sRNA Antitoxins: More than One Way to Repress a Toxin

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Founding member (plasmid or bacterium) a | Genetic organization b | Mode of antitoxin action c | |
|---|---|---|---|---|
| Type I | Hok/Sok | Plasmid R1 (63) |  | Inhibit protein synthesis |
| RNAI/RNAII | pAD1 (48) |  | Inhibit protein synthesis | |
| Ldr/Rdl | Escherichia coli (34) | | Inhibit protein synthesis | |
| TisB/IstR1 | Escherichia coli (39) |  | Inhibit protein synthesis | |
| TxpA/RatA | Bacillus subtilis (15) | | Stimulate mRNA degradation | |
| SymE/SymR | Escherichia coli (55) |  | Inhibit protein synthesis | |
| Ibs/Sib | Escherichia coli (41) |  | Inhibit protein synthesis | |
| ShoB/OhsC | Escherichia coli (41) | | Inhibit protein synthesis | |
| BsrG/SR4 | Bacillus subtilis (58) | | Stimulate degradation and inhibit translation | |
| Zor/Orz | Escherichia coli (5, 43) | | Inhibit protein synthesis | |
| RalR/RalA | Escherichia coli (56) | | Inhibit protein synthesis | |
| DinQ/AgrB | Escherichia coli (42) | | Inhibit protein synthesis | |
| Type III | ToxN/ToxI | pECA1039 (59) |  | Protein sequestration |
| ABIQ/antiQ | pSRQ900 (62) |  | Protein sequestration | |
2. Repression by RNA Antitoxins and Toxin Function
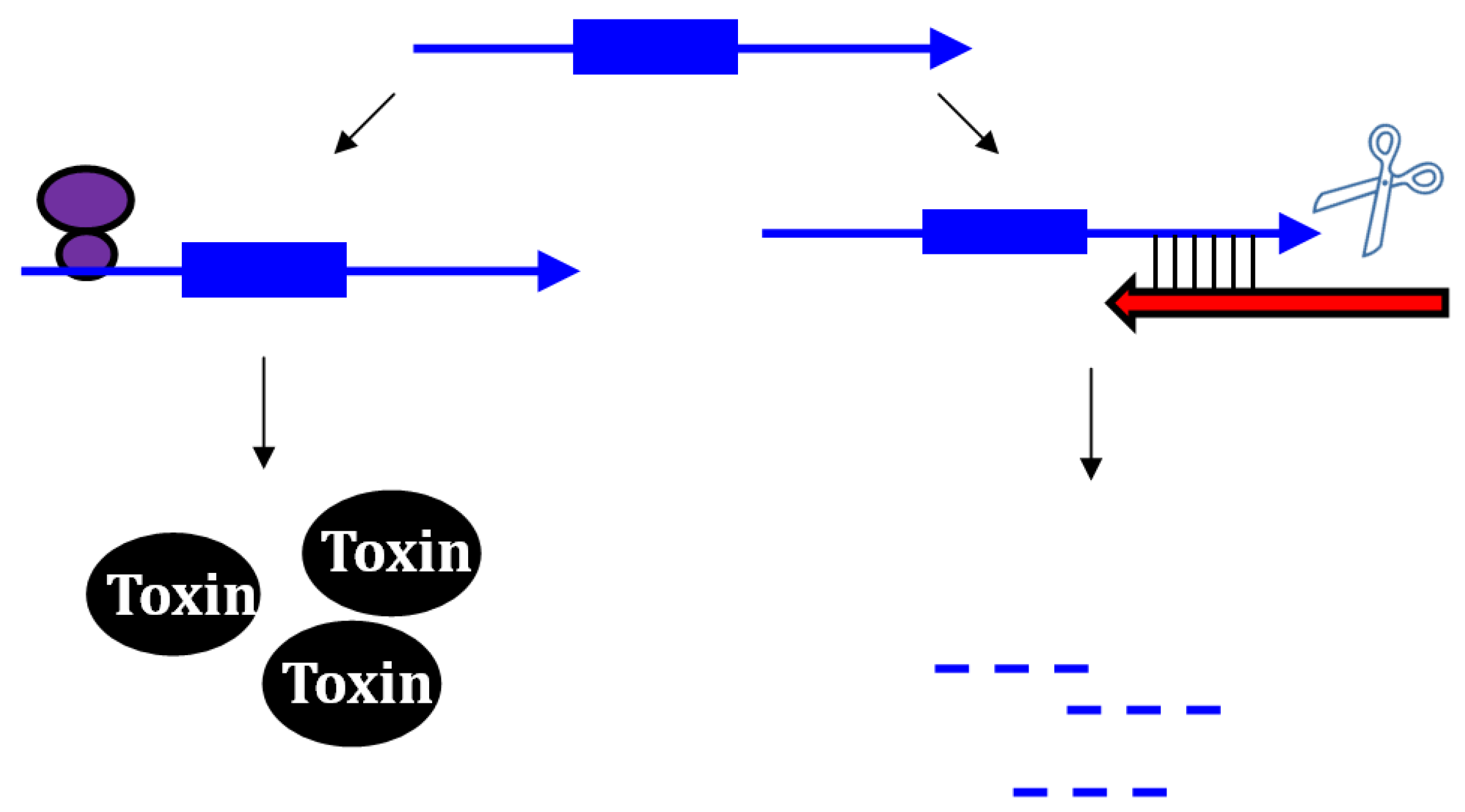
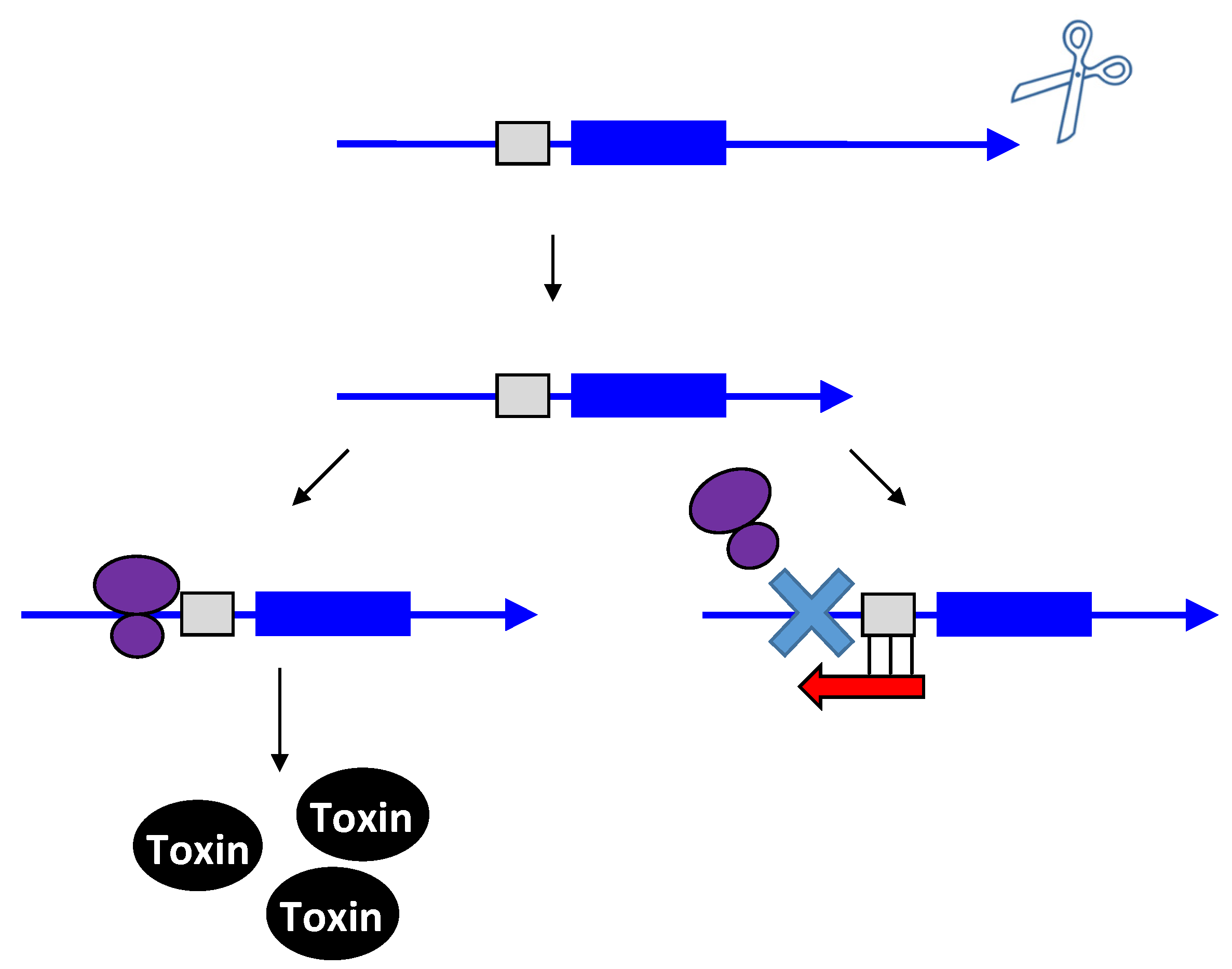
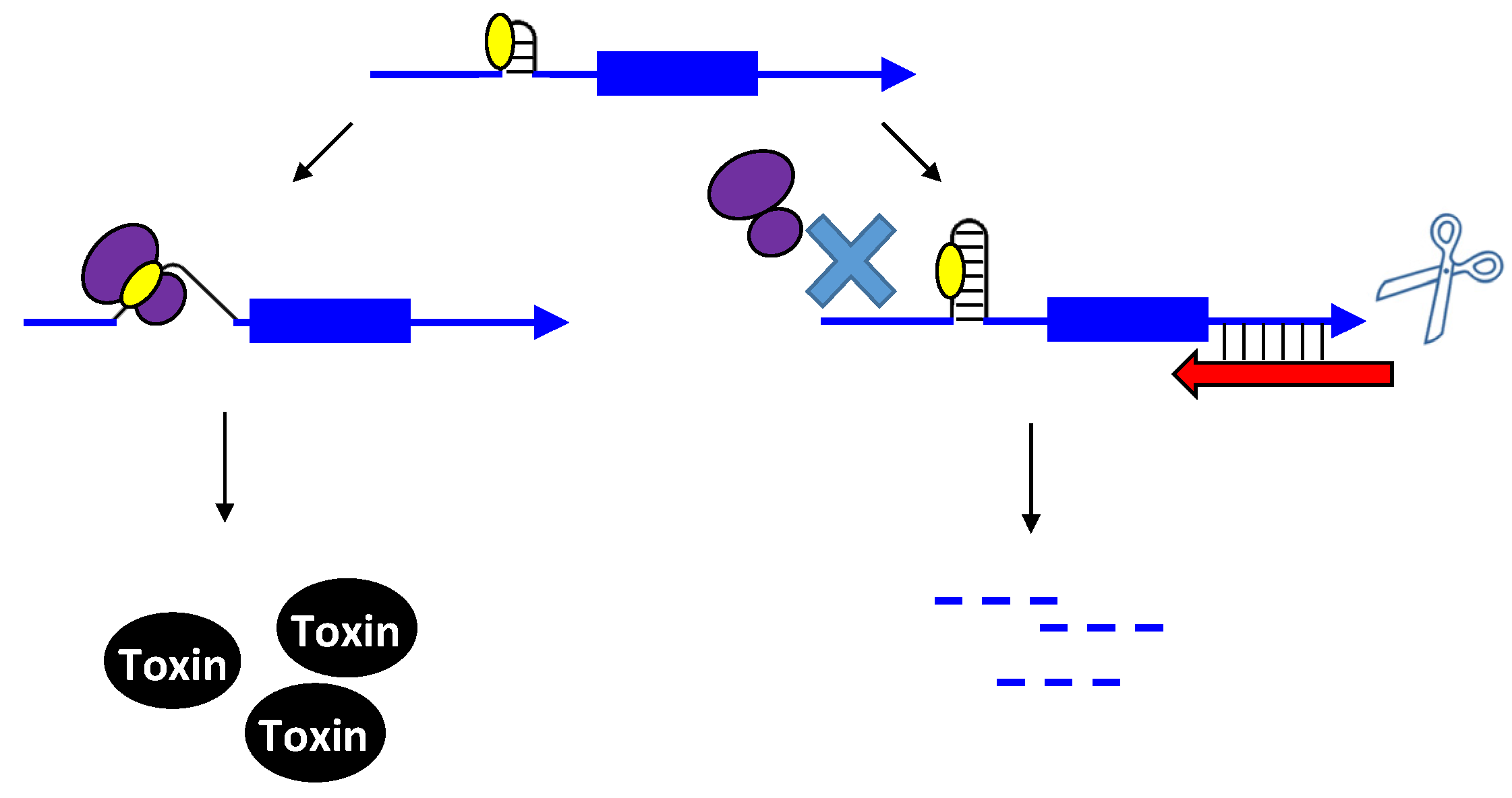
2.1. Type I Antitoxins: Repression through Base Pairing Interactions
2.1.1. Repressing Type I Toxins: Controlling mRNA Stability

2.1.2. Repressing Type I Toxins: Controlling Protein Synthesis



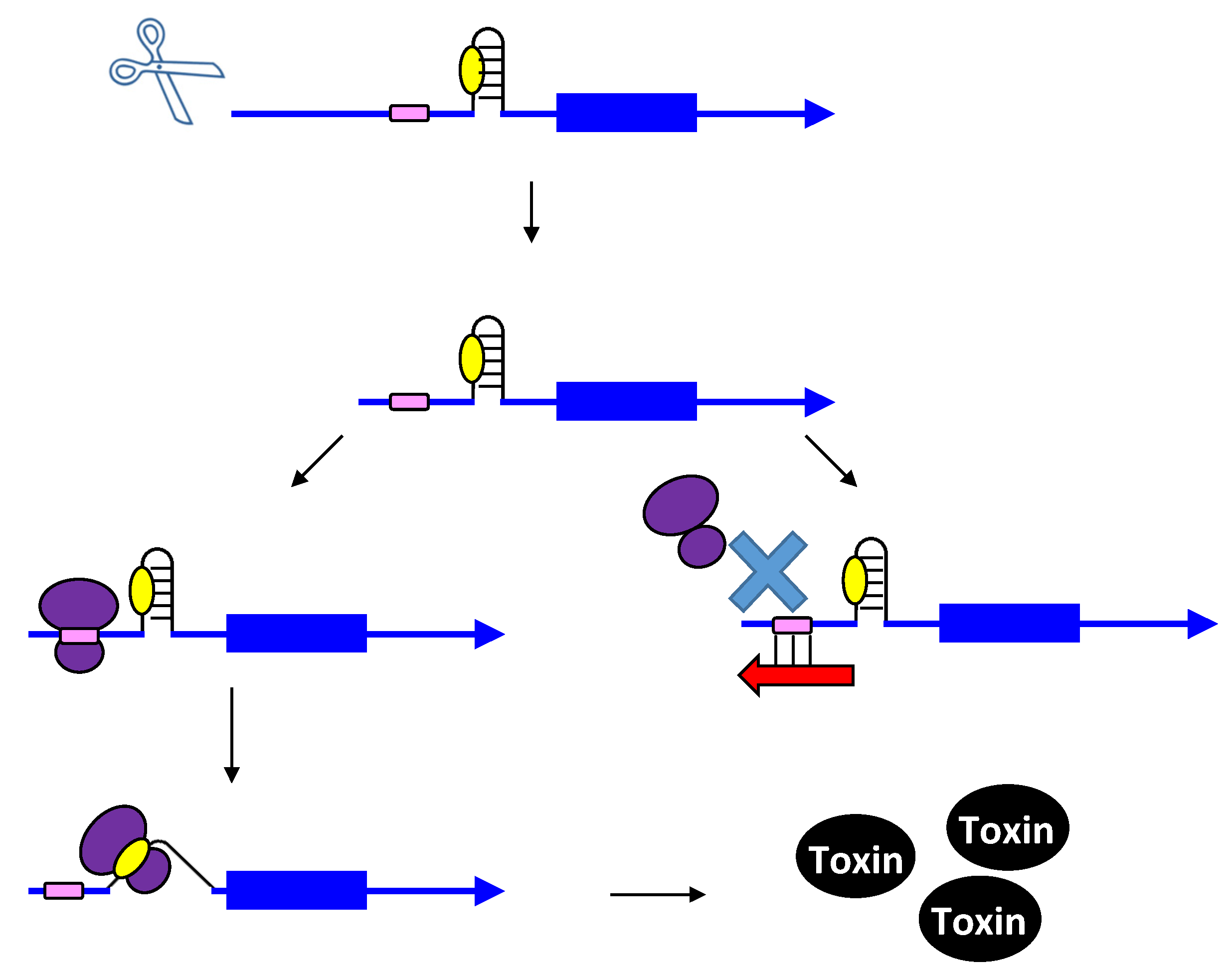
2.2. Repressing Type I Toxins: Controlling Degradation and Translation

2.2.1. Controlling Protein Expression: mRNA Degradation versus Translation Inhibition
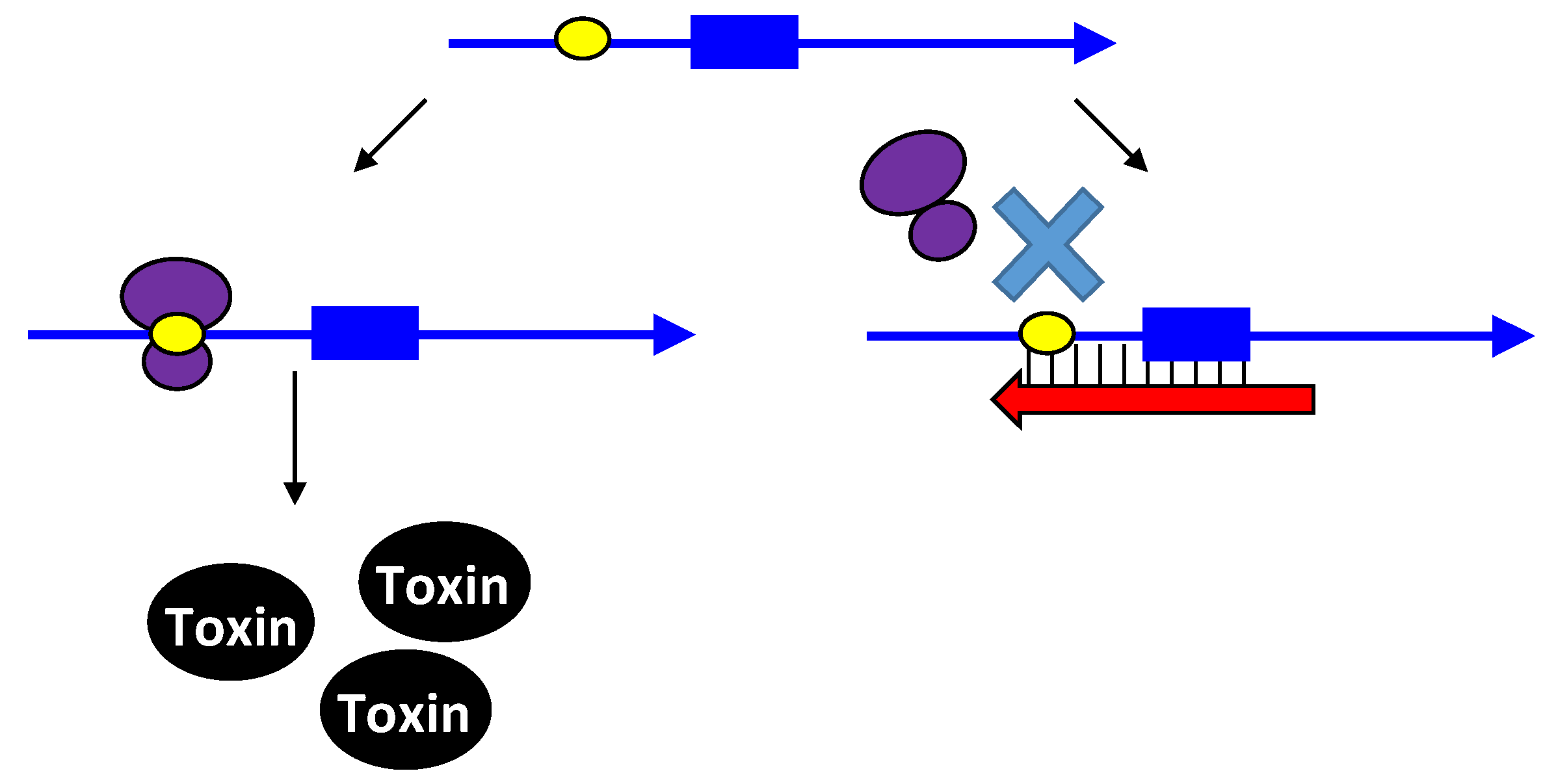
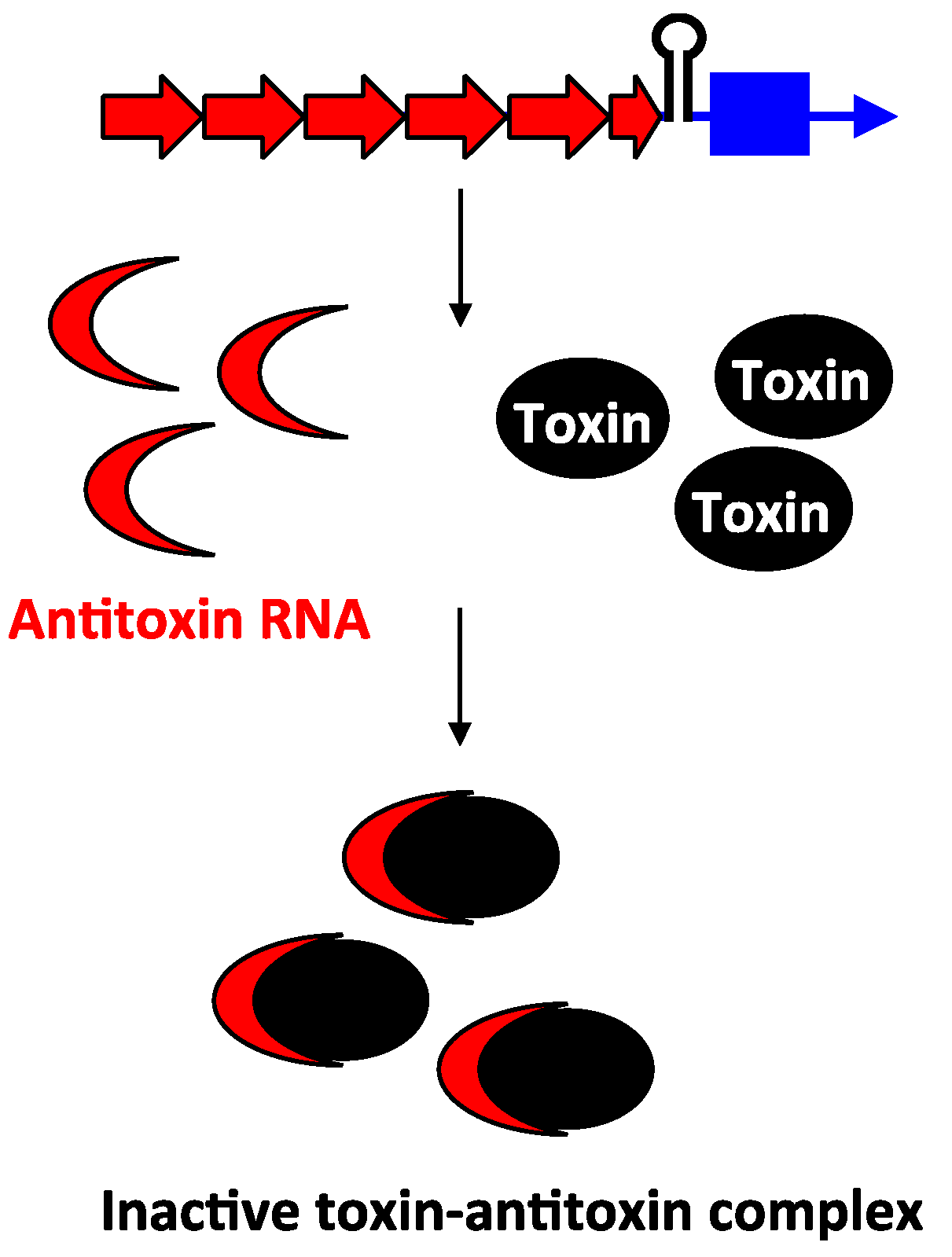
2.3. Type III Antitoxins: Repression through Protein Sequestration

2.4. Function of Type I and Type III Gene Pairs
2.4.1. Plasmid Remnants?
2.4.2. Impairment of Chromosomal Structure
2.4.3. Persister Formation
2.4.4. Chromosomal Stabilization and Recombination
2.4.5. Protection from Foreign DNA
2.4.6. Inhibition of Competitors
2.4.7. Nucleic Acid Cleavage
3. Regulating the Toxin mRNA versus the Protein
3.1. Type I Antitoxins: Base Pairing to the Toxin mRNA
3.2. Type III Antitoxins: Binding to the Toxic Proteins
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goeders, N.; van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins 2014, 6, 304–324. [Google Scholar] [CrossRef]
- Kroll, J.; Klinter, S.; Schneider, C.; Voss, I.; Steinbuchel, A. Plasmid addiction systems: Perspectives and applications in biotechnology. Microb. Biotechnol. 2010, 3, 634–657. [Google Scholar] [CrossRef]
- Weaver, K.E. Emerging plasmid-encoded antisense RNA regulated systems. Curr. Opin. Microbiol. 2007, 10, 110–116. [Google Scholar] [CrossRef]
- Zielenkiewicz, U.; Ceglowski, P. Mechanisms of plasmid stable maintenance with special focus on plasmid addiction systems. Acta Biochim. Pol. 2001, 48, 1003–1023. [Google Scholar]
- Fozo, E.M.; Makarova, K.S.; Shabalina, S.A.; Yutin, N.; Koonin, E.V.; Storz, G. Abundance of type I toxin-antitoxin systems in bacteria: Searches for new candidates and discovery of novel families. Nucleic Acids Res. 2010, 38, 3743–3759. [Google Scholar] [CrossRef]
- Pedersen, K.; Gerdes, K. Multiple hok genes on the chromosome of Escherichia coli. Mol. Microbiol. 1999, 32, 1090–1102. [Google Scholar] [CrossRef]
- Weaver, K.E.; Reddy, S.G.; Brinkman, C.L.; Patel, S.; Bayles, K.W.; Endres, J.L. Identification and characterization of a family of toxin-antitoxin systems related to the Enterococcus faecalis plasmid pAD1 par addiction module. Microbiology 2009, 155, 2930–2940. [Google Scholar] [CrossRef]
- Leplae, R.; Geeraerts, D.; Hallez, R.; Guglielmini, J.; Dreze, P.; van Melderen, L. Diversity of bacterial type II toxin-antitoxin systems: A comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011, 39, 5513–5525. [Google Scholar]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 2009, 4, 19. [Google Scholar]
- Makarova, K.S.; Wolf, Y.I.; Snir, S.; Koonin, E.V. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 2011, 193, 6039–6056. [Google Scholar]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar]
- Lalaouna, D.; Simoneau-Roy, M.; Lafontaine, D.; Masse, E. Regulatory RNAs and target mRNA decay in prokaryotes. Biochim. Biophys. Acta 2013, 1829, 742–747. [Google Scholar]
- Wagner, E.G. Cycling of RNAs on Hfq. RNA Biol. 2013, 10, 619–626. [Google Scholar]
- Durand, S.; Gilet, L.; Condon, C. The essential function of B. subtilis RNase III is to silence foreign toxin genes. PLoS Genet. 2012, 8, e1003181. [Google Scholar]
- Silvaggi, J.M.; Perkins, J.B.; Losick, R. Small untranslated RNA antitoxin in Bacillus subtilis. J. Bacteriol. 2005, 187, 6641–6650. [Google Scholar]
- Lee, J.M.; Zhang, S.; Saha, S.; Santa Anna, S.; Jiang, C.; Perkins, J. RNA expression analysis using an antisense Bacillus subtilis genome array. J. Bacteriol. 2001, 183, 7371–7380. [Google Scholar]
- Franch, T.; Gerdes, K. Programmed cell death in bacteria: Translational repression by mRNA end-pairing. Mol. Microbiol. 1996, 21, 1049–1060. [Google Scholar]
- Franch, T.; Gultyaev, A.P.; Gerdes, K. Programmed cell death by hok/sok of plasmid R1: Processing at the hok mRNA 3'-end triggers structural rearrangements that allow translation and antisense RNA binding. J. Mol. Biol. 1997, 273, 38–51. [Google Scholar]
- Moller-Jensen, J.; Franch, T.; Gerdes, K. Temporal translational control by a metastable RNA structure. J. Biol. Chem. 2001, 276, 35707–35713. [Google Scholar] [CrossRef]
- Gerdes, K.; Thisted, T.; Martinussen, J. Mechanism of post-segregational killing by the hok/sok system of plasmid R1: Sok antisense RNA regulates formation of a hok mRNA species correlated with killing of plasmid-free cells. Mol. Microbiol. 1990, 4, 1807–1818. [Google Scholar] [CrossRef]
- Thisted, T.; Gerdes, K. Mechanism of post-segregational killing by the hok/sok system of plasmid R1. Sok antisense RNA regulates hok gene expression indirectly through the overlapping mok gene. J. Mol. Biol. 1992, 223, 41–54. [Google Scholar] [CrossRef]
- Gerdes, K.; Nielsen, A.; Thorsted, P.; Wagner, E.G. Mechanism of killer gene activation. Antisense RNA-dependent RNase III cleavage ensures rapid turn-over of the stable hok, srnB and pndA effector messenger RNAs. J. Mol. Biol. 1992, 226, 637–649. [Google Scholar] [CrossRef]
- Akimoto, S.; Ohnishi, Y. R483 and F plasmid genes promoting RNA degradation: Comparative restriction mapping. Microbiol. Immunol. 1982, 26, 779–793. [Google Scholar]
- Gerdes, K.; Poulsen, L.K.; Thisted, T.; Nielsen, A.K.; Martinussen, J.; Andreasen, P.H. The hok killer gene family in gram-negative bacteria. New Biol. 1990, 2, 946–956. [Google Scholar]
- Ito, R.; Ohnishi, Y. The roles of RNA polymerase and RNase I in stable RNA degradation in Escherichia coli carrying the srnb+ gene. Biochim. Biophys. Acta 1983, 739, 27–34. [Google Scholar] [CrossRef]
- Loh, S.M.; Cram, D.S.; Skurray, R.A. Nucleotide sequence and transcriptional analysis of a third function (flm) involved in F-plasmid maintenance. Gene 1988, 66, 259–268. [Google Scholar] [CrossRef]
- Nielsen, A.K.; Thorsted, P.; Thisted, T.; Wagner, E.G.; Gerdes, K. The rifampicin-inducible genes srnB from F and pnd from R483 are regulated by antisense RNAs and mediate plasmid maintenance by killing of plasmid-free segregants. Mol. Microbiol. 1991, 5, 1961–1973. [Google Scholar] [CrossRef]
- Ohnishi, Y.; Akimoto, S. I-like R plasmids promote degradation of stable ribonucleic acid in Escherichia coli. J. Bacteriol. 1980, 144, 833–835. [Google Scholar]
- Ohnishi, Y.; Iguma, H.; Ono, T.; Nagaishi, H.; Clark, A.J. Genetic mapping of the F plasmid gene that promotes degradation of stable ribonucleic acid in Escherichia coli. J. Bacteriol. 1977, 132, 784–789. [Google Scholar]
- Ohnishi, Y. F factor promotes turnover of stable RNA in Escherichia coli. Science 1975, 24, 257–258. [Google Scholar]
- Fozo, E.M.; Hemm, M.R.; Storz, G. Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 2008, 72, 579–589. [Google Scholar]
- Kawano, M. Divergently overlapping cis-encoded antisense RNA regulating toxin-antitoxin systems from E. coli: hok/sok, ldr/rdl, symE/symR. RNA Biol. 2012, 9, 1520–1527. [Google Scholar] [CrossRef]
- Kawano, M.; Kanaya, S.; Oshima, T.; Masuda, Y.; Ara, T.; Mori, H. Distribution of repetitive sequences on the leading and lagging strands of the Escherichia coli genome: Comparative study of Long Direct Repeat (LDR) sequences. DNA Res. 2002, 9, 1–10. [Google Scholar]
- Kawano, M.; Oshima, T.; Kasai, H.; Mori, H. Molecular characterization of Long Direct Repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli. Mol. Microbiol. 2002, 45, 333–349. [Google Scholar] [CrossRef]
- Rudd, K.E. Novel intergenic repeats of Escherichia coli K-12. Res. Microbiol. 1999, 150, 653–664. [Google Scholar] [CrossRef]
- Gerdes, K.; Wagner, E.G. RNA antitoxins. Curr. Opin. Microbiol. 2007, 10, 117–124. [Google Scholar] [CrossRef]
- Argaman, L.; Hershberg, R.; Vogel, J.; Bejerano, G.; Wagner, E.G.; Margalit, H.; Altuvia, S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 2001, 11, 941–950. [Google Scholar] [CrossRef]
- Fernandez de Henestrosa, A.R.; Ogi, T.; Aoyagi, S.; Chafin, D.; Hayes, J.J.; Ohmori, H.; Woodgate, R. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol. Microbiol. 2000, 35, 1560–1572. [Google Scholar]
- Vogel, J.; Argaman, L.; Wagner, E.G.; Altuvia, S. The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr. Biol. 2004, 14, 2271–2276. [Google Scholar] [CrossRef]
- Darfeuille, F.; Unoson, C.; Vogel, J.; Wagner, E.G. An antisense RNA inhibits translation by competing with standby ribosomes. Mol. Cell 2007, 26, 381–392. [Google Scholar] [CrossRef]
- Fozo, E.M.; Kawano, M.; Fontaine, F.; Kaya, Y.; Mendieta, K.S.; Jones, K.L.; Ocampo, A.; Rudd, K.E.; Storz, G. Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol. Microbiol. 2008, 70, 1076–1093. [Google Scholar] [CrossRef]
- Weel-Sneve, R.; Kristiansen, K.I.; Odsbu, I.; Dalhus, B.; Booth, J.; Rognes, T.; Skarstad, K.; Bjoras, M. Single transmembrane peptide DinQ modulates membrane-dependent activities. PLoS Genet. 2013, 9, e1003260. [Google Scholar] [CrossRef]
- Wen, J.; Won, D.; Fozo, E.M. The zorO-orzO type I toxin-antitoxin locus: Repression by the OrzO antitoxin. Nucleic Acids Res. 2014, 42, 1930–1946. [Google Scholar] [CrossRef]
- Weaver, K.E.; Jensen, K.D.; Colwell, A.; Sriram, S.I. Functional analysis of the Enterococcus faecalis plasmid pAD1-encoded stability determinant par. Mol. Microbiol. 1996, 20, 53–63. [Google Scholar] [CrossRef]
- Weaver, K.E.; Tritle, D.J. Identification and characterization of an Enterococcus faecalis plasmid pAD1-encoded stability determinant which produces two small RNA molecules necessary for its function. Plasmid 1994, 32, 168–181. [Google Scholar] [CrossRef]
- Weaver, K.E.; Weaver, D.M.; Wells, C.L.; Waters, C.M.; Gardner, M.E.; Ehli, E.A. Enterococcus faecalis plasmid pAD1-encoded Fst toxin affects membrane permeability and alters cellular responses to lantibiotics. J. Bacteriol. 2003, 185, 2169–2177. [Google Scholar] [CrossRef]
- Greenfield, T.J.; Ehli, E.; Kirshenmann, T.; Franch, T.; Gerdes, K.; Weaver, K.E. The antisense RNA of the par locus of pAD1 regulates the expression of a 33-amino-acid toxic peptide by an unusual mechanism. Mol. Microbiol. 2000, 37, 652–660. [Google Scholar]
- Greenfield, T.J.; Franch, T.; Gerdes, K.; Weaver, K.E. Antisense RNA regulation of the par post-segregational killing system: Structural analysis and mechanism of binding of the antisense RNA, RNA II and its target, RNA I. Mol. Microbiol. 2001, 42, 527–537. [Google Scholar] [CrossRef]
- Greenfield, T.J.; Weaver, K.E. Antisense RNA regulation of the pAD1 par post-segregational killing system requires interaction at the 5' and 3' ends of the RNAs. Mol. Microbiol. 2000, 37, 661–670. [Google Scholar] [CrossRef]
- Weaver, K.E.; Ehli, E.A.; Nelson, J.S.; Patel, S. Antisense RNA regulation by stable complex formation in the Enterococcus faecalis plasmid pAD1 par addiction system. J. Bacteriol. 2004, 186, 6400–6408. [Google Scholar] [CrossRef]
- Koyanagi, S.; Levesque, C.M. Characterization of a Streptococcus mutans intergenic region containing a small toxic peptide and its cis-encoded antisense small RNA antitoxin. PLoS One 2013, 8, e54291. [Google Scholar] [CrossRef]
- Sayed, N.; Jousselin, A.; Felden, B. A cis-antisense RNA acts in trans in Staphylococcus aureus to control translation of a human cytolytic peptide. Nat. Struct. Mol. Biol. 2012, 19, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Sayed, N.; Nonin-Lecomte, S.; Rety, S.; Felden, B. Functional and structural insights of a Staphylococcus aureus apoptotic-like membrane peptide from a toxin-antitoxin module. J. Biol. Chem. 2012, 287, 43454–43463. [Google Scholar] [CrossRef]
- Han, K.; Kim, K.S.; Bak, G.; Park, H.; Lee, Y. Recognition and discrimination of target mRNAs by Sib RNAs, a cis-encoded sRNA family. Nucleic Acids Res. 2010, 38, 5851–5866. [Google Scholar] [CrossRef]
- Kawano, M.; Aravind, L.; Storz, G. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol. Microbiol. 2007, 64, 738–754. [Google Scholar] [CrossRef]
- Guo, Y.; Quiroga, C.; Chen, Q.; McAnulty, M.J.; Benedik, M.J.; Wood, T.K.; Wang, X. RalR (a DNase) and RalA (a small RNA) form a type I toxin-antitoxin system in Escherichia coli. Nucleic Acids Res. 2014, 42, 6448–6462. [Google Scholar] [CrossRef]
- Jahn, N.; Brantl, S. One antitoxin—Two functions: SR4 controls toxin mRNA decay and translation. Nucleic Acids Res. 2013, 41, 9870–9880. [Google Scholar] [CrossRef]
- Jahn, N.; Preis, H.; Wiedemann, C.; Brantl, S. BsrG/SR4 from Bacillus subtilis—The first temperature-dependent type I toxin-antitoxin system. Mol. Microbiol. 2012, 83, 579–598. [Google Scholar] [CrossRef]
- Fineran, P.C.; Blower, T.R.; Foulds, I.J.; Humphreys, D.P.; Lilley, K.S.; Salmond, G.P. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc. Natl. Acad. Sci. USA 2009, 106, 894–899. [Google Scholar]
- Blower, T.R.; Pei, X.Y.; Short, F.L.; Fineran, P.C.; Humphreys, D.P.; Luisi, B.F.; Salmond, G.P. A processed noncoding RNA regulates an altruistic bacterial antiviral system. Nat. Struct. Mol. Biol. 2011, 18, 185–190. [Google Scholar] [CrossRef]
- Blower, T.R.; Short, F.L.; Rao, F.; Mizuguchi, K.; Pei, X.Y.; Fineran, P.C.; Luisi, B.F.; Salmond, G.P. Identification and classification of bacterial type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 2012, 40, 6158–6173. [Google Scholar] [CrossRef]
- Samson, J.E.; Spinelli, S.; Cambillau, C.; Moineau, S. Structure and activity of AbiQ, a lactococcal endoribonuclease belonging to the type III toxin-antitoxin system. Mol. Microbiol. 2013, 87, 756–768. [Google Scholar] [CrossRef]
- Gerdes, K.; Bech, F.W.; Jorgensen, S.T.; Lobner-Olesen, A.; Rasmussen, P.B.; Atlung, T.; Boe, L.; Karlstrom, O.; Molin, S.; von Meyenburg, K. Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. EMBO J. 1986, 5, 2023–2029. [Google Scholar]
- Pecota, D.C.; Osapay, G.; Selsted, M.E.; Wood, T.K. Antimicrobial properties of the Escherichia coli R1 plasmid host killing peptide. J. Biotechnol. 2003, 100, 1–12. [Google Scholar] [CrossRef]
- Weaver, K.E. Enterococcus faecalis plasmid pAD1 replication and maintenance. Dev. Biol. Stand. 1995, 85, 89–98. [Google Scholar]
- Weaver, K.E. The par toxin-antitoxin system from Enterococcus faecalis plasmid pAD1 and its chromosomal homologs. RNA Biol. 2012, 9, 1498–1503. [Google Scholar] [CrossRef]
- Brinkman, C.L.; Bumgarner, R.; Kittichotirat, W.; Dunman, P.M.; Kuechenmeister, L.J.; Weaver, K.E. Characterization of the effects of an RpoC mutation that confers resistance to the Fst peptide toxin-antitoxin system toxin. J. Bacteriol. 2013, 195, 156–166. [Google Scholar] [CrossRef]
- Patel, S.; Weaver, K.E. Addiction toxin Fst has unique effects on chromosome segregation and cell division in Enterococcus faecalis and Bacillus subtilis. J. Bacteriol. 2006, 188, 5374–5384. [Google Scholar] [CrossRef]
- Michaux, C.; Hartke, A.; Martini, C.; Reiss, S.; Albrecht, D.; Budin-Verneuil, A.; Sanguinetti, M.; Engelmann, S.; Hain, T.; Verneuil, N.; et al. Involvement of Enterococcus faecalis sRNAs in the stress response and virulence. Infect. Immun. 2014. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef]
- Lewis, K. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immun. 2008, 322, 107–131. [Google Scholar]
- Gefen, O.; Balaban, N.Q. The importance of being persistent: Heterogeneity of bacterial populations under antibiotic stress. FEMS Microbiol. Rev. 2009, 33, 704–717. [Google Scholar] [CrossRef]
- Hooper, D.C. Mechanisms of action of antimicrobials: Focus on fluoroquinolones. Clin. Infect. Dis. 2001, 32, S9–S15. [Google Scholar] [CrossRef]
- Dorr, T.; Vulic, M.; Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010, 8, e1000317. [Google Scholar] [CrossRef]
- Wagner, E.G.; Unoson, C. The toxin-antitoxin system tisB-istr1: Expression, regulation, and biological role in persister phenotypes. RNA Biol. 2012, 9, 1513–1519. [Google Scholar] [CrossRef]
- Unoson, C.; Wagner, E.G. A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli. Mol. Microbiol. 2008, 70, 258–270. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Prock, S.; Reichert, J.; Wadhwani, P.; Zimpfer, B.; Burck, J.; Berditsch, M.; Elstner, M.; Ulrich, A.S. Peptide-lipid interactions of the stress-response peptide TisB that induces bacterial persistence. Biophys. J. 2012, 103, 1460–1469. [Google Scholar] [CrossRef]
- Gurnev, P.A.; Ortenberg, R.; Dorr, T.; Lewis, K.; Bezrukov, S.M. Persister-promoting bacterial toxin TisB produces anion-selective pores in planar lipid bilayers. FEBS Lett. 2012, 586, 2529–2534. [Google Scholar] [CrossRef]
- Dorr, T.; Lewis, K.; Vulic, M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 2009, 5, e1000760. [Google Scholar] [CrossRef]
- Maisonneuve, E.; Shakespeare, L.J.; Jørgensen, M.G.; Gerdes, K. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. USA 2011, 108, 13206–13211. [Google Scholar]
- Kwan, B.W.; Valenta, J.A.; Benedik, M.J.; Woods, T.K. Arrested protein synthesis increases persister-like cell formation. Antimicrob. Agents Chemother. 2013, 57, 1468–1473. [Google Scholar] [CrossRef]
- Rotem, E.; Loinger, A.; Ronin, I.; Levin-Reisman, I.; Gabay, C.; Shoresh, N.; Biham, O.; Balaban, N.Q. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc. Natl. Acad. Sci. USA 2010, 107, 12541–12546. [Google Scholar] [CrossRef]
- Irnov, I.; Sharma, C.M.; Vogel, J.; Winkler, W.C. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res. 2010, 38, 6637–6651. [Google Scholar] [CrossRef]
- Saito, S.; Kakeshita, H.; Nakamura, K. Novel small RNA-encoding genes in the intergenic regions of Bacillus subtilis. Gene 2009, 428, 2–8. [Google Scholar] [CrossRef]
- Tovar-Rojo, F.; Setlow, P. Effects of mutant small, acid-soluble spore proteins from Bacillus subtilis on DNA in vivo and in vitro. J. Bacteriol. 1991, 173, 4827–4835. [Google Scholar]
- Yamamoto, T.; Obana, N.; Yee, L.M.; Asai, K.; Nomura, N.; Nakamura, K. SP10 infectivity is aborted after bacteriophage SP10 infection induces nonA transcription on the prophage SPβ region of the bacillus subtilis genome. J. Bacteriol. 2014, 196, 693–706. [Google Scholar] [CrossRef]
- Kunkel, B.; Losick, R.; Stragier, P. The Bacillus subtilis gene for the development transcription factor sigma k is generated by excision of a dispensable DNA element containing a sporulation recombinase gene. Genes Dev. 1990, 4, 525–535. [Google Scholar] [CrossRef]
- Sato, T.; Kobayashi, Y. The ars operon in the skin element of Bacillus subtilis confers resistance to arsenate and arsenite. J. Bacteriol. 1998, 180, 1655–1661. [Google Scholar]
- Durand, S.; Jahn, N.; Condon, C.; Brantl, S. Type I toxin-antitoxin systems in Bacillus subtilis. RNA Biol. 2012, 9, 1491–1497. [Google Scholar] [CrossRef]
- Pecota, D.C.; Wood, T.K. Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. 1996, 178, 2044–2050. [Google Scholar]
- Samson, J.E.; Magadan, A.H.; Sabri, M.; Moineau, S. Revenge of the phages: Defeating bacterial defences. Nat. Rev. Microbiol. 2013, 11, 675–687. [Google Scholar] [CrossRef]
- Emond, E.; Dion, E.; Walker, S.A.; Vedamuthu, E.R.; Kondo, J.K.; Moineau, S. AbiQ, an abortive infection mechanism from Lactococcus lactis. Appl. Environ. Microbiol. 1998, 64, 4748–4756. [Google Scholar]
- Pinel-Marie, M.L.; Brielle, R.; Felden, B. Dual toxic-peptide-coding Staphylococcus aureus RNA under antisense regulation targets host cells and bacterial rivals unequally. Cell Rep. 2014, 7, 424–435. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.D.; Vivas, E.I.; Walsh, C.T.; Kolter, R. MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli. J. Bacteriol. 1995, 177, 4194–4197. [Google Scholar]
- Nichols, R.J.; Sen, S.; Choo, Y.J.; Beltrao, P.; Zietek, M.; Chaba, R.; Lee, S.; Kazmierczak, K.M.; Lee, K.J.; Wong, A.; et al. Phenotypic landscape of a bacterial cell. Cell 2011, 144, 143–156. [Google Scholar] [CrossRef]
- Lima-Mendez, G.; Toussaint, A.; Leplae, R. Analysis of the phage sequence space: The benefit of structured information. Virology 2007, 365, 241–249. [Google Scholar] [CrossRef]
- Chibani-Chennoufi, S.; Bruttin, A.; Dillmann, M.L.; Brussow, H. Phage-host interaction: An ecological perspective. J. Bacteriol. 2004, 186, 3677–3686. [Google Scholar] [CrossRef]
- Better, M.; Freifelder, D. Studies on the replication of Escherichia coli phage Lambda DNA. I. The kinetics of DNA replication and requirements for the generation of rolling circles. Virology 1983, 126, 168–182. [Google Scholar] [CrossRef]
- Ploss, M.; Kuhn, A. Kinetics of filamentous phage assembly. Phys. Biol. 2010, 7, 045002. [Google Scholar] [CrossRef]
- Jorgensen, M.G.; Thomason, M.K.; Havelund, J.; Valentin-Hansen, P.; Storz, G. Dual function of the McaS small RNA in controlling biofilm formation. Genes Dev. 2013, 27, 1132–1145. [Google Scholar] [CrossRef]
- Thomason, M.K.; Fontaine, F.; de Lay, N.; Storz, G. A small RNA that regulates motility and biofilm formation in response to changes in nutrient availability in Escherichia coli. Mol. Microbiol. 2012, 84, 17–35. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Awano, N.; Masuda, H.; Park, J.-H.; Inouye, M. Transcriptional repressor HipB regulates the multiple promoters in Escherichia coli. J. Mol. Microbiol. Biotechnol. 2013, 23, 440–447. [Google Scholar] [CrossRef]
- Wang, X.; Kim, Y.; Hong, S.H.; Ma, Q.; Brown, B.L.; Pu, M.; Tarone, A.M.; Benedik, M.J.; Peti, W.; Page, R.; et al. Antitoxin MqsA helps mediate the bacterial general stress response. Nat. Chem. Biol. 2011, 7, 359–366. [Google Scholar] [CrossRef]
- Soo, V.W.; Wood, T.K. Antitoxin MqsA represses curli formation through the master biofilm regulator CsgD. Sci. Rep. 2013, 3, 3186. [Google Scholar]
- Kim, Y.; Wang, X.; Zhang, X.S.; Grigoriu, S.; Page, R.; Peti, W.; Wood, T.K. Escherichia coli toxin/antitoxin pair MqsR/MqsA regulate toxin CspD. Environ. Microbiol. 2010, 12, 1105–1121. [Google Scholar] [CrossRef]
- Hu, Y.; Benedik, M.J.; Wood, T.K. Antitoxin DinJ influences the general stress response through transcript stabilizer CspE. Environ. Microbiol. 2012, 14, 669–679. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wen, J.; Fozo, E.M. sRNA Antitoxins: More than One Way to Repress a Toxin. Toxins 2014, 6, 2310-2335. https://doi.org/10.3390/toxins6082310
Wen J, Fozo EM. sRNA Antitoxins: More than One Way to Repress a Toxin. Toxins. 2014; 6(8):2310-2335. https://doi.org/10.3390/toxins6082310
Chicago/Turabian StyleWen, Jia, and Elizabeth M. Fozo. 2014. "sRNA Antitoxins: More than One Way to Repress a Toxin" Toxins 6, no. 8: 2310-2335. https://doi.org/10.3390/toxins6082310
APA StyleWen, J., & Fozo, E. M. (2014). sRNA Antitoxins: More than One Way to Repress a Toxin. Toxins, 6(8), 2310-2335. https://doi.org/10.3390/toxins6082310