corrects azotemia-dependent priming of the neutrophil oxidative burst. Am. J. Kidney Dis. 1999, 33, 483–491. [Google Scholar] [CrossRef]
- Condliffe, A.M.; Kitchen, E.; Chilvers, E.R. Neutrophil priming: Pathophysiological consequences and underlying mechanisms. Clin. Sci. (Lond.) 1998, 94, 461–471. [Google Scholar]
- Sela, S.; Shurtz-Swirski, R.; Cohen-Mazor, M.; Mazor, R.; Chezar, J.; Shapiro, G.; Hassan, K.; Shkolnik, G.; Geron, R.; Kristal, B. Primed peripheral polymorphonuclear leukocyte: A culprit underlying chronic low-grade inflammation and systemic oxidative stress in chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2431–2438. [Google Scholar] [CrossRef]
- Ward, R.A.; Ouseph, R.; McLeish, K.R. Effects of high-flux hemodialysis on oxidant stress. Kidney Int. 2003, 63, 353–359. [Google Scholar]
- Filep, J.G.; El Kebir, D. Neutrophil apoptosis: A target for enhancing the resolution of inflammation. J. Cell. Biochem. 2009, 108, 1039–1046. [Google Scholar] [CrossRef]
- Cohen, G.; Rudnicki, M.; Hörl, W.H. Uremic toxins modulate the spontaneous apoptotic cell death and essential functions of neutrophils. Kidney Int. 2001, 78, S48–S52. [Google Scholar]
- Glorieux, G.; Vanholder, R.; Lameire, N. Uraemic retention and apoptosis: what is the balance for the inflammatory status in uraemia? Eur. J. Clin. Invest. 2003, 33, 631–634. [Google Scholar] [CrossRef]
- Trevani, A.S.; Andonegui, G.; Giordano, M.; Lopez, D.H.; Gamberale, R.; Minucci, F.; Geffner, J.R. Extracellular acidification induces human neutrophil activation. J. Immunol. 1999, 162, 4849–4857. [Google Scholar]
- Cohen, G.; Rudnicki, M.; Deicher, R.; Hörl, W.H. Immunoglobulin light chains modulate polymorphonuclear leucocyte apoptosis. Eur. J. Clin. Invest. 2003, 33, 669–676. [Google Scholar]
- Cohen, G.; Rudnicki, M.; Walter, F.; Niwa, T.; Hörl, W.H. Glucose-modified proteins modulate essential functions and apoptosis of polymorphonuclear leukocytes. J. Am. Soc. Nephrol. 2001, 12, 1264–1271. [Google Scholar]
- Cohen, G.; Raupachova, J.; Hörl, W.H. The uraemic toxin phenylacetic acid contributes to inflammation by priming polymorphonuclear leucocytes. Nephrol. Dial. Transplant. 2012. [Google Scholar]
- Cohen, G.; Raupachova, J.; Wimmer, T.; Deicher, R.; Hörl, W.H. The uraemic retention solute para-hydroxy-hippuric acid attenuates apoptosis of polymorphonuclear leukocytes from healthy subjects but not from haemodialysis patients. Nephrol. Dial. Transplant. 2008, 23, 2512–2519. [Google Scholar] [CrossRef]
- Jankowski, J.; Tepel, M.; Stephan, N.; van der Giet, M.; Breden, V.; Zidek, W.; Schluter, H. Characterization of p-hydroxy-hippuric acid as an inhibitor of Ca2+-ATPase in end-stage renal failure. Kidney Int. 2001, 78, S84–S88. [Google Scholar]
- Perianayagam, M.C.; Balakrishnan, V.S.; King, A.J.; Pereira, B.J.; Jaber, B.L. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int. 2002, 61, 456–463. [Google Scholar] [CrossRef]
- Perianayagam, M.C.; Balakrishnan, V.S.; Pereira, B.J.; Jaber, B.L. C5a delays apoptosis of human neutrophils via an extracellular signal-regulated kinase and Bad-mediated signalling pathway. Eur. J. Clin. Invest. 2004, 34, 50–56. [Google Scholar]
- Kettritz, R.; Falk, R.J.; Jennette, J.C.; Gaido, M.L. Neutrophil superoxide release is required for spontaneous and FMLP-mediated but not for TNF alpha-mediated apoptosis. J. Am. Soc. Nephrol. 1997, 8, 1091–1100. [Google Scholar]
- Haag-Weber, M.; Hörl, W.H. Calcium-dependent neutrophil activation. Contrib. Nephrol. 1992, 100, 269–285. [Google Scholar]
- Hörl, W.H.; Haag-Weber, M.; Mai, B.; Massry, S.G. Verapamil reverses abnormal [Ca2+]i and carbohydrate metabolism of PMNL of dialysis patients. Kidney Int. 1995, 47, 1741–1745. [Google Scholar] [CrossRef]
- Lucas, M.; Diaz, P. Thapsigargin-induced calcium entry and apoptotic death of neutrophils are blocked by activation of protein kinase C. Pharmacology 2001, 63, 191–196. [Google Scholar]
- Hu, T.H.; Bei, L.; Huang, Y.F.; Shen, X. The relationship between fMLP induced neutrophil respiratory burst and the apoptosis of neutrophil. Shi Yan Sheng Wu Xue Bao 1999, 32, 359–366. [Google Scholar]
- Carracedo, J.; Ramirez, R.; Soriano, S.; Alvarez de Lara, M.A.; Rodriguez, M.; Martin-Malo, A.; Aljama, P. Monocytes from dialysis patients exhibit characteristics of senescent cells: Does it really mean inflammation? Contrib. Nephrol. 2005, 149, 208–218. [Google Scholar]
- Galli, F.; Ghibelli, L.; Buoncristiani, U.; Bordoni, V.; D’Intini, V.; Benedetti, S.; Canestrari, F.; Ronco, C.; Floridi, A. Mononuclear leukocyte apoptosis in haemodialysis patients: The role of cell thiols and vitamin E. Nephrol. Dial. Transplant. 2003, 18, 1592–1600. [Google Scholar] [CrossRef]
- Fernandez-Fresnedo, G.; Ramos, M.A.; Gonzalez-Pardo, M.C.; de Francisco, A.L.; Lopez-Hoyos, M.; Arias, M. B lymphopenia in uremia is related to an accelerated in vitro apoptosis and dysregulation of Bcl-2. Nephrol. Dial. Transplant. 2000, 15, 502–510. [Google Scholar]
- Meier, P.; Dayer, E.; Blanc, E.; Wauters, J.P. Early T cell activation correlates with expression of apoptosis markers in patients with end-stage renal disease. J. Am. Soc. Nephrol. 2002, 13, 204–212. [Google Scholar]
- Sardenberg, C.; Suassuna, P.; Andreoli, M.C.; Watanabe, R.; Dalboni, M.A.; Manfredi, S.R.; dos Santos, O.P.; Kallas, E.G.; Draibe, S.A.; Cendoroglo, M. Effects of uraemia and dialysis modality on polymorphonuclear cell apoptosis and function. Nephrol. Dial. Transplant. 2006, 21, 160–165. [Google Scholar]
- Soriano, S.; Martin-Malo, A.; Carracedo, J.; Ramirez, R.; Rodriguez, M.; Aljama, P. Lymphocyte apoptosis: Role of uremia and permeability of dialysis membrane. Nephron. Clin. Pract. 2005, 100, C71–C77. [Google Scholar]
- D’Intini, V.; Bordoni, V.; Bolgan, I.; Bonello, M.; Brendolan, A.; Crepaldi, C.; Gastaldon, F.; Levin, N.W.; Bellomo, R.; Ronco, C. Monocyte apoptosis in uremia is normalized with continuous blood purification modalities. Blood Purif. 2004, 22, 9–12. [Google Scholar] [CrossRef]
- Pesanti, E.L. Immunologic defects and vaccination in patients with chronic renal failure. Infect. Dis. Clin. North. Am. 2001, 15, 813–832. [Google Scholar]
- Macdougall, I.C.; Cooper, A.C. Erythropoietin resistance: The role of inflammation and pro-inflammatory cytokines. Nephrol. Dial. Transplant. 2002, 17 Suppl. 11, 39–43. [Google Scholar] [CrossRef]
- Deicher, R.; Ziai, F.; Cohen, G.; Mullner, M.; Hörl, W.H. High-dose parenteral iron sucrose depresses neutrophil intracellular killing capacity. Kidney Int. 2003, 64, 728–736. [Google Scholar] [CrossRef]
- Sengoelge, G.; Sunder-Plassmann, G.; Hörl, W.H. Potential risk for infection and atherosclerosis due to iron therapy. J. Ren. Nutr. 2005, 15, 105–110. [Google Scholar]
- Patruta, S.I.; Hörl, W.H. Iron and infection. Kidney Int. 1999, 69, S125–S130. [Google Scholar] [CrossRef]
- Deicher, R.; Hörl, W.H. New insights into the regulation of iron homeostasis. Eur. J. Clin. Invest. 2006, 36, 301–309. [Google Scholar]
- Atanasiu, V.; Manolescu, B.; Stoian, I. Hepcidin the link between inflammation and anemia in chronic renal failure. Rom. J. Intern. Med. 2006, 44, 25–33. [Google Scholar]
- Ashby, D.R.; Gale, D.P.; Busbridge, M.; Murphy, K.G.; Duncan, N.D.; Cairns, T.D.; Taube, D.H.; Bloom, S.R.; Tam, F.W.; Chapman, R.S.; et al. Plasma hepcidin levels are elevated but responsive to erythropoietin therapy in renal disease. Kidney Int. 2009, 75, 976–981. [Google Scholar] [CrossRef]
- Rocchetta, F.; Solini, S.; Mister, M.; Mele, C.; Cassis, P.; Noris, M.; Remuzzi, G.; Aiello, S. Erythropoietin enhances immunostimulatory properties of immature dendritic cells. Clin. Exp. Immunol. 2011, 165, 202–210. [Google Scholar] [CrossRef]
- Reichel, H.; Recker, A.; Deppisch, R.; Stier, E.; Ritz, E. 25-Hydroxyvitamin D3 metabolism in vitro by mononuclear cells from hemodialysis patients. Nephron 1992, 62, 404–412. [Google Scholar]
- Schomig, M.; Ritz, E. Management of disturbed calcium metabolism in uraemic patients: 1. Use of vitamin D metabolites. Nephrol. Dial. Transplant. 2000, 15 Suppl. 5, 18–24. [Google Scholar] [CrossRef]
- Glorieux, G.; Vanholder, R. Blunted response to vitamin D in uremia. Kidney Int. 2001, 78, S182–S185. [Google Scholar]
- Shroff, R.; Wan, M.; Rees, L. Can vitamin D slow down the progression of chronic kidney disease? Pediatr. Nephrol. 2011. [Google Scholar]
- Szeto, F.L.; Reardon, C.A.; Yoon, D.; Wang, Y.; Wong, K.E.; Chen, Y.; Kong, J.; Liu, S.Q.; Thadhani, R.; Getz, G.S.; et al. Vitamin d receptor signaling inhibits atherosclerosis in mice. Mol. Endocrinol. 2012, 26, 1091–1101. [Google Scholar] [CrossRef]
- Wahl, P.; Wolf, M. FGF23 in chronic kidney disease. Adv. Exp. Med. Biol. 2012, 728, 107–125. [Google Scholar]
- Sinha, M.D.; Turner, C.; Dalton, R.N.; Rasmussen, P.; Waller, S.; Booth, C.J.; Goldsmith, D.J. Investigating FGF-23 concentrations and its relationship with declining renal function in paediatric patients with pre-dialysis CKD Stages 3–5. Nephrol. Dial. Transplant. 2012. [Google Scholar]
- Kendrick, J.; Cheung, A.K.; Kaufman, J.S.; Greene, T.; Roberts, W.L.; Smits, G.; Chonchol, M. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J. Am. Soc. Nephrol. 2011, 22, 1913–1922. [Google Scholar] [CrossRef]
- Heine, G.H.; Seiler, S.; Fliser, D. FGF-23: The rise of a novel cardiovascular risk marker in CKD. Nephrol. Dial. Transplant. 2012, 27, 3072–3081. [Google Scholar]
- Seiler, S.; Reichart, B.; Roth, D.; Seibert, E.; Fliser, D.; Heine, G.H. FGF-23 and future cardiovascular events in patients with chronic kidney disease before initiation of dialysis treatment. Nephrol. Dial. Transplant. 2010, 25, 3983–3989. [Google Scholar] [CrossRef]
- Deicher, R.; Kirsch, B.; Mullner, M.; Kaczirek, K.; Niederle, B.; Hörl, W.H. Impact of parathyroidectomy on neutrophil cytosolic calcium in chronic kidney disease patients: A prospective parallel group trial. J. Intern. Med. 2005, 258, 67–76. [Google Scholar]
- Karpati, I.; Seres, I.; Matyus, J.; Ben, T.; Paragh, G.; Varga, Z.; Kakuk, G. Which parameters affect cytosolic free calcium in polymorphonuclear leukocytes of haemodialysis patients? Nephrol. Dial. Transplant. 2001, 16, 1409–1415. [Google Scholar] [CrossRef]
- Koorts, A.M.; Kruger, M.C.; Potgieter, C.D.; Viljoen, M. Intracellular free calcium in the neutrophils of maintenance haemodialysis patients. Clin. Physiol. Funct. Imaging 2002, 22, 285–294. [Google Scholar]
- Massry, S.; Smogorzewski, M. Dysfunction of polymorphonuclear leukocytes in uremia: Role of parathyroid hormone. Kidney Int. 2001, 78, S195–S196. [Google Scholar]
- Hansch, G.M.; Karnaoukhova, S.; Chang, S.H.; Rus, H.; Nicolescu, F.; Deppisch, R.; Meissner, C.; Ludwig, H.; Ritz, E. Activation of human neutrophils after contact with cellulose-based haemodialysis membranes: Intracellular calcium signalling in single cells. Nephrol. Dial. Transplant. 1996, 11, 2453–2460. [Google Scholar]
- Smogorzewski, M.; Massry, S.G. Defects in B-cell function and metabolism in uremia: Role of parathyroid hormone. Kidney Int. 2001, 78, S186–S189. [Google Scholar] [CrossRef]
- Griveas, I.; Visvardis, G.; Papadopoulou, D.; Mitsopoulos, E.; Kyriklidou, P.; Manou, E.; Meimaridou, D.; Ginikopoulou, E.; Sakellariou, G.; Fleva, A.; et al. Cellular immunity and levels of parathyroid hormone in uremic patients receiving hemodialysis. Ren. Fail. 2005, 27, 275–278. [Google Scholar]
- Riancho, J.A.; Zarrabeitia, M.T.; de Francisco, A.L.; Amado, J.A.; Napal, J.; Arias, M.; Gonzalez-Macias, J. Vitamin D therapy modulates cytokine secretion in patients with renal failure. Nephron 1993, 65, 364–368. [Google Scholar]
- Jurewicz, M.; McDermott, D.H.; Sechler, J.M.; Tinckam, K.; Takakura, A.; Carpenter, C.B.; Milford, E.; Abdi, R. Human T and natural killer cells possess a functional renin-angiotensin system: Further mechanisms of angiotensin II-induced inflammation. J. Am. Soc. Nephrol. 2007, 18, 1093–1102. [Google Scholar] [CrossRef]
- Nataraj, C.; Oliverio, M.I.; Mannon, R.B.; Mannon, P.J.; Audoly, L.P.; Amuchastegui, C.S.; Ruiz, P.; Smithies, O.; Coffman, T.M. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J. Clin. Invest. 1999, 104, 1693–1701. [Google Scholar]
- El Bekay, R.; Alvarez, M.; Monteseirin, J.; Alba, G.; Chacon, P.; Vega, A.; Martin-Nieto, J.; Jimenez, J.; Pintado, E.; Bedoya, F.J.; et al. Oxidative stress is a critical mediator of the angiotensin II signal in human neutrophils: involvement of mitogen-activated protein kinase, calcineurin, and the transcription factor NF-kappaB. Blood 2003, 102, 662–671. [Google Scholar] [CrossRef]
- Suzuki, Y.; Gomez-Guerrero, C.; Shirato, I.; Lopez-Franco, O.; Hernandez-Vargas, P.; Sanjuan, G.; Ruiz-Ortega, M.; Sugaya, T.; Okumura, K.; Tomino, Y.; et al. Susceptibility to T cell-mediated injury in immune complex disease is linked to local activation of renin-angiotensin system: The role of NF-AT pathway. J. Immunol. 2002, 169, 4136–4146. [Google Scholar]
- Vanholder, R.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; Descamps-Latscha, B.; Henle, T.; et al. Uremic toxicity: present state of the art. Int. J. Artif. Organs 2001, 24, 695–725. [Google Scholar]
- Schepers, E.; Glorieux, G.; Vanholder, R. The gut: the forgotten organ in uremia? Blood Purif. 2010, 29, 130–136. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and Pathologic Concentrations of Uremic Toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Neirynck, N.; Glorieux, G. An update on protein-bound uremic retention solutes. J. Ren. Nutr. 2012, 22, 90–94. [Google Scholar] [CrossRef]
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J. A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar]
- Cohen, G.; Glorieux, G.; Thornalley, P.; Schepers, E.; Meert, N.; Jankowski, J.; Jankowski, V.; Argiles, A.; Anderstam, B.; Brunet, P.; et al. Review on uraemic toxins III: Recommendations for handling uraemic retention solutes in vitro—towards a standardized approach for research on uraemia. Nephrol. Dial. Transplant. 2007, 22, 3381–3390. [Google Scholar] [CrossRef]
- Meert, N.; Schepers, E.; De Smet, R.; Argiles, A.; Cohen, G.; Deppisch, R.; Drueke, T.; Massy, Z.; Spasovski, G.; Stegmayr, B.; et al. Inconsistency of reported uremic toxin concentrations. Artif. Organs 2007, 31, 600–611. [Google Scholar] [CrossRef]
- Vanholder, R.; Meert, N.; Schepers, E.; Glorieux, G.; Argiles, A.; Brunet, P.; Cohen, G.; Drueke, T.; Mischak, H.; Spasovski, G.; et al. Review on uraemic solutes II—variability in reported concentrations: causes and consequences. Nephrol. Dial. Transplant. 2007, 22, 3115–3121. [Google Scholar]
- Schmidt, S.; Westhoff, T.H.; Krauser, P.; Ignatius, R.; Jankowski, J.; Jankowski, V.; Zidek, W.; van der Giet, M. The uraemic toxin phenylacetic acid impairs macrophage function. Nephrol. Dial. Transplant. 2008, 23, 3485–3493. [Google Scholar] [CrossRef]
- Schepers, E.; Glorieux, G.; Jankowski, V.; Dhondt, A.; Jankowski, J.; Vanholder, R. Dinucleoside polyphosphates: Newly detected uraemic compounds with an impact on leucocyte oxidative burst. Nephrol. Dial. Transplant. 2010, 25, 2636–2644. [Google Scholar]
- Hirayama, A.; Noronha-Dutra, A.A.; Gordge, M.P.; Neild, G.H.; Hothersall, J.S. Inhibition of neutrophil superoxide production by uremic concentrations of guanidino compounds. J. Am. Soc. Nephrol. 2000, 11, 684–689. [Google Scholar]
- Glorieux, G.L.; Dhondt, A.W.; Jacobs, P.; Van Langeraert, J.; Lameire, N.H.; De Deyn, P.P.; Vanholder, R.C. In vitro study of the potential role of guanidines in leukocyte functions related to atherogenesis and infection. Kidney Int. 2004, 65, 2184–2192. [Google Scholar] [CrossRef]
- Schepers, E.; Glorieux, G.; Dhondt, A.; Leybaert, L.; Vanholder, R. Role of symmetric dimethylarginine in vascular damage by increasing ROS via store-operated calcium influx in monocytes. Nephrol. Dial. Transplant. 2009, 24, 1429–1435. [Google Scholar]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar]
- Postea, O.; Krotz, F.; Henger, A.; Keller, C.; Weiss, N. Stereospecific and redox-sensitive increase in monocyte adhesion to endothelial cells by homocysteine. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 508–513. [Google Scholar]
- Stopper, H.; Treutlein, A.T.; Bahner, U.; Schupp, N.; Schmid, U.; Brink, A.; Perna, A.; Heidland, A. Reduction of the genomic damage level in haemodialysis patients by folic acid and vitamin B12 supplementation. Nephrol. Dial. Transplant. 2008, 23, 3272–3279. [Google Scholar]
- Perna, A.F.; Capasso, R.; Lombardi, C.; Acanfora, F.; Satta, E.; Ingrosso, D. Hyperhomocysteinemia and macromolecule modifications in uremic patients. Clin. Chem. Lab. Med. 2005, 43, 1032–1038. [Google Scholar]
- Nakayama, M.; Nakayama, K.; Zhu, W.J.; Shirota, Y.; Terawaki, H.; Sato, T.; Kohno, M.; Ito, S. Polymorphonuclear leukocyte injury by methylglyoxal and hydrogen peroxide: A possible pathological role for enhanced oxidative stress in chronic kidney disease. Nephrol. Dial. Transplant. 2008, 23, 3096–3102. [Google Scholar]
- Ward, R.A.; McLeish, K.R. Methylglyoxal: A stimulus to neutrophil oxygen radical production in chronic renal failure? Nephrol. Dial. Transplant. 2004, 19, 1702–1707. [Google Scholar] [CrossRef]
- Okado, A.; Kawasaki, Y.; Hasuike, Y.; Takahashi, M.; Teshima, T.; Fujii, J.; Taniguchi, N. Induction of apoptotic cell death by methylglyoxal and 3-deoxyglucosone in macrophage-derived cell lines. Biochem. Biophys. Res. Commun. 1996, 225, 219–224. [Google Scholar]
- Cohen, G.; Haag-Weber, M.; Mai, B.; Deicher, R.; Hörl, W.H. Effect of immunoglobulin light chains from hemodialysis and continuous ambulatory peritoneal dialysis patients on polymorphonuclear leukocyte functions. J. Am. Soc. Nephrol. 1995, 6, 1592–1599. [Google Scholar]
- Cohen, G.; Hörl, W.H. Retinol binding protein isolated from acute renal failure patients inhibits polymorphonuclear leucocyte functions. Eur. J. Clin. Invest. 2004, 34, 774–781. [Google Scholar]
- Cohen, G.; Raupachova, J.; Ilic, D.; Werzowa, J.; Hörl, W.H. Effect of leptin on polymorphonuclear leucocyte functions in healthy subjects and haemodialysis patients. Nephrol. Dial. Transplant. 2011, 26, 2271–2281. [Google Scholar]
- Cohen, G.; Ilic, D.; Raupachova, J.; Hörl, W.H. Resistin inhibits essential functions of polymorphonuclear leukocytes. J. Immunol. 2008, 181, 3761–3768. [Google Scholar]
- Wimmer, T.; Cohen, G.; Säemann, M.D.; Hörl, W.H. Effects of Tamm-Horsfall protein on polymorphonuclear leukocyte function. Nephrol. Dial. Transplant. 2004, 19, 2192–2197. [Google Scholar]
- Tolle, M.; Huang, T.; Schuchardt, M.; Jankowski, V.; Prufer, N.; Jankowski, J.; Tietge, U.J.; Zidek, W.; van der Giet, M. High-density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-inflammatory-serum amyloid A. Cardiovasc. Res. 2012, 94, 154–162. [Google Scholar]
- Weichhart, T.; Kopecky, C.; Kubicek, M.; Haidinger, M.; Doller, D.; Katholnig, K.; Suarna, C.; Eller, P.; Tolle, M.; Gerner, C.; et al. Serum amyloid A in uremic HDL promotes inflammation. J. Am. Soc. Nephrol. 2012, 23, 934–947. [Google Scholar] [CrossRef]
- Glorieux, G.; Helling, R.; Henle, T.; Brunet, P.; Deppisch, R.; Lameire, N.; Vanholder, R. In vitro evidence for immune activating effect of specific AGE structures retained in uremia. Kidney Int. 2004, 66, 1873–1880. [Google Scholar] [CrossRef]
- Rashid, G.; Korzets, Z.; Bernheim, J. Advanced glycation end products stimulate tumor necrosis factor-alpha and interleukin-1 beta secretion by peritoneal macrophages in patients on continuous ambulatory peritoneal dialysis. Isr. Med. Assoc. J. 2006, 8, 36–39. [Google Scholar]
- Kirstein, M.; Brett, J.; Radoff, S.; Ogawa, S.; Stern, D.; Vlassara, H. Advanced protein glycosylation induces transendothelial human monocyte chemotaxis and secretion of platelet-derived growth factor: Role in vascular disease of diabetes and aging. Proc. Natl. Acad. Sci. USA 1990, 87, 9010–9014. [Google Scholar]
- Toure, F.; Zahm, J.M.; Garnotel, R.; Lambert, E.; Bonnet, N.; Schmidt, A.M.; Vitry, F.; Chanard, J.; Gillery, P.; Rieu, P. Receptor for advanced glycation end-products (RAGE) modulates neutrophil adhesion and migration on glycoxidated extracellular matrix. Biochem. J. 2008, 416, 255–261. [Google Scholar]
- Witko-Sarsat, V.; Gausson, V.; Nguyen, A.T.; Touam, M.; Drueke, T.; Santangelo, F.; Descamps-Latscha, B. AOPP-induced activation of human neutrophil and monocyte oxidative metabolism: A potential target for N-acetylcysteine treatment in dialysis patients. Kidney Int. 2003, 64, 82–91. [Google Scholar] [CrossRef]
- Nguyen-Khoa, T.; Massy, Z.A.; Witko-Sarsat, V.; Canteloup, S.; Kebede, M.; Lacour, B.; Drueke, T.; Descamps-Latscha, B. Oxidized low-density lipoprotein induces macrophage respiratory burst via its protein moiety: A novel pathway in atherogenesis? Biochem. Biophys. Res. Commun. 1999, 263, 804–809. [Google Scholar] [CrossRef]
- Sedgwick, J.B.; Hwang, Y.S.; Gerbyshak, H.A.; Kita, H.; Busse, W.W. Oxidized low density lipoprotein activates migration and degranulation of human granulocytes. Am. J. Respir. Cell. Mol. Biol. 2003, 30, 30. [Google Scholar]
- Meier, P.; Golshayan, D.; Blanc, E.; Pascual, M.; Burnier, M. Oxidized LDL modulates apoptosis of regulatory T cells in patients with ESRD. J. Am. Soc. Nephrol. 2009, 20, 1368–1384. [Google Scholar]
- Capasso, R.; Sambri, I.; Cimmino, A.; Salemme, S.; Lombardi, C.; Acanfora, F.; Satta, E.; Puppione, D.L.; Perna, A.F.; Ingrosso, D. Homocysteinylated albumin promotes increased monocyte-endothelial cell adhesion and up-regulation of MCP1, Hsp60 and ADAM17. PLoS One 2012, 7, e31388. [Google Scholar]
- Jankowski, J.; van der Giet, M.; Jankowski, V.; Schmidt, S.; Hemeier, M.; Mahn, B.; Giebing, G.; Tolle, M.; Luftmann, H.; Schluter, H.; et al. Increased plasma phenylacetic acid in patients with end-stage renal failure inhibits iNOS expression. J. Clin. Invest. 2003, 112, 256–264. [Google Scholar]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients—a prospective cohort study. Nephrol. Dial. Transplant. 2011, 27, 1169–1175. [Google Scholar]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transpl. 2009, 25, 1183–1191. [Google Scholar]
- Chauveau, P.; Chadefaux, B.; Coude, M.; Aupetit, J.; Hannedouche, T.; Kamoun, P.; Jungers, P. Increased plasma homocysteine concentration in patients with chronic renal failure. Miner. Electrolyte Metab. 1992, 18, 196–198. [Google Scholar]
- Beltowski, J. Protein homocysteinylation: A new mechanism of atherogenesis? Postep. Hig. Med. Dosw. 2005, 59, 392–404. [Google Scholar]
- Hannam-Harris, A.C.; Gordon, J.; Smith, J.L. Immunoglobulin synthesis by neoplastic B lymphocytes: Free light chain synthesis as a marker of B cell differentiation. J. Immunol. 1980, 125, 2177–2181. [Google Scholar]
- Hutchison, C.A.; Harding, S.; Hewins, P.; Mead, G.P.; Townsend, J.; Bradwell, A.R.; Cockwell, P. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1684–1690. [Google Scholar]
- Cohen, G.; Rudnicki, M.; Schmaldienst, S.; Hörl, W.H. Effect of dialysis on serum/plasma levels of free immunoglobulin light chains in end-stage renal disease patients. Nephrol. Dial. Transplant. 2002, 17, 879–883. [Google Scholar]
- Hutchison, C.A.; Cockwell, P.; Reid, S.; Chandler, K.; Mead, G.P.; Harrison, J.; Hattersley, J.; Evans, N.D.; Chappell, M.J.; Cook, M.; et al. Efficient removal of immunoglobulin free light chains by hemodialysis for multiple myeloma: In vitro and in vivo studies. J. Am. Soc. Nephrol. 2007, 18, 886–895. [Google Scholar] [CrossRef]
- Lepreux, S.; Bioulac-Sage, P.; Gabbiani, G.; Sapin, V.; Housset, C.; Rosenbaum, J.; Balabaud, C.; Desmouliere, A. Cellular retinol-binding protein-1 expression in normal and fibrotic/cirrhotic human liver: Different patterns of expression in hepatic stellate cells and (myo)fibroblast subpopulations. J. Hepatol. 2004, 40, 774–780. [Google Scholar]
- Melhus, H.; Li, Q.; Nordlinder, H.; Farnebo, L.O.; Grimelius, L. Expression of cellular retinol- and retinoic acid-binding proteins in normal and pathologic human parathyroid glands. Endocr. Pathol. 2001, 12, 423–427. [Google Scholar]
- Busch, C.; Siegenthaler, G.; Vahlquist, A.; Nordlinder, H.; Sundelin, J.; Saksena, P.; Eriksson, U. Expression of cellular retinoid-binding proteins during normal and abnormal epidermal differentiation. J. Invest. Dermatol. 1992, 99, 795–802. [Google Scholar]
- Ong, D.E.; Page, D.L. Cellular retinol-binding protein (type two) is abundant in human small intestine. J. Lipid Res. 1987, 28, 739–745. [Google Scholar]
- Scarpioni, L.; Dall’aglio, P.P.; Poisetti, P.G.; Buzio, C. Retinol binding protein in serum and in urine of glomerular and tubular nephropathies. Clin. Chim. Acta 1976, 68, 107–113. [Google Scholar]
- Bankson, D.D.; Rifai, N.; Silverman, L.M. Serum retinol-binding protein and creatinine in onset of and recovery from acute renal failure. Clin. Chem. 1987, 33, 1942. [Google Scholar]
- Axelsson, J.; O’Byrne, S.M.; Blaner, W.S.; Carrero, J.J.; Bruchfeld, A.; Heimburger, O.; Barany, P.; Lindholm, B.; Stenvinkel, P. Serum retinol-binding protein concentration and its association with components of the uremic metabolic syndrome in nondiabetic patients with chronic kidney disease stage 5. Am. J. Nephrol. 2008, 29, 447–453. [Google Scholar]
- DeLany, J. Leptin hormone and other biochemical influences on systemic inflammation. J. Bodyw. Mov. Ther. 2008, 12, 121–132. [Google Scholar]
- Teta, D. Adipokines as uremic toxins. J. Ren. Nutr. 2012, 22, 81–85. [Google Scholar]
- Kopp, A.; Buechler, C.; Neumeier, M.; Weigert, J.; Aslanidis, C.; Scholmerich, J.; Schaffler, A. Innate immunity and adipocyte function: Ligand-specific activation of multiple Toll-like receptors modulates cytokine, adipokine, and chemokine secretion in adipocytes. Obesity (Silver Spring) 2009, 17, 648–656. [Google Scholar] [CrossRef]
- Diez, J.J.; Iglesias, P.; Fernandez-Reyes, M.J.; Aguilera, A.; Bajo, M.A.; Alvarez-Fidalgo, P.; Codoceo, R.; Selgas, R. Serum concentrations of leptin, adiponectin and resistin, and their relationship with cardiovascular disease in patients with end-stage renal disease. Clin. Endocrinol. (Oxf.) 2005, 62, 242–249. [Google Scholar] [CrossRef]
- Widjaja, A.; Kielstein, J.T.; Horn, R.; von zur Muhlen, A.; Kliem, V.; Brabant, G. Free serum leptin but not bound leptin concentrations are elevated in patients with end-stage renal disease. Nephrol. Dial. Transplant. 2000, 15, 846–850. [Google Scholar]
- Aminzadeh, M.A.; Pahl, M.V.; Barton, C.H.; Doctor, N.S.; Vaziri, N.D. Human uraemic plasma stimulates release of leptin and uptake of tumour necrosis factor-alpha in visceral adipocytes. Nephrol. Dial. Transplant. 2009, 24, 3626–3631. [Google Scholar]
- Curat, C.A.; Wegner, V.; Sengenes, C.; Miranville, A.; Tonus, C.; Busse, R.; Bouloumie, A. Macrophages in human visceral adipose tissue: increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 2006, 49, 744–747. [Google Scholar]
- Kunnari, A.M.; Savolainen, E.R.; Ukkola, O.H.; Kesaniemi, Y.A.; Jokela, M.A. The expression of human resistin in different leucocyte lineages is modulated by LPS and TNFalpha. Regul. Pept. 2009, 157, 57–63. [Google Scholar]
- Axelsson, J.; Bergsten, A.; Qureshi, A.R.; Heimburger, O.; Barany, P.; Lonnqvist, F.; Lindholm, B.; Nordfors, L.; Alvestrand, A.; Stenvinkel, P. Elevated resistin levels in chronic kidney disease are associated with decreased glomerular filtration rate and inflammation, but not with insulin resistance. Kidney Int. 2006, 69, 596–604. [Google Scholar]
- Nusken, K.D.; Kratzsch, J.; Wienholz, V.; Stohr, W.; Rascher, W.; Dotsch, J. Circulating resistin concentrations in children depend on renal function. Nephrol. Dial. Transplant. 2006, 21, 107–112. [Google Scholar]
- Palanivel, R.; Maida, A.; Liu, Y.; Sweeney, G. Regulation of insulin signalling, glucose uptake and metabolism in rat skeletal muscle cells upon prolonged exposure to resistin. Diabetologia 2006, 49, 183–190. [Google Scholar]
- Bostrom, E.A.; Tarkowski, A.; Bokarewa, M. Resistin is stored in neutrophil granules being released upon challenge with inflammatory stimuli. Biochim. Biophys. Acta 2009, 1793, 1894–1900. [Google Scholar] [CrossRef]
- Walcher, D.; Hess, K.; Berger, R.; Aleksic, M.; Heinz, P.; Bach, H.; Durst, R.; Hausauer, A.; Hombach, V.; Marx, N. Resistin: A newly identified chemokine for human CD4-positive lymphocytes. Cardiovasc. Res. 2010, 85, 167–174. [Google Scholar]
- Saemann, M.D.; Weichhart, T.; Hörl, W.H.; Zlabinger, G.J. Tamm-Horsfall protein: A multilayered defence molecule against urinary tract infection. Eur. J. Clin. Invest. 2005, 35, 227–235. [Google Scholar]
- Prajczer, S.; Heidenreich, U.; Pfaller, W.; Kotanko, P.; Lhotta, K.; Jennings, P. Evidence for a role of uromodulin in chronic kidney disease progression. Nephrol. Dial. Transplant. 2010, 25, 1896–1903. [Google Scholar]
- Saemann, M.D.; Weichhart, T.; Zeyda, M.; Staffler, G.; Schunn, M.; Stuhlmeier, K.M.; Sobanov, Y.; Stulnig, T.M.; Akira, S.; von Gabain, A.; et al. Tamm-Horsfall glycoprotein links innate immune cell activation with adaptive immunity via a Toll-like receptor-4-dependent mechanism. J. Clin. Invest. 2005, 115, 468–475. [Google Scholar]
- Murphy, A.J.; Woollard, K.J.; Suhartoyo, A.; Stirzaker, R.A.; Shaw, J.; Sviridov, D.; Chin-Dusting, J.P. Neutrophil activation is attenuated by high-density lipoprotein and apolipoprotein A–I in in vitro and in vivo models of inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1333–1341. [Google Scholar] [CrossRef]
- Murphy, A.J.; Woollard, K.J.; Hoang, A.; Mukhamedova, N.; Stirzaker, R.A.; McCormick, S.P.; Remaley, A.T.; Sviridov, D.; Chin-Dusting, J. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2071–2077. [Google Scholar]
- Liao, X.L.; Lou, B.; Ma, J.; Wu, M.P. Neutrophils activation can be diminished by apolipoprotein A–I. Life Sci. 2005, 77, 325–335. [Google Scholar]
- Reiser, K.M. Nonenzymatic glycation of collagen in aging and diabetes. Proc. Soc. Exp. Biol. Med. 1998, 218, 23–37. [Google Scholar]
- Thornalley, P. The clinical significance of glycation. Clin. Lab. 1999, 45, 263–273. [Google Scholar]
- Miyata, T.; Maeda, K.; Kurokawa, K.; van Ypersele de Strihou, C. Oxidation conspires with glycation to generate noxious advanced glycation end products in renal failure. Nephrol. Dial. Transplant. 1997, 12, 255–258. [Google Scholar]
- Makita, Z.; Radoff, S.; Rayfield, E.J.; Yang, Z.; Skolnik, E.; Delaney, V.; Friedman, E.A.; Cerami, A.; Vlassara, H. Advanced glycosylation end products in patients with diabetic nephropathy. N. Engl. J. Med. 1991, 325, 836–842. [Google Scholar]
- Capeillere-Blandin, C.; Gausson, V.; Nguyen, A.T.; Descamps-Latscha, B.; Drueke, T.; Witko-Sarsat, V. Respective role of uraemic toxins and myeloperoxidase in the uraemic state. Nephrol. Dial. Transplant. 2006, 21, 1555–1563. [Google Scholar]
- Kormoczi, G.F.; Wolfel, U.M.; Rosenkranz, A.R.; Hörl, W.H.; Oberbauer, R.; Zlabinger, G.J. Serum proteins modified by neutrophil-derived oxidants as mediators of neutrophil stimulation. J. Immunol. 2001, 167, 451–460. [Google Scholar]
- Donadio, C.; Tognotti, D.; Donadio, E. Albumin modification and fragmentation in renal disease. Clin. Chim. Acta 2012, 413, 391–395. [Google Scholar]
- Mera, K.; Anraku, M.; Kitamura, K.; Nakajou, K.; Maruyama, T.; Tomita, K.; Otagiri, M. Oxidation and carboxy methyl lysine-modification of albumin: Possible involvement in the progression of oxidative stress in hemodialysis patients. Hypertens. Res. 2005, 28, 973–980. [Google Scholar]
- Kraus, L.M.; Kraus, A.P., Jr. Carbamoylation of amino acids and proteins in uremia. Kidney Int. 2001, 78, S102–S107. [Google Scholar]
- Kraus, L.M.; Elberger, A.J.; Handorf, C.R.; Pabst, M.J.; Kraus, A.P., Jr. Urea-derived cyanate forms epsilon-amino-carbamoyl-lysine (homocitrulline) in leukocyte proteins in patients with end-stage renal disease on peritoneal dialysis. J. Lab. Clin. Med. 1994, 123, 882–891. [Google Scholar]
- Pavone, B.; Sirolli, V.; Giardinelli, A.; Bucci, S.; Forli, F.; Di Cesare, M.; Sacchetta, P.; Di Pietro, N.; Pandolfi, A.; Urbani, A.; et al. Plasma protein carbonylation in chronic uremia. J. Nephrol. 2011, 24, 453–464. [Google Scholar] [CrossRef]
- Verkade, M.A.; van Druningen, C.J.; Vaessen, L.M.; Hesselink, D.A.; Weimar, W.; Betjes, M.G. Functional impairment of monocyte-derived dendritic cells in patients with severe chronic kidney disease. Nephrol. Dial. Transplant. 2007, 22, 128–138. [Google Scholar]
- Lim, W.H.; Kireta, S.; Leedham, E.; Russ, G.R.; Coates, P.T. Uremia impairs monocyte and monocyte-derived dendritic cell function in hemodialysis patients. Kidney Int. 2007, 72, 1138–1148. [Google Scholar]
- Panzer, U.; Kurts, C. T cell cross-talk with kidney dendritic cells in glomerulonephritis. J. Mol. Med. 2010, 88, 19–26. [Google Scholar]
- Zeyda, M.; Kirsch, B.M.; Geyeregger, R.; Stuhlmeier, K.M.; Zlabinger, G.J.; Hörl, W.H.; Saemann, M.D.; Stulnig, T.M. Inhibition of human dendritic cell maturation and function by the novel immunosuppressant FK778. Transplantation 2005, 80, 1105–1111. [Google Scholar] [CrossRef]
- Zeyda, M.; Geyeregger, R.; Poglitsch, M.; Weichhart, T.; Zlabinger, G.J.; Koyasu, S.; Hörl, W.H.; Stulnig, T.M.; Watschinger, B.; Saemann, M.D. Impairment of T cell interactions with antigen-presenting cells by immunosuppressive drugs reveals involvement of calcineurin and NF-kappaB in immunological synapse formation. J. Leukoc. Biol. 2007, 81, 319–327. [Google Scholar]
- Stenvinkel, P.; Ekstrom, T.J. Does the uremic milieu affect the epigenotype? J. Ren. Nutr. 2009, 19, 82–85. [Google Scholar] [CrossRef]
- Chmielewski, M.; Lindholm, B.; Stenvinkel, P.; Ekstrom, J.T. The role of epigenetics in kidney diseases. Prilozi 2011, 32, 45–54. [Google Scholar]
- Jimenez, R.; Carracedo, J.; Santamaria, R.; Soriano, S.; Madueno, J.A.; Ramirez, R.; Rodriguez, M.; Martin-Malo, A.; Aljama, P. Replicative senescence in patients with chronic kidney failure. Kidney Int. 2005, 99, S11–S15. [Google Scholar]
- Stoyanova, E.; Sandoval, S.B.; Zuniga, L.A.; El-Yamani, N.; Coll, E.; Pastor, S.; Reyes, J.; Andres, E.; Ballarin, J.; Xamena, N.; et al. Oxidative DNA damage in chronic renal failure patients. Nephrol. Dial. Transplant. 2010, 25, 879–885. [Google Scholar]
- Buemi, M.; Floccari, F.; Costa, C.; Caccamo, C.; Belghity, N.; Campo, S.; Pernice, F.; Bonvissuto, G.; Coppolino, G.; Barilla, A.; et al. Dialysis-related genotoxicity: Sister chromatid exchanges and DNA lesions in T and B lymphocytes of uremic patients. Genomic damage in patients on hemodiafiltration. Blood Purif. 2006, 24, 569–574. [Google Scholar] [CrossRef]
- Sebekova, K.; Wagner, Z.; Schupp, N.; Boor, P. Genomic damage and malignancy in end-stage renal failure: Do advanced glycation end products contribute? Kidney Blood Press. Res. 2007, 30, 56–66. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Ekstrom, T.J. Epigenetics and the uremic phenotype: A matter of balance. Contrib. Nephrol. 2008, 161, 55–62. [Google Scholar]
- Fink, K.; Brink, A.; Vienken, J.; Heidland, A.; Stopper, H. Homocysteine exerts genotoxic and antioxidative effects in vitro. Toxicol In Vitro 2007, 21, 1402–1408. [Google Scholar] [CrossRef]
- Andreini, B.; Panichi, V.; Cirami, C.; Migliori, M.; De Pietro, S.; Taccola, D.; Aloisi, M.; Antonelli, A.; Giusti, R.; Rindi, P.; et al. ANCA in dialysis patients: A role for bioincompatibility? Int. J. Artif. Organs 2000, 23, 97–103. [Google Scholar]
- Schreiber, A.; Xiao, H.; Jennette, J.C.; Schneider, W.; Luft, F.C.; Kettritz, R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 289–298. [Google Scholar] [CrossRef]
- Schreiber, A.; Rolle, S.; Peripelittchenko, L.; Rademann, J.; Schneider, W.; Luft, F.C.; Kettritz, R. Phosphoinositol 3-kinase-gamma mediates antineutrophil cytoplasmic autoantibody-induced glomerulonephritis. Kidney Int. 2010, 77, 118–128. [Google Scholar]
- Harper, L.; Cockwell, P.; Adu, D.; Savage, C.O. Neutrophil priming and apoptosis in anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Kidney Int. 2001, 59, 1729–1738. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cohen, G.; Hörl, W.H. Immune Dysfunction in Uremia—An Update. Toxins 2012, 4, 962-990. https://doi.org/10.3390/toxins4110962
Cohen G, Hörl WH. Immune Dysfunction in Uremia—An Update. Toxins. 2012; 4(11):962-990. https://doi.org/10.3390/toxins4110962
Chicago/Turabian StyleCohen, Gerald, and Walter H. Hörl. 2012. "Immune Dysfunction in Uremia—An Update" Toxins 4, no. 11: 962-990. https://doi.org/10.3390/toxins4110962
APA StyleCohen, G., & Hörl, W. H. (2012). Immune Dysfunction in Uremia—An Update. Toxins, 4(11), 962-990. https://doi.org/10.3390/toxins4110962