A qPCR Assay to Detect and Quantify Shiga Toxin-Producing E. coli (STEC) in Cattle and on Farms: A Potential Predictive Tool for STEC Culture-Positive Farms


Abstract
:1. Introduction
2. Results
2.1. Method Development and Testing

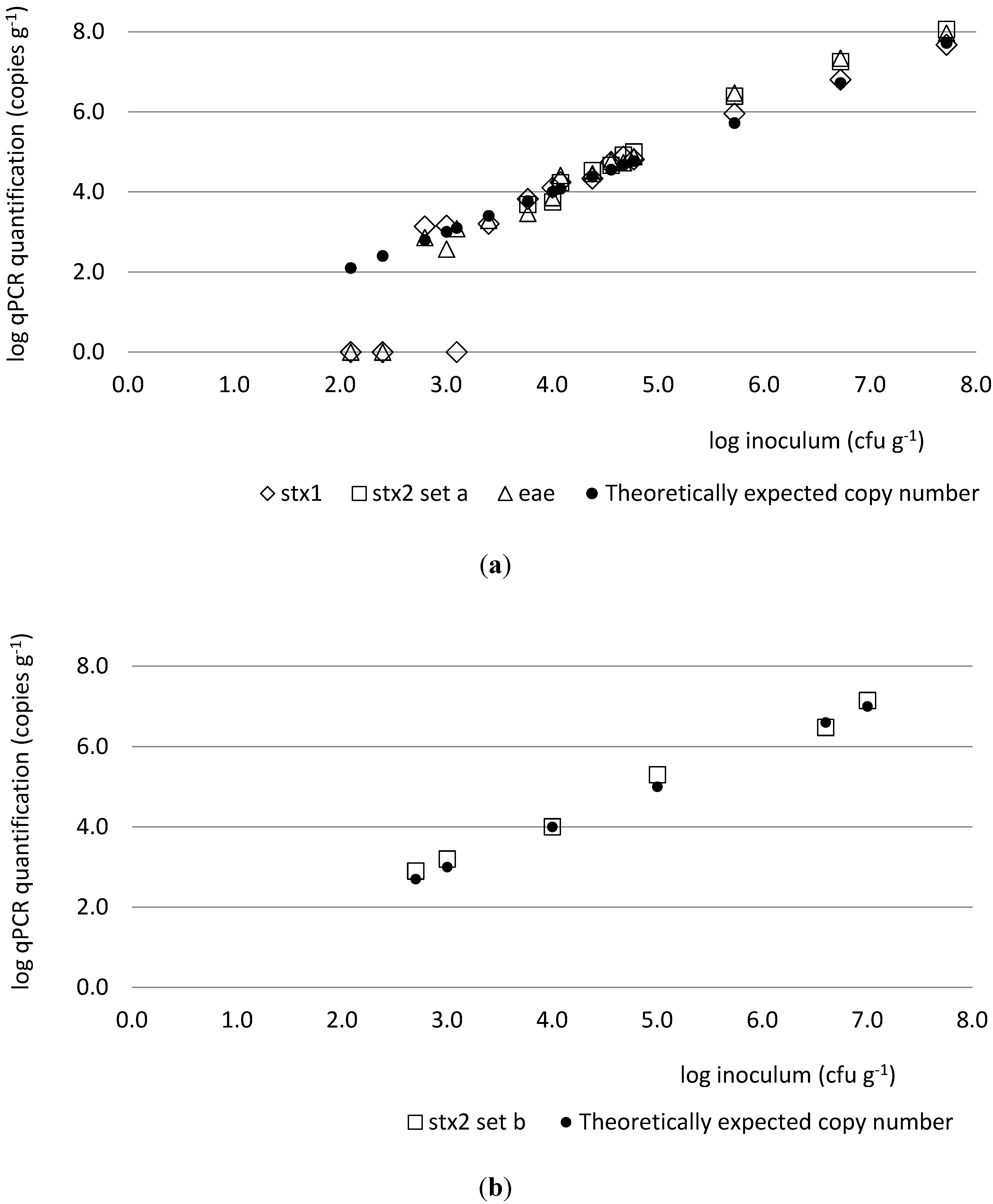{kind=link}
{kind=link}
| Gene | Primer or probe ∂ | Sequence (5’- 3’) † | Position (5’- 3’) | Accession number |
|---|---|---|---|---|
| stx1 | stx1-F | GAC GCA GTC TGT IGC AAG AG | 516-535 | Z36899 |
| stx1-R | cga aaa cgi aaa gct tca gct g | 581-560 | Z36899 | |
| stx1-P Ф | ATG TTA CGG TTT GTT ACT GTG | 538-558 | Z36899 | |
| stx2 | stx2-F | TCA GGC AIA TAC AGA GAG AAT TTC G | 578-602 | AY443044 |
| stx2-Ra | ccg gig tca tcg tat aca cag | 646-626 | AY443044 | |
| stx2-Rb | ccg gig tca tcg tat aaa cag | 646-626 | AY443044 | |
| stx2-P Ф | CAC TGT CTG AAA CTG CT | 608-624 | AY443044 | |
| eae | eae-F | GGA AGC CAA AGC GCA CAA | 1507-1524 | AF025311 |
| eae-R | ggc icg agc igt cac ttt ata a | 1593-1572 | AF025311 | |
| eae-P § | TAC CAG GCT ATT TTG CCI GCT TAT GTG C | 1528-1555 | AF025311 |
| Target gene | LOQ ∂ (copies/reaction) | Cq Ф for 105 copies/reaction | Efficiency | R2 (Regression coefficient) |
|---|---|---|---|---|
| stx1 | 1 to 10 | 24.3 | 90% | 0.9997 |
| stx2 (using primer set a) | 10 to 100 | 22.8 | 94% | 0.9983 |
| stx2(using primer set b) | 1 to 10 | 23.9 | 87% | 0.9997 |
| eae | 1 to 10 | 23.9 | 98% | 0.9992 |

| Species orbacterial subgroup | Strain | Serotype | Virulence genes | qPCR detection | ||||
|---|---|---|---|---|---|---|---|---|
| stx1 | stx2 | eae | stx1 | stx2 | eae | |||
| EPEC ∂ | MB3885 † | O157 | - | - | eae γ1 | - | - | + |
| MB3886 | O157 | - | - | eae γ1 | - | - | + | |
| STEC Ф | MB3892 | O91 | stx1ab | stx2b | - | + | + | - |
| MB3900 | O175 | - | stx2 | - | - | + | - | |
| MB3957 | O146 | stx1ab, stx1c | stx2b | - | + | + | - | |
| MB3963 | O128ab | stx1ab, stx1c | stx2b | - | + | + | - | |
| MB3986 | O181 | stx1ab | - | - | + | - | - | |
| MB4213 | no info | - | Stx2d, stx2e, stx2g | - | - | + | - | |
| MB4376 (EH250) | O118 | - | stx2b | - | - | + | - | |
| MB4377 | no info | stx1d | - | - | + | - | - | |
| MB4378 | O138 | - | stx2e | - | - | + | - | |
| MB4380 | no info | stx1c | stx2b | - | + | + | - | |
| MB3893 | O145 | stx1ab | - | eae γ1 | + | - | + | |
| MB3920 | O157 | - | stx2 | eae γ1 | - | + | + | |
| MB3936 | O26 | stx1ab | stx2 | eae β1 | + | + | + | |
| MB3938 | O145 | - | stx2d | eae γ1 | - | + | + | |
| MB4033 | O111 | stx1ab | stx2 | eae γ2 | + | + | + | |
| MB4074 | O26 | stx1ab | - | eae β1 | + | - | + | |
| MB4108 | O111 | stx1ab | stx2 | eae γ2 | + | + | + | |
| MB4117 | O103 | stx1ab | - | eae ε | + | - | + | |
| MB4141 | O103 | stx1ab | stx2d | eae ε | + | + | + | |
| MB4208 | O157 | stx1ab | stx2c | eae γ1 | + | + | + | |
| MB4379 | Orough | - | stx2f | eae | - | - | + | |
| ETEC § | MB1520 | - | - | - | - | - | - | - |
| Escherichia coli | MB544 (LMG2092T) | - | - | - | - | - | - | - |
| Shigella dysenteriae | MB 4436 (CIP 57.28) | - | stx1ab | - | - | + | - | - |
| Citrobacter rodentium | MB4471 (ATCC 51116) | - | - | - | eae | - | - | + |
| Escherichia albertii | MB4434 (LMG 20972) | - | - | - | eae | - | - | + |
| Enterobacter aerogenes | MB260 | - | - | - | - | - | - | - |
| Citrobacter diversus | MB423 | - | - | - | - | - | - | - |
| Hafnia alvei | MB291 | - | - | - | - | - | - | - |
| Klebsiella pneumoniae | MB263 | - | - | - | - | - | - | - |
| Salmonella Dublin | MB1145 | - | - | - | - | - | - | - |
| Salmonella Typhimurium | MB1135 | - | - | - | - | - | - | - |
| Serratia proteamaculans | MB262 | - | - | - | - | - | - | - |
| Shigella boydii | MB4435 | - | - | - | - | - | - | - |
| Yersinia enterocolitica | MB868 | - | - | - | - | - | - | - |
| Campylobacter jejuni | MB1263 | - | - | - | - | - | - | - |
| Pseudomonas aeruginosa | MB289 | - | - | - | - | - | - | - |
| Bacillus subtilis | MB3611 | - | - | - | - | - | - | - |
| Clostridium perfringens | MB128 | - | - | - | - | - | - | - |
| Enterococcus faecalis | MB30 | - | - | - | - | - | - | - |
| Listeria monocytogenes | MB38 | - | - | - | - | - | - | - |
| Staphylococcus aureus | MB4038 | - | - | - | - | - | - | - |
| Streptococcus thermophilus | MB1654 | - | - | - | - | - | - | - |
2.2. Study of Molecular Method on Native Cattle Fecal Samples
| Sample | Farm | Serogroup ∂ | Virulence gene isolate | Enumeration of virulence genes in fecal sample (log copies g−1) | |||||
|---|---|---|---|---|---|---|---|---|---|
| stx1 | stx2 (using primer set a) | stx2b (using primer set b) | eae | ||||||
| A1 | A | O157 | stx1 | stx2 | eae | 0.0 | 4.7 | 0.0 | 4.2 |
| A7 | A | O157 | stx1 | stx2 | eae | 0.0 | 4.2 | 0.0 | 3.5 |
| A8 | A | O157 | stx1 | stx2 | eae | 0.0 | 3.5 | 0.0 | 0.0 |
| A9 | A | O157 | stx1 | stx2 | eae | 0.0 | 2.8 | 0.0 | 0.0 |
| B9 | B | - | - | stx2 | - | 0.0 | 4.4 | 0.0 | 0.0 |
| C1 | C | - | stx1 | stx2 | - | 0.0 | 0.0 | 0.0. | 0.0 |
| C3 | C | - | - | stx2 | - | 0.0 | 4.4 | 0.0 | 0.0 |
| C4 | C | O26 | stx1 | - | eae | 4.8 | 5.6 | 5.1 | 0.0 |
| C6 | C | O26 | stx1 | - | eae | 4.3 | 4.4 | 0.0 | 0.0 |
| C9 | C | O26 | stx1 | - | eae | 0.0 | 5.2 | 5.0 | 0.0 |

| Result culture method:STEC culture-positive farm | Total | |||
|---|---|---|---|---|
| Negative | Positive | |||
| Result qPCR method: stx detection in the individual fecal sample | Negative | 23 | 5 | 28 |
| Positive | 7 | 24 | 31 | |
| Total | 30 | 29 | 59 | |
3. Discussion
4. Experimental Section
4.1. Method Development and Testing
4.1.1. Bacterial Isolates
4.1.2. Cattle Fecal Samples
Sample Preparation and DNA Extraction
Artificial Contamination with STEC
4.1.3. Real-Time PCR (qPCR)
Preparation of DNA Standards
Primers and Probes
qPCR Using SYBR Green Fluorescence
qPCR Using Hydrolysis Probes
Quantification of STEC Virulence Genes in an Artificially Contaminated Cattle Fecal Sample
4.2. Study of the qPCR Assays on Native Cattle Feces Samples
4.2.1. Quantification of STEC Virulence Genes in Native Fecal Samples
4.2.2. Classical Culture for Isolation of STEC
4.3. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Beutin, L.; Krause, G.; Zimmermann, S.; Kaulfuss, S.; Gleier, K. Characterization of Shiga toxin-producing Escherichia coli strains isolated from human patients in Germany over a 3-year period. J. Clin. Microbiol. 2004, 42, 1099–1108. [Google Scholar] [CrossRef]
- Karmali, M.A. Infection by verocytotoxin-producing Escherichia coli. Clin. Microbiol. Rev. 1989, 2, 15–38. [Google Scholar]
- Vaz, T.M.; Irino, K.; Nishimura, L.S.; Cergole-Novella, M.C.; Guth, B.E. Genetic heterogeneity of Shiga toxin-producing Escherichia coli strains isolated in Sao Paulo, Brazil, from 1976 through 2003, as revealed by pulsed-field gel electrophoresis. J. Clin. Microbiol. 2006, 44, 798–804. [Google Scholar] [CrossRef]
- Verstraete, K.; de Reu, K.; van Weyenberg, S.; Piérard, D.; de Zutter, L.; Herman, L.; Robyn, J.; Heyndrickx, M. Genetic characteristics of Shiga toxin-producing E. coli O157, O26, O103, O111, and O145 isolates from humans, food, and cattle in Belgium. Epidemiol. Infect. 2012, 141, 2503–2515. [Google Scholar]
- Ramachandran, V.; Brett, K.; Hornitzky, M.A.; Dowton, M.; Bettelheim, K.A.; Walker, M.J.; Djordjevic, S.P. Distribution of intimin subtypes among Escherichia coli isolates from ruminant and human sources. J. Clin. Microbiol. 2003, 41, 5022–5032. [Google Scholar] [CrossRef]
- Persson, S.; Olsen, K.E.P.; Ethelberg, S.; Scheutz, F. Subtyping method for Escherichia coli Shiga toxin (verocytotoxin) 2 variants and correlations to clinical manifestations. J. Clin. Microbiol. 2007, 45, 2020–2024. [Google Scholar] [CrossRef]
- Boerlin, P. Evolution of virulence factors in Shiga-toxin-producing Escherichia coli. Cell. Mol. Life Sci. 1999, 56, 735–741. [Google Scholar] [CrossRef]
- Bettelheim, K.A. Non-O157 verotoxin-producing Escherichia coli: A problem, paradox, and paradigm. Exp. Biol. Med. 2003, 228, 333–344. [Google Scholar]
- Blanco, M.; Padola, N.L.; Kruger, A.; Sanz, M.E.; Blanco, J.E.; Gonzalez, E.A.; Dahbi, G.; Mora, A.; Bernardez, M.I.; Etcheverria, A.I.; et al. Virulence genes and intimin types of Shiga-toxin-producing Escherichia coli isolated from cattle and beef products in Argentina. Int. Microbiol. 2004, 7, 269–276. [Google Scholar]
- European Food Safety Authority (EFSA). The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in the in 2009. EFSA J. 2011, 9, 200–209. [Google Scholar]
- Fukushima, H.; Seki, R. High numbers of Shiga toxin-producing Escherichia coli found in bovine faeces collected at slaughter in Japan. FEMS Microbiol. Lett. 2004, 238, 189–197. [Google Scholar]
- Verstraete, K.; de Zutter, L.; Messens, W.; Herman, L.; Heyndrickx, M.; de Reu, K. Effect of the enrichment time and immunomagnetic separation on the detection of Shiga toxin-producing Escherichia coli O26, O103, O111, O145 and sorbitol positive O157 from artificially inoculated cattle faeces. Vet. Microbiol. 2010, 145, 106–112. [Google Scholar] [CrossRef]
- Possé, B.; de Zutter, L.; Heyndrickx, M.; Herman, L. Quantitative isolation efficiency of O26, O103, O111, O145 and O157 STEC serotypes from artificially contaminated food and cattle feces samples using a new isolation protocol. J. Appl. Microbiol. 2008, 105, 227–235. [Google Scholar] [CrossRef]
- Arthur, T.M.; Keen, J.E.; Bosilevac, J.M.; Brichta-Harhay, D.M.; Kalchayanand, N.; Shackelford, S.D.; Wheeler, T.L.; Nou, X.; Koohmaraie, M. Longitudinal study of Escherichia coli O157:H7 in a beef cattle feedlot and role of high-level shedders in hide contamination. Appl. Environ. Microbiol. 2009, 75, 6515–6523. [Google Scholar] [CrossRef]
- Fratamico, P.M.; Bagi, L.K.; Pepe, T. A multiplex polymerase chain reaction assay for rapid detection and identification of Escherichia coli O157:H7 in foods and bovine feces. J. Food Prot. 2000, 63, 1032–1037. [Google Scholar]
- Ibekwe, A.M.; Grieve, C.M. Detection and quantification of Escherichia coli O157:H7 in environmental samples by real-time PCR. J. Appl. Microbiol. 2003, 94, 421–431. [Google Scholar] [CrossRef]
- Ibekwe, A.M.; Watt, P.M.; Grieve, C.M.; Sharma, V.K.; Lyons, S.R. Multiplex fluorogenic real-time PCR for detection and quantification of Escherichia coli O157:H7 in dairy wastewater wetlands. Appl. Environ. Microbiol. 2002, 68, 4853–4862. [Google Scholar] [CrossRef]
- Jacob, M.E.; Shi, X.; An, B.; Nagaraja, T.G.; Bai, J. Evaluation of a multiplex real-time polymerase chain reaction for the quantification of Escherichia coli O157 in cattle feces. Foodborne Pathog. Dis. 2012, 9, 79–85. [Google Scholar] [CrossRef]
- Isobe, J.; Kimata, K.; Shimojima, M.; Hosorogi, S.; Tanaka, D.; Gyobu, Y. Isolation of Escherichia coli O128:HNM harboring stx2f gene from diarrhea patients. Kansenshogaku Zasshi 2004, 78, 1000–1005. [Google Scholar]
- Kawano, K.; Okada, M.; Haga, T.; Maeda, K.; Goto, Y. Relationship between pathogenicity for humans and stx genotype in Shiga toxin-producing Escherichia coli serotype O157. Eur. J. Clin. Microbiol. Infect. Dis. 2008, 27, 227–232. [Google Scholar] [CrossRef]
- Werbrouck, H.; Botteldoorn, N.; Uyttendaele, M.; Herman, L.; van Coillie, E. Quantification of gene expression of Listeria monocytogenes by real-time reverse transcription PCR: Optimization, evaluation and pitfalls. J. Microbiol. Methods 2007, 69, 306–314. [Google Scholar] [CrossRef]
- Belanger, S.D.; Boissinot, M.; Menard, C.; Picard, F.J.; Bergeron, M.G. Rapid detection of Shiga toxin-producing bacteria in feces by multiplex PCR with molecular beacons on the smart cycler. J. Clin. Microbiol. 2002, 40, 1436–1440. [Google Scholar] [CrossRef]
- Fratamico, P.M.; DebRoy, C.; Miyamoto, T.; Liu, Y.H. PCR Detection of Enterohemorrhagic Escherichia coli O145 in food by targeting genes in the E. coli O145 O-Antigen gene cluster and the Shiga Toxin 1 and Shiga Toxin 2 genes. Foodborne Pathog. Dis. 2009, 6, 605–611. [Google Scholar] [CrossRef]
- Grys, T.E.; Sloan, L.M.; Rosenblatt, J.E.; Patel, R. Rapid and sensitive detection of Shiga toxin-producing Escherichia coli from nonenriched stool specimens by real-time PCR in comparison to enzyme immunoassay and culture. J. Clin. Microbiol. 2009, 47, 2008–2012. [Google Scholar] [CrossRef]
- Iijima, Y.; Asako, N.T.; Aihara, M.; Hayashi, K. Improvement in the detection rate of diarrhoeagenic bacteria in human stool specimens by a rapid realtime PCR assay. J. Med. Microbiol. 2004, 53, 617–622. [Google Scholar] [CrossRef]
- Jinneman, K.C.; Yoshitomi, K.J.; Weagant, S.D. Multiplex real-time PCR method to identify Shiga toxin genes stx1 and stx2 and Escherichia coli O157:H7/H- serotype. Appl. Environ. Microbiol. 2003, 69, 6327–6333. [Google Scholar] [CrossRef]
- O’Hanlon, K.A.; Catarame, T.M.G.; Duffy, G.; Blair, I.S.; McDowell, D.A. Rapid detection and quantification of E. coli O157/O26/O111 in minced beef by real-time PCR. J. Appl. Microbiol. 2004, 96, 1013–1023. [Google Scholar] [CrossRef]
- Perelle, S.; Dilasser, F.; Grout, J.L.; Fach, P. Detection by 5’-nuclease PCR of Shiga-toxin producing Escherichia coli O26, O55, O91, O103, O111, O113, O145 and O157:H7, associated with the world's most frequent clinical cases. Mol. Cell. Probes 2004, 18, 185–192. [Google Scholar] [CrossRef]
- Reischl, U.; Youssef, M.T.; Kilwinski, J.; Lehn, N.; Zhang, W.L.; Karch, H.; Strockbine, N.A. Real-time fluorescence PCR assays for detection and characterization of Shiga toxin, intimin, and enterohemolysin genes from Shiga toxin-producing Escherichia coli. J. Clin. Microbiol. 2002, 40, 2555–2565. [Google Scholar] [CrossRef]
- Sharma, V.K.; Dean-Nystrom, E.A.; Casey, T.A. Semi-automated fluorogenic PCR assays (TaqMan) for rapid detection of Escherichia coli O157:H7 and other Shiga toxigenic E. coli. Mol. Cell. Probes 1999, 13, 291–302. [Google Scholar] [CrossRef]
- Sharma, V.K. Detection and quantitation of enterohemorrhagic Escherichia coli O157, O111, and O26 in beef and bovine feces by real-time polymerase chain reaction. J. Food Prot. 2002, 65, 1371–1380. [Google Scholar]
- Bellin, T.; Pulz, M.; Matussek, A.; Hempen, H.G.; Gunzer, F. Rapid detection of enterohemorrhagic Escherichia coli by real-time PCR with fluorescent hybridization probes. J. Clin. Microbiol. 2001, 39, 370–374. [Google Scholar] [CrossRef]
- Fitzmaurice, J.; Glennon, M.; Duffy, G.; Sheridan, J.J.; Carroll, C.; Maher, M. Application of real-time PCR and RT-PCR assays for the detection and quantitation of VT1 and VT2 toxin genes in E. coli O157:H7. Mol. Cell. Probes 2004, 18, 123–132. [Google Scholar] [CrossRef]
- Hoorfar, J.; Malorny, B.; Abdulmawjood, A.; Cook, N.; Wagner, M.; Fach, P. Practical considerations in design of internal amplification controls for diagnostic PCR assays. J. Clin. Microbiol. 2004, 42, 1863–1868. [Google Scholar] [CrossRef]
- Verstraete, K.; de Zutter, L.; Robyn, J.; Daube, G.; Herman, L.; Heyndrickx, M.; de Schaetzen, M.A.; de Reu, K. Validation of a method for simultaneous isolation of Shiga toxin-producing Escherichia coli O26, O103, O111 and O145 from minced beef by an international ring-trial. Foodborne Pathog. Dis. 2012, 9, 412–417. [Google Scholar] [CrossRef] [Green Version]
- Verstraete, K.; Robyn, J.; Del-Favero, J.; de Rijk, P.; Joris, A.; Herman, L.; Heyndrickx, M.; de Zutter, L.; de Reu, K. Evaluation of a multiplex-PCR detection in combination with an isolation method for STEC O26, O103, O111, O145 and sorbitol fermenting O157 in food. Food Microbiol. 2012, 29, 49–55. [Google Scholar] [CrossRef]
- Menrath, A.; Wieler, L.H.; Heidemanns, K.; Semmler, T.; Fruth, A.; Kemper, N. Shiga toxin producing Escherichia coli: Identification of non-O157:H7-super-shedding cows ans related risk factors. Gut Pathog. 2010, 2, 1–9. [Google Scholar] [CrossRef]
- Bae, S.; Wuertz, S. Discrimination of viable and dead fecal Bacteroidales bacteria by quantitative PCR with propidium monoazide. Appl. Environ. Microbiol. 2009, 75, 2940–2944. [Google Scholar] [CrossRef]
- Hornitzky, M.A.; Mercieca, K.; Bettelheim, K.A.; Djordjevic, S.P. Bovine feces from animals with gastrointestinal infections are a source of serologically diverse atypical enteropathogenic Escherichia coli and Shiga toxin-producing E. coli strains that commonly possess intimin. Appl. Environ. Microbiol. 2005, 71, 3405–3412. [Google Scholar] [CrossRef]
- Blanco, M.; Schumacher, S.; Tasara, T.; Zweifel, C.; Blanco, J.E.; Dahbi, G.; Blanco, J.; Stephan, R. Serotypes, intimin variants and other virulence factors of eae positive Escherichia coli strains isolated from healthy cattle in Switzerland. Identification of a new intimin variant gene (eae-eta2). BMC Microbiol. 2005, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Flamm, R.K.; Hinrichs, D.J.; Thomashow, M.F. Introduction of Pam-Beta-1 into Listeria monocytogenes by conjugation and homology between native L. monocytogenes plasmids. Infect. Immun. 1984, 44, 157–161. [Google Scholar]
- Perna, N.T.; Plunkett, G.; Burland, V.; Mau, B.; Glasner, J.D.; Rose, D.J.; Mayhew, G.F.; Evans, P.S.; Gregor, J.; Kirkpatrick, H.A.; et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 2001, 410, 240–240. [Google Scholar]
- Knutsson, R.; Lofstrom, C.; Grage, H.; Hoorfar, J.; Radstrom, P. Modeling of 5’ nuclease real-time responses for optimization of a high-throughput enrichment PCR procedure for Salmonella enterica. J. Clin. Microbiol. 2002, 40, 52–60. [Google Scholar] [CrossRef]
- International Organization for Standardization (ISO). ISO 16140: Microbiology of Food and Animal Feeding Stuffs—Protocol for the Validation of Alternative Methods. International Organisation for Standardisation. Available online: http://www.iso.org/iso/home/store/catalogue_tc/catalogue_detail.htm?csnumber=30158 (accessed on 24 March 2014).
- Possé, B.; de Zutter, L.; Heyndrickx, M.; Herman, L. Novel differential and confirmation plating media for Shiga toxin-producing Escherichia coli serotypes O26, O103, O111, O145 and sorbitol-positive and -negative O157. FEMS Microbiol. Lett. 2008, 282, 124–131. [Google Scholar] [CrossRef]
- Botteldoorn, N.; Heyndrickx, M.; Rijpens, N.; Herman, L. Detection and characterization of verotoxigenic Escherichia coli by a VTEC/EHEC multiplex PCR in porcine faeces and pig carcass swabs. Res. Microbiol. 2003, 154, 97–104. [Google Scholar] [CrossRef]
- DebRoy, C.; Roberts, E.; Kundrat, J.; Davis, M.A.; Briggs, C.E.; Fratamico, P.M. Detection of Escherichia coli serogroups O26 and O113 by PCR amplification of the wzx and wzy genes. Appl. Environ. Microbiol. 2004, 70, 1830–1832. [Google Scholar] [CrossRef]
- Fratamico, P.M.; DebRoy, C.; Strobaugh, T.P.; Chen, C.Y. DNA sequence of the Escherichia coli O103O antigen gene cluster and detection of enterohemorrhagic E. coli O103 by PCR amplification of the wzx and wzy genes. Can. J. Microbiol. 2005, 51, 515–522. [Google Scholar] [CrossRef]
- Paton, A.W.; Paton, J.C. Detection and characterization of shiga toxigenic Escherichia coli by using multiplex PCR assays for stx(1), stx(2), eaeA, enterohemorrhagic E-coli hlyA, rfb(O111), and rfb(O.157). J. Clin. Microbiol. 1998, 36, 598–602. [Google Scholar]
- Feng, L.; Senchenkova, S.N.; Tao, J.; Shashkov, A.S.; Liu, B.; Shevelev, S.D.; Reeves, P.R.; Xu, J.G.; Knirel, Y.A.; Wang, L. Structural and genetic characterization of enterohemorrhagic Escherichia coli O145O antigen and development of an O145 serogroup-specific PCR assay. J. Bacteriol. 2005, 187, 758–764. [Google Scholar] [CrossRef]
- Perelle, S.; Dilasser, F.; Grout, J.; Fach, P. Identification of the O-antigen biosynthesis genes of Escherichia coli O91 and development of a O91 PCR serotyping test. J. Appl. Microbiol. 2002, 93, 758–764. [Google Scholar] [CrossRef]
- Fratamico, P.M.; Briggs, C.E.; Needle, D.; Chen, C.Y.; DebRoy, C. Sequence of the Escherichia coli O121 O-antigen gene cluster and detection of enterohemorrhagic E. coli O121 by PCR amplification of the wzx and wzy genes. J. Clin. Microbiol. 2003, 41, 3379–3383. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Verstraete, K.; Van Coillie, E.; Werbrouck, H.; Van Weyenberg, S.; Herman, L.; Del-Favero, J.; De Rijk, P.; De Zutter, L.; Joris, M.-A.; Heyndrickx, M.; et al. A qPCR Assay to Detect and Quantify Shiga Toxin-Producing E. coli (STEC) in Cattle and on Farms: A Potential Predictive Tool for STEC Culture-Positive Farms. Toxins 2014, 6, 1201-1221. https://doi.org/10.3390/toxins6041201
Verstraete K, Van Coillie E, Werbrouck H, Van Weyenberg S, Herman L, Del-Favero J, De Rijk P, De Zutter L, Joris M-A, Heyndrickx M, et al. A qPCR Assay to Detect and Quantify Shiga Toxin-Producing E. coli (STEC) in Cattle and on Farms: A Potential Predictive Tool for STEC Culture-Positive Farms. Toxins. 2014; 6(4):1201-1221. https://doi.org/10.3390/toxins6041201
Chicago/Turabian StyleVerstraete, Karen, Els Van Coillie, Hadewig Werbrouck, Stephanie Van Weyenberg, Lieve Herman, Jurgen Del-Favero, Peter De Rijk, Lieven De Zutter, Maria-Adelheid Joris, Marc Heyndrickx, and et al. 2014. "A qPCR Assay to Detect and Quantify Shiga Toxin-Producing E. coli (STEC) in Cattle and on Farms: A Potential Predictive Tool for STEC Culture-Positive Farms" Toxins 6, no. 4: 1201-1221. https://doi.org/10.3390/toxins6041201
APA StyleVerstraete, K., Van Coillie, E., Werbrouck, H., Van Weyenberg, S., Herman, L., Del-Favero, J., De Rijk, P., De Zutter, L., Joris, M. -A., Heyndrickx, M., & DeReu, K. (2014). A qPCR Assay to Detect and Quantify Shiga Toxin-Producing E. coli (STEC) in Cattle and on Farms: A Potential Predictive Tool for STEC Culture-Positive Farms. Toxins, 6(4), 1201-1221. https://doi.org/10.3390/toxins6041201