Structural Insights into Bacillus thuringiensis Cry, Cyt and Parasporin Toxins

Abstract
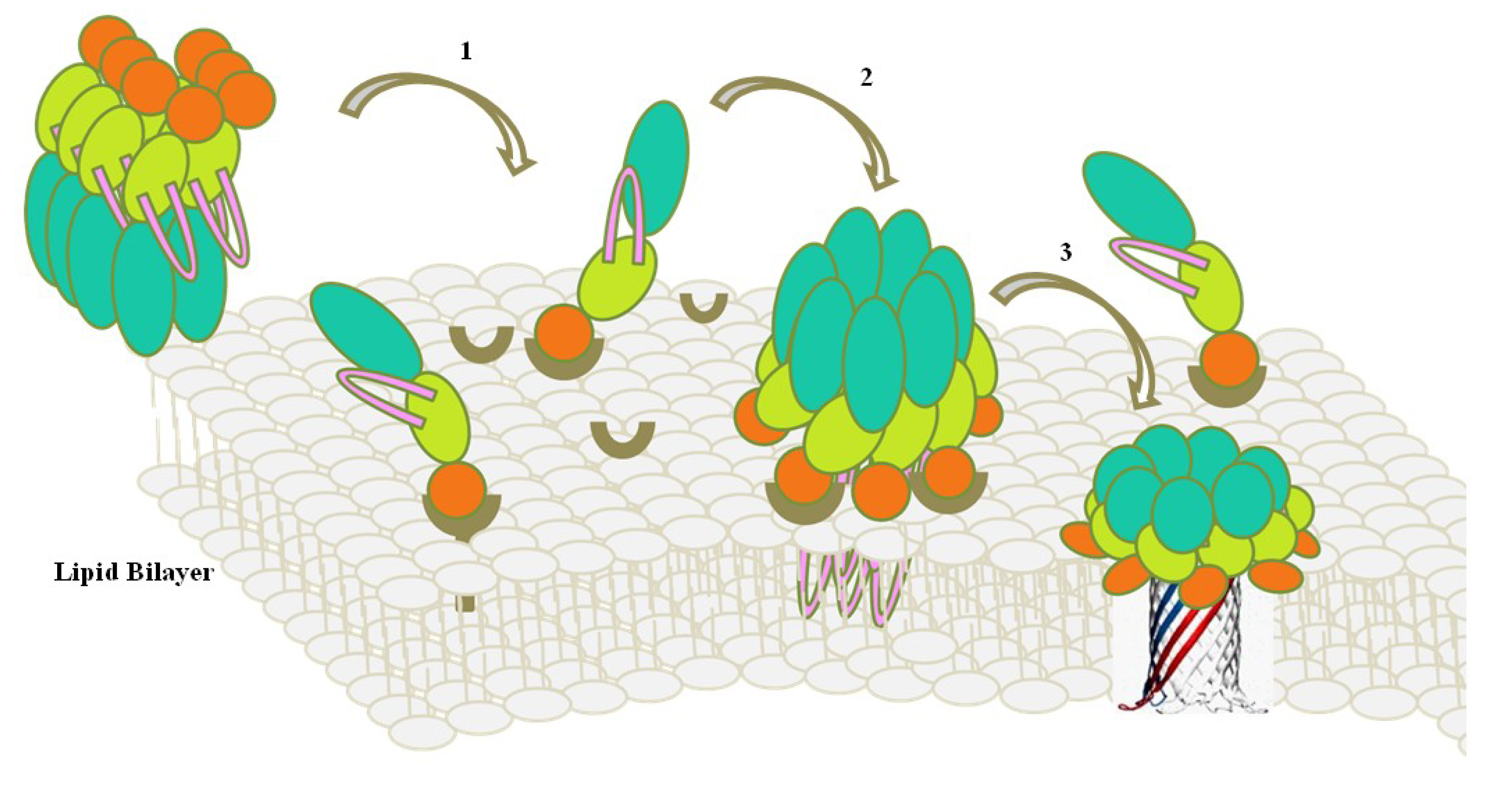
:1. Introduction
2. Cry Toxins
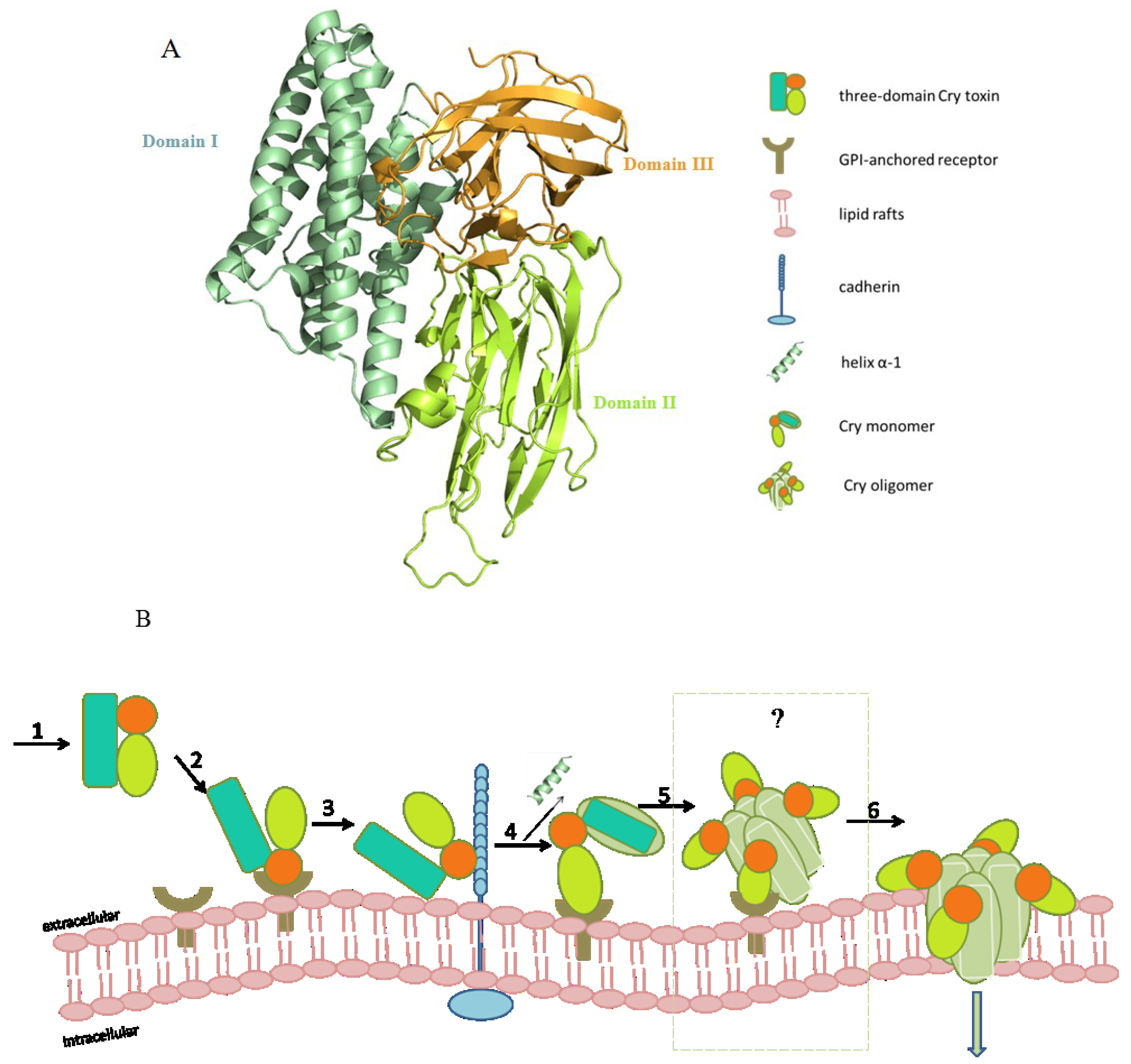
2.1. Structure of Three-Domain Cry Toxins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
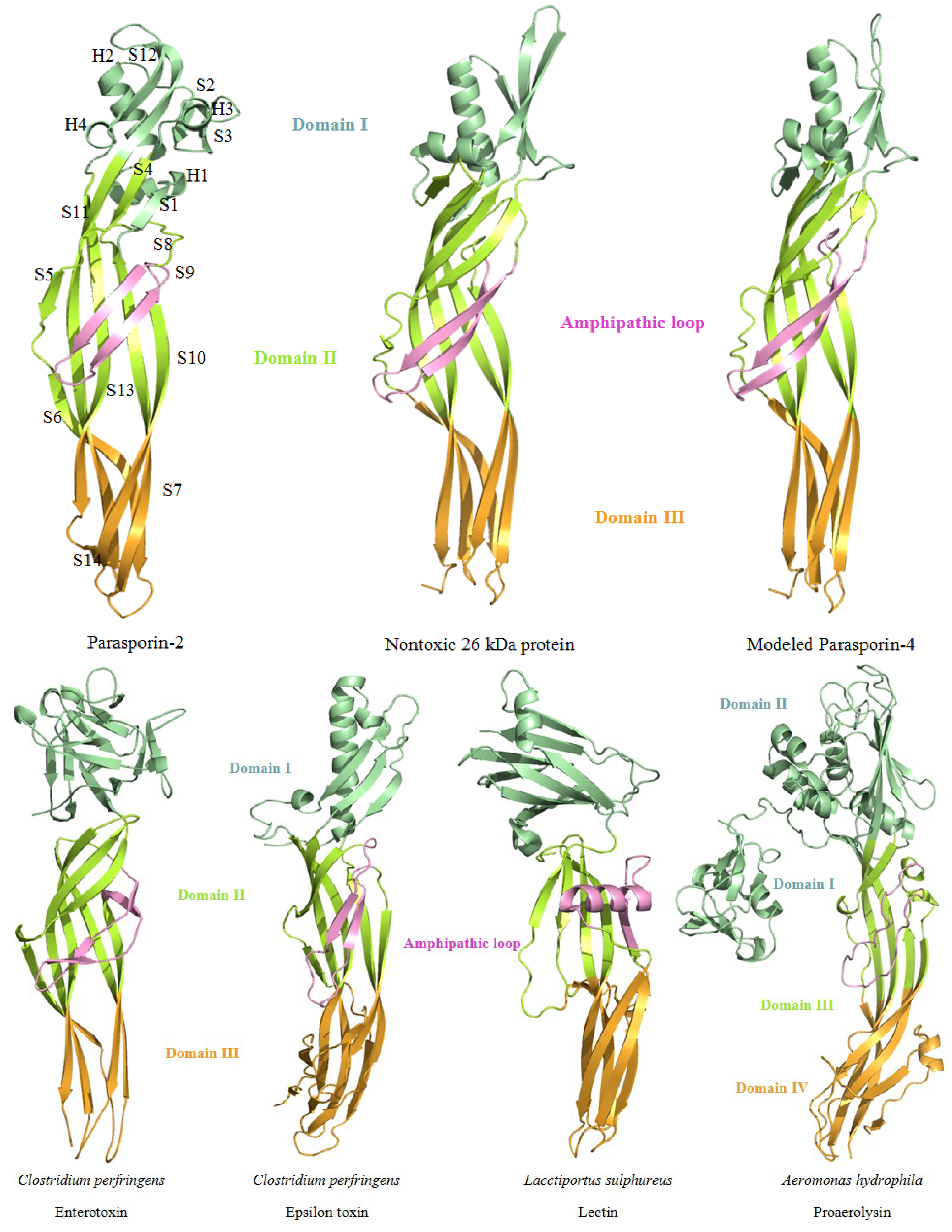{kind=link}
{kind=link}
| Toxin | Target Insects | Expressing Strain | Activation | Resolution | PDB ID | Domain I | Domain II | Domain III | References |
|---|---|---|---|---|---|---|---|---|---|
| Cry1Aa | Lepidoptera | Bt kurstaki HD-1 | Trypsin 65 kDa | 2.25 Å | 1CIY | resi 33–253 | resi 265–461 | resi 463–609, resi 254264 | [23] |
| Cry1Ac * | Lepidoptera | Bt kurstaki HD73 | Trypsin 65 kDa | 2.35 Å | 4ARX | resi 31–263 | resi 255–462 | resi 463–609 | [24] |
| Cry2Aa | Diptera Lepidoptera | Bt kurstaki HD-1 | Protoxin 62 kDa | 2.2 Å | 1I5P | resi 1–272 | resi 273–473 | resi 474–633 | [25] |
| Cry3Aa | Coleoptera | Bt tenebrionis 1911 | Papain 67 kDa | 2.5 Å | 1DLC | resi 61–290 | resi 291–500 | resi 501–644 | [26] |
| Cry3Bb | Coleoptera | E. coli EG7231 | 67.2 kDa | 2.4Å | 1JI6 | resi 64–294 | resi 295–503 | resi 504–652 | [27] |
| Cry4Aa | Diptera | Bt israelensis | Trypsin 65 kDa | 2.8 Å | 2C9K | resi 68–321 | resi 322–524 | resi 525–679 | [28] |
| Cry4Ba | Diptera | Bt israelensis | Chymotrypsin 68 kDa | 1.75 Å | 1W99 | resi 84–282 | resi 283–466 | resi 467–641 | [29] |
| Cry8Ea * | Coleoptera | Bt185 | Chymotrypsin 66.2 kDa | 2.2 Å | 3EB7 | resi 64–290 | resi 291–500 | resi 501–652 | [30] |
| Cry5B | Nematode | Bt kurstaki HD-1(Native) E.coli M15(Sel-Met) | Elastase 66.14 kDa | 2.3 Å | 4D8M | resi 112–328 | resi 341–541 | resi 541–698 | [31] |

2.1.1. Domain I of the Three-Domain Cry Toxins

2.1.2. Domain II of Three-Domain Cry Toxins

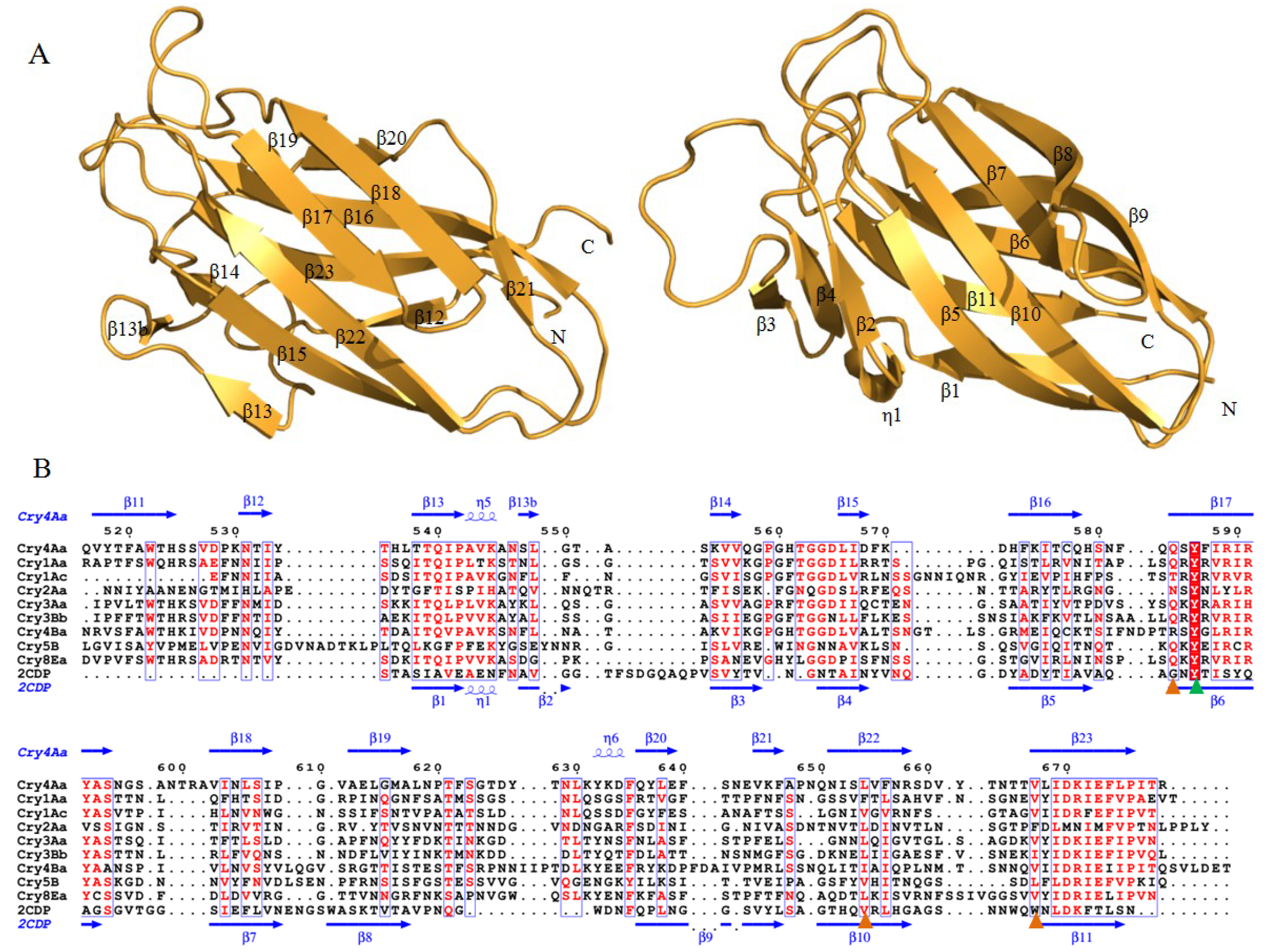
2.1.3. Domain III of Three-Domain Cry Toxins

2.1.4. Structure of Three-Domain Cry Toxins in Parasporin
2.2. Comparisons with Other Structure Known Toxins or Modules
2.3. Mechanism of the Three-Domain Cry Toxin
2.3.1. The Pore Formation Model
2.3.2. The Signaling Pathway Model
3. Cyt Toxin
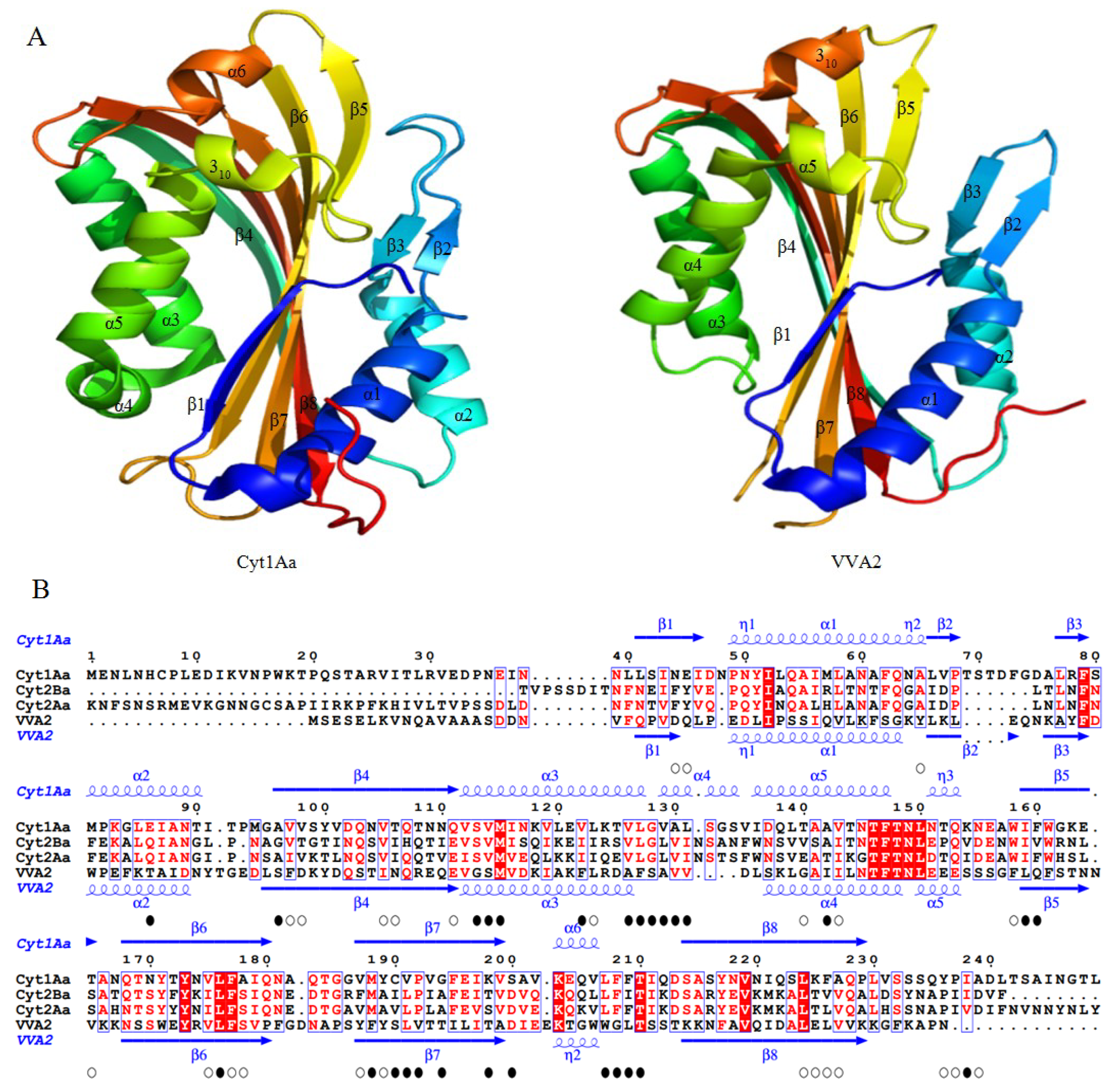
3.1. Structure of the Cyt Toxins
3.2. Comparisons with Other Structure Known PFTs
3.3. Binding Mechanism of the Cyt Toxin
3.4. Mechanism of Synergism with the Cry Toxins

4. Parasporin Toxins
4.1. Definition and Classification
4.2. Cytotoxicity and Action Mechanism
4.2.1. Parasporin-1
4.2.2. Parasporin-2
4.2.3. Parasporin-3
4.2.4. Parasporin-4
4.2.5. Other Parasporins
4.3. Structure of Parasporins and Other Aerolysin Family Members
4.3.1. Structure of the Aerolysin-Type Parasporin
4.3.1.1. Domain I of the Aerolysin-Type Parasporin
4.3.1.2. Domain II of the β-PFT Type Parasporin
4.3.1.3. Domain III of the β-PFT Type Parasporin

4.3.2. Comparisons with Other Structure Known Toxins in Aerolysin Family
4.3.2.1. The Most Diverse Domain
| Toxins | Strain | PDB ID | Native Size in kDa | Cytotoxicity | Receptor Characteristics | Pore Formation | Oligomerization (number of monomers) | Toxin Type |
|---|---|---|---|---|---|---|---|---|
| Parasporin-1 | B. thuringiensis A1190 | unkown | 81 | Hela, MOLT-4, Hep G2, HL-60 | Beclin-1 | unknown | unknown | three-domain-type Cry toxin |
| Parasporin-2 | B. thuringiensis A1547 | 2ZTB | 37 | MOLT-4, Jurkat, Sawano, HL-60, and HepG2 | require GPI-anchored protein | possibly | unknown | Aerolysin-type β-PFT |
| Parasporin-3 | B. thuringiensis A1462 | - | 88 | HL-60, HepG2 | unknown | unknown | unknown | three-domain-type Cry toxin |
| Parasporin-4 | B. thuringiensis A1470 | - | 34 | MOLT-4, HL-60, HepH2, Caco-2, Sawano, TCS | cholesterol-dependent | possibly | unknown | cholesterol-independent β-PFT |
| nontoxic 26 kDa protein | B. thuringiensis A1470 | 2D42 | 32 | nontoxic | unknown | unknown | unknown | nontoxic Aerolysin-type β-PFT |
| Aerolysin | Aeromonas hydrophila | 1PRE | 52 | broad range with GPI-anchored epithelia cells | GPI-anchored receptors | yes | 7 | Aerolysin-type β-PFT |
| Epsilon | Clostridium perfringens | 1UYJ | 32.5 | limit cell lines as MDCK, G-402 | non GPI-anchored membrane protein | yes | 7 | Aerolysin-type β-PFT |
| Entertoxin | Clostridium perfringens | 2XH6 | 35 | intestinal epithelia cells | Claudi | yes | 3/6 | Aerolysin-typeβ-PFT |
| LSL | Laetiportus sulphureus | 1W3A | 35 | unknown | glycoproteins | possibly | 4/6 | Aerolysin-type β-PFT |
4.3.2.2. The Pore-Forming Domain
4.3.2.3. Membrane Insertion Mechanism

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Didelot, X.; Barker, M.; Falush, D.; Priest, F.G. Evolution of pathogenicity in the Bacillus cereus group. Syst. Appl. Microbiol. 2009, 32, 81–90. [Google Scholar] [CrossRef]
- Schinepf, E.; Crickmore, N.; van Rie, J.; Lereclus, D.; Baum, B.; Feitelson, J.; Zeigler, D.R.; Dean, D.H. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol. Mol. Biol. Rev. 1998, 62, 775–806. [Google Scholar]
- Nester, E.W.; Thomashow, L.S.; Metz, M. 100 Years of Bacillus thuringiensis: A Critical Scientific Assessment; American Society for Microbiology (ASM): Washington, DC, USA, 2002. [Google Scholar]
- Khyami-Horani, H.; Hajaij, M.; Charles, J.F. Characterization of Bacillus thuringiensis ser. jordanica (Serotype H71), a novel serovariety isolated in Jordan. Curr. Microbiol. 2003, 47, 26–31. [Google Scholar] [CrossRef]
- De Barjac, H.; Bonnefoi, A. Essai de classification biochimique et sérologique de 24 souches de Bacillus du type B. thuringiensis. Entomophaga 1962, 7, 5–31. (In French) [Google Scholar] [CrossRef]
- Crickmore, N.; Zeigler, D.R.; Schnepf, E.; van Rie, J.; Lereclus, D.; Baum, J.; Bravo, A.; Dean, D.H. Bacillus thuringiensis Toxin Nomenclature. Available online: http://www.lifesci.sussex.ac.uk/home/Neil_Crickmore/Bt (accessed on 3 September 2014).
- Gao, M.; Li, R.; Dai, S.; Wu, Y.; Yi, D. Diversity of Bacillus thuringiensis strains from soil in China and their pesticidal activities. Biol. Control 2008, 44, 380–388. [Google Scholar] [CrossRef]
- Zhong, C.; Ellar, D.J.; Bishop, A.; Johnson, C.; Lin, S.; Hart, E.R. Characterization of a Bacillus thuringiensis δ-endotoxin which is toxic to insects in three orders. J. Invertebr. Pathol. 2000, 76, 131–139. [Google Scholar] [CrossRef]
- Haider, M.Z.; Ward, E.S.; Ellar, D.J. Cloning and heterologous expression of an insecticidal delta-endotoxin gene from Bacillus thuringiensis var. aizawai ICI toxic to both lepidoptera and diptera. Gene 1987, 52, 285–290. [Google Scholar] [CrossRef]
- Crickmore, N.; Zeigler, D.R.; Feitelson, J.; Schnepf, E.; van Rie, J.; Lereclus, D.; Baum, J.; Dean, D.H. Revision of the nomenclature for the Bacillus thuringiensis pesticidal crystal proteins. Microbiol. Mol. Biol. Rev. 1998, 62, 807–813. [Google Scholar]
- Gill, S.S.; Hornung, J.M. Cytolytic activity of Bacillus thuringiensis proteins to insect and mammalian-cell lines. J. Invertebr. Pathol. 1987, 50, 16–25. [Google Scholar] [CrossRef]
- Mizuki, E.; Park, Y.S.; Saitoh, H.; Yamashita, S.; Akao, T.; Higuchi, K.; Ohba, M. Parasporin, a human leukemic cell-recognizing parasporal protein of Bacillus thuringiensis. Clin. Diagn. Lab. Immunol. 2000, 7, 625–634. [Google Scholar]
- Okumura, S.; Ohba, M.; Mizuki, E.; Crickmore, N.; Côté, J.C.; Nagamatsu, Y.; Kitada, S.; Sakai, H.; Harata, K.; Shin, T. Parasporin nomenclature. 2010. Available online: http://parasporin.fitc.pref.fukuoka.jp/ (accessed on 3 September 2014).
- De Maagd, R.A.; Bravo, A.; Berry, C.; Crickmore, N.; Schnepf, H.E. Structure, diversity, and evolution of protein toxins from spore-forming entomopathogenic bacteria. Annu. Rev. Genet. 2003, 37, 409–433. [Google Scholar] [CrossRef]
- Estruch, J.J.; Warren, G.W.; Mullins, W.A.; Nye, G.J.; Craig, J.A.; Koziel, M.G. Vip3A, a novel Bacillus thuringiensis vegetative insecticidal protein with a wide spectrum of activities against lepidopteran insects. Proc. Natl. Acad. Sci. USA 1996, 93, 5389–5394. [Google Scholar]
- Andrews, R.E.; Bibilos, M.M.; Bulla, L.A. Protease activation of the entomocidal protoxin of Bacillus thuringiensis subsp. kurstaki. Appl. Environ. Microbiol. 1985, 50, 737–742. [Google Scholar]
- Zhang, X.; Candas, M.; Griko, N.B.; Rose-Young, L.; Bulla, L.A. Cytotoxicity of Bacillus thuringiensis Cry1Ab toxin depends on specific binding of the toxin to the cadherin receptor BT-R1 expressed in insect cells. Cell. Death. Differ. 2005, 12, 1407–1416. [Google Scholar] [CrossRef]
- Bravo, A.; Gómez, I.; Conde, J.; Munoz-Garay, C.; Sanchez, J.; Miranda, R.; Zhuang, M.; Gill, S.S.; Soberón, M. Oligomerization triggers binding of a Bacillus thuringiensis Cry1Ab pore-forming toxin to aminopeptidase N receptor leading to insertion into membrane microdomains. Biochim. Biophys. Acta 2004, 1667, 38–46. [Google Scholar]
- Pardo-Lopez, L.; Munoz-Garay, C.; Porta, H.; Rodriguez-Almazan, C.; Soberon, M.; Bravo, A. Strategies to improve the insecticidal activity of Cry toxins from Bacillus thuringiensis. Peptides 2009, 30, 589–595. [Google Scholar] [CrossRef]
- Gonzalez, M.R.; Bischofberger, M.; Pernot, L.; van der Goot, F.G.; Freche, B. Bacterial pore-forming toxins: The (w)hole story? Cell. Mol. Life. Sci. 2008, 65, 493–507. [Google Scholar] [CrossRef]
- Feil, S.C.; Polekhina, G.; Gorman, M.A.; Parker, M.W. Proteins membrane binding and pore formation introduction. Adv. Exp. Med. Biol. 2010, 677, 1–13. [Google Scholar] [CrossRef]
- Geny, B.; Popoff, M.R. Bacterial protein toxins and lipids: Pore formation or toxin entry into cells. Biol. Cell. 2006, 98, 667–678. [Google Scholar]
- Grochulski, P.; Masson, L.; Borisova, S.; Pusztaicarey, M.; Schwartz, J.L.; Brousseau, R.; Cygler, M. Bacillus thuringiensis CrylA(a) insecticidal toxin: Crystal structure and channel formation. J. Mol. Biol. 1995, 254, 447–464. [Google Scholar] [CrossRef]
- Derbyshire, D.J.; Ellar, D.J.; Li, J. Crystallization of the Bacillus thuringiensis toxin Cry1Ac and its complex with the receptor ligand N-acetyl-d-galactosamine. Acta Crystallogr. Sect. D: Biol. Crystallogr 2001, 57, 1938–1944. [Google Scholar] [CrossRef]
- Morse, R.J.; Yamamoto, T.; Stroud, R.M. Structure of Cry2Aa suggests an unexpected receptor binding epitope. Structure 2001, 9, 409–417. [Google Scholar] [CrossRef]
- Li, J.D.; Carroll, J.; Ellar, D.J. Crystal structure of insecticidal δ-endotoxin from Bacillus thuringiensis at 2.5 Å resolution. Nature 1991, 353, 815–821. [Google Scholar] [CrossRef]
- Galitsky, N.; Cody, V.; Wojtczak, A.; Ghosh, D.; Luft, J.R.; Pangborn, W.; English, L. Structure of the insecticidal bacterial delta-endotoxin Cry3Bb1 of Bacillus thuringiensis. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2001, 57, 1101–1109. [Google Scholar] [CrossRef]
- Boonserm, P.; Mo, M.; Angsuthanasombat, C.; Lescar, J. Structure of the functional form of the mosquito larvicidal Cry4Aa toxin from Bacillus thuringiensis at a 2.8-Angstrom resolution. J. Bacteriol. 2006, 188, 3391–3401. [Google Scholar] [CrossRef]
- Boonserm, P.; Davis, P.; Ellar, D.J.; Li, J. Crystal structure of the mosquito-larvicidal toxin Cry4Ba and its biological implications. J. Mol. Biol. 2005, 348, 363–382. [Google Scholar] [CrossRef]
- Guo, S.Y.; Ye, S.; Liu, Y.F.; Wei, L.; Xue, J.; Wu, H.F.; Song, F.P.; Zhang, J.; Wu, X.A.; Huang, D.F.; et al. Crystal structure of Bacillus thuringiensis Cry8Ea1: An insecticidal toxin toxic to underground pests, the larvae of Holotrichia parallela. J. Struct. Biol. 2009, 168, 259–266. [Google Scholar] [CrossRef]
- Hui, F.; Scheib, U.; Hu, Y.; Sommer, R.J.; Aroian, R.V.; Ghosh, P. Structure and glycolipid binding properties of the nematicidal protein Cry5B. Biochemistry 2012, 51, 9911–9921. [Google Scholar] [CrossRef]
- de Maagd, R.A.; Bravo, A.; Crickmore, N. How Bacillus thuringiensis has evolved specific toxins to colonize the insect world. Trends Genet. 2001, 17, 193–199. [Google Scholar] [CrossRef]
- Gouet, P.; Robert, X.; Courcelle, E. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucl. Acids Res. 2003, 31, 3320–3323. [Google Scholar] [CrossRef]
- Soberón, M.; Pardo-Lopez, L.; Lopez, I.; Gómez, I.; Tabashnik, B.E.; Bravo, A. Engineering modified Bt toxins to counter insect resistance. Science 2007, 318, 1640–1642. [Google Scholar] [CrossRef]
- Porta, H.; Jimenez, G.; Cordoba, E.; Leon, P.; Soberón, M.; Bravo, A. Tobacco plants expressing the Cry1AbMod toxin suppress tolerance to Cry1Ab toxin of Manduca sexta cadherin-silenced larvae. Insect Biochem. Mol. Biol. 2011, 41, 513–519. [Google Scholar] [CrossRef]
- Vachon, V.; Prefontaine, G.; Rang, C.; Coux, F.; Juteau, M.; Schwartz, J.L.; Brousseau, R.; Frutos, R.; Laprade, R.; Masson, L. Helix 4 mutants of the Bacillus thuringiensis insecticidal toxin Cry1Aa display altered pore-forming abilities. Appl. Environ. Microbiol. 2004, 70, 6123–6130. [Google Scholar] [CrossRef]
- Tigue, N.J.; Jacoby, J.; Ellar, D.J. The α-helix 4 residue, Asn135, is involved in the oligomerization of Cry1Ac1 and Cry1Ab5 Bacillus thuringiensis toxins. Appl. Environ. Microbiol. 2001, 67, 5715–5720. [Google Scholar] [CrossRef]
- Kanintronkul, Y.; Sramala, I.; Katzenmeier, G.; Panyim, S.; Angsuthanasombat, C. Specific mutations within the α4-α5 loop of the Bacillus thuringiensis Cry4B toxin reveal a crucial role for Asn-166 and Tyr-170. Mol. Biotechnol. 2003, 24, 11–19. [Google Scholar] [CrossRef]
- Pornwiroon, W.; Katzenmeier, G.; Panyim, S.; Angsuthanasombat, C. Aromaticity of Tyr-202 in the α4-α5 loop is essential for toxicity of the Bacillus thuringiensis Cry4A toxin. J. Biochem. Mol. Biol. 2004, 37, 292–297. [Google Scholar] [CrossRef]
- Gazit, E.; la Rocca, P.; Sansom, M.S.P.; Shai, Y. The structure and organization within the membrane of the helices composing the pore-forming domain of Bacillus thuringiensis delta-endotoxin are consistent with an “umbrella-like” structure of the pore. Proc. Natl. Acad. Sci. USA 1998, 95, 12289–12294. [Google Scholar] [CrossRef]
- Gazit, E.; Shai, Y. The assembly and organization of the α5 and α7 helices from the pore-forming domain of Bacillus thuringiensis δ-endotoxin: Relevance to a functional model. J. Biol. Chem. 1995, 270, 2571–2578. [Google Scholar] [CrossRef]
- Park, H.-W.; Federici, B. Effect of specific mutations in helix α7 of domain I on the stability and crystallization of Cry3A in Bacillus thuringiensis. Mol. Biotechnol. 2004, 27, 89–100. [Google Scholar] [CrossRef]
- Tiewsiri, K.; Fischer, W.B.; Angsuthanasombat, C. Lipid-induced conformation of helix 7 from the pore-forming domain of the Bacillus thuringiensis Cry4Ba toxin: Implications for toxicity mechanism. Arch. Biochem. Biophys. 2009, 482, 17–24. [Google Scholar] [CrossRef]
- Rajamohan, F.; Lee, M.K.; Dean, D.H. Bacillus thuringiensis insecticidal proteins: Molecular mode of action. Prog. Nucleic Acid. Res. Mol. Biol. 1998, 60, 1–27. [Google Scholar] [CrossRef]
- Gómez, I.; Miranda-Rios, J.; Rudino-Pinera, E.; Oltean, D.I.; Gill, S.S.; Bravo, A.; Soberon, M. Hydropathic complementarity determines interaction of epitope 869HITDTNNK876 in Manduca sexta Bt-R1 receptor with loop 2 of domain II of Bacillus thuringiensis Cry1A toxins. J. Biol. Chem. 2002, 277, 30137–30143. [Google Scholar]
- Gómez, I.; Arenas, I.; Benitez, I.; Miranda-Rios, J.; Becerril, B.; Grande, R.; Almagro, J.C.; Bravo, A.; Soberón, M. Specific epitopes of domains II and III of Bacillus thuringiensis Cry1Ab toxin involved in the sequential interaction with cadherin and aminopeptidase-N receptors in Manduca sexta. J. Biol. Chem. 2006, 281, 34032–34039. [Google Scholar] [CrossRef]
- Rajamohan, F.; Alzate, O.; Cotrill, J.A.; Curtiss, A.; Dean, D.H. Protein engineering of Bacillus thuringiensis δ-endotoxin: Mutations at domain II of CryIAb enhance receptor affinity and toxicity toward gypsy moth larvae. Proc. Natl. Acad. Sci. USA 1996, 93, 14338–14343. [Google Scholar] [CrossRef]
- Rajamohan, F.; Hussain, S.R.A.; Cotrill, J.A.; Gould, F.; Dean, D.H. Mutations at domain II, loop 3, of Bacillus thuringiensis CryIAa and CryIAb δ-endotoxins suggest loop 3 is involved in initial binding to lepidopteran midguts. J. Biol. Chem. 1996, 271, 25220–25226. [Google Scholar] [CrossRef]
- Lee, M.K.; Rajamohan, F.; Jenkins, J.L.; Curtiss, A.S.; Dean, D.H. Role of two arginine residues in domain II, loop 2 of Cry1Ab and Cry1Ac Bacillus thuringiensis δ-endotoxin in toxicity and binding to Manduca sexta and Lymantria dispar aminopeptidase N. Mol Microbiol 2000, 38, 289–298. [Google Scholar] [CrossRef]
- Arenas, I.; Bravo, A.; Soberón, M.; Gómez, I. Role of alkaline phosphatase from Manduca sexta in the mechanism of action of Bacillus thuringiensis Cry1Ab toxin. J. Biol. Chem. 2010, 285, 12497–12503. [Google Scholar] [CrossRef]
- Gómez, I.; Dean, D.H.; Bravo, A.; Soberon, M. Molecular basis for Bacillus thuringiensis Cry1Ab toxin specificity: Two structural determinants in the Manduca sexta Bt-R1 receptor interact with loops α8 and 2 in domain II of Cy1Ab toxin. Biochemistry 2003, 42, 10482–10489. [Google Scholar] [CrossRef]
- Atsumi, S.; Inoue, Y.; Ishizaka, T.; Mizuno, E.; Yoshizawa, Y.; Kitami, M.; Sato, R. Location of the Bombyx mori 175 kDa cadherin-like protein-binding site on Bacillus thuringiensis Cry1Aa toxin. FEBS J. 2008, 275, 4913–4926. [Google Scholar] [CrossRef]
- Xie, R.Y.; Zhuang, M.B.; Ross, L.S.; Gomez, I.; Oltean, D.I.; Bravo, A.; Soberon, M.; Gill, S.S. Single amino acid mutations in the cadherin receptor from Heliothis virescens affect its toxin binding ability to Cry1A toxins. J. Biol. Chem. 2005, 280, 8416–8425. [Google Scholar]
- Pacheco, S.; Gomez, I.; Arenas, I.; Saab-Rincon, G.; Rodriguez-Almazan, C.; Gill, S.S.; Bravo, A.; Soberon, M. Domain II loop 3 of Bacillus thuringiensis Cry1Ab toxin is involved in a “ping pong” binding mechanism with Manduca sexta aminopeptidase-N and cadherin receptors. J. Biol. Chem. 2009, 284, 32750–32757. [Google Scholar] [CrossRef]
- Chen, J.; Brown, M.R.; Hua, G.; Adang, M.J. Comparison of the localization of Bacillus thuringiensis Cry1A δ-endotoxins and their binding proteins in larval midgut of tobacco hornworm, Manduca sexta. Cell. Tissue. Res. 2005, 321, 123–129. [Google Scholar] [CrossRef]
- Aronson, A.I.; Wu, D.; Zhang, C.L. Mutagenesis of specificity and toxicity regions of a Bacillus thuringiensis protoxin gene. J. Bacteriol. 1995, 177, 4059–4065. [Google Scholar]
- De Maagd, R.A.; Kwa, M.S.G.; van der Klei, H.; Yamatoto, T.; Schipper, B.; Vlak, J.M.; Stiekema, W.; Bosch, D. Domain III substitution in Bacillus thuringiensis δ-endotoxin CryIA(b) results in superior toxicity for Spodoptera exigua and altered membrane protein recognition. Appl. Environ. Microbiol. 1996, 62, 1537–1543. [Google Scholar]
- Lee, M.K.; Young, B.A.; Dean, D.H. Domain III exchanges of Bacillus thuringiensis CryIA toxins affect binding to different gypsy moth midgut receptors. Biochem. Biophys. Res. Commun. 1995, 216, 306–312. [Google Scholar] [CrossRef]
- Masson, L.; Lun, Y.J.; Mazza, A.; Brousseau, R.; Adang, M.J. The CryIA(c) receptor purified from Manduca sexta displays mltiple specificities. J. Biol. Chem. 1995, 270, 20309–20315. [Google Scholar]
- Burton, S.L.; Ellar, D.J.; Li, J.; Derbyshire, D.J. N-acetylgalactosamine on the putative insect receptor aminopeptidase N is recognised by a site on the domain III lectin-like fold of a Bacillus thuringiensis insecticidal toxin. J. Mol. Biol. 1999, 287, 1011–1022. [Google Scholar] [CrossRef]
- Shan, S.; Zhang, Y.; Ding, X.; Hu, S.; Sun, Y.; Yu, Z.; Liu, S.; Zhu, Z.; Xia, L. A Cry1Ac toxin variant generated by directed evolution has enhanced toxicity against lepidopteran insects. Curr. Microbiol. 2011, 62, 358–365. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Q.; Wang, F.; Ding, X.; Xia, L. Residue 544 in Domain III of the Bacillus thuringiensis Cry1Ac toxin is involved in protein structure stability. Protein J. 2010, 29, 440–444. [Google Scholar] [CrossRef]
- Tabashnik, B.E.; Liu, Y.-B.; de Maagd, R.A.; Dennehy, T.J. Cross-resistance of pink bollworm (Pectinophora gossypiella) to Bacillus thuringiensis toxins. Appl. Environ. Microbiol. 2000, 66, 4582–4584. [Google Scholar] [CrossRef]
- Akiba, T.; Ichimatsu, T.; Katayama, H.; Akao, T.; Nakamura, O.; Mizuki, E.; Ohba, M.; Harata, K. Structure of parasporin-1, a novel bacterial cytotoxin against human cancer cells. Acta Crystallogr. Sect. A 2005, A61, C250. [Google Scholar]
- Katayama, H.; Kusaka, Y.; Yokota, H.; Akao, T.; Kojima, M.; Nakamura, O.; Mekada, E.; Mizuki, E. Parasporin-1, a novel cytotoxic protein from Bacillus thuringiensis, induces Ca2+ influx and a sustained elevation of the cytoplasmic Ca2+ concentration in toxin-sensitive cells. J. Biol. Chem. 2007, 282, 7742–7752. [Google Scholar]
- Yamashita, S.; Katayama, H.; Saitoh, H.; Akao, T.; Park, Y.S.; Mizuki, E.; Ohba, M.; Ito, A. Typical three-domain Cry proteins of Bacillus thuringiensis strain A1462 exhibit cytocidal activity on limited human cancer cells. J. Biochem. 2005, 138, 663–672. [Google Scholar] [CrossRef]
- Vidisha, K. Investigation of Parasporins, the Cytotoxic Proteins from the Bacterium Bacillus thuringiensis. Ph.D. Thesis, University of Sussex, Brighton, UK, 2013. [Google Scholar]
- Nagamatsu, Y.; Okamura, S.; Saitou, H.; Akao, T.; Mizuki, E. Three Cry toxins in two types from Bacillus thuringiensis strain M019 preferentially kill human hepatocyte cancer and uterus cervix cancer cells. Biosci. Biotechnol. Biochem. 2010, 74, 494–498. [Google Scholar] [CrossRef]
- Iacovache, I.; van der Goot, F.G.; Pernot, L. Pore formation: An ancient yet complex form of attack. Biochim. Biophys. Acta 2008, 1778, 1611–1623. [Google Scholar] [CrossRef]
- Parker, M.W.; Postma, J.P.M.; Pattus, F.; Tucker, A.D.; Tsernoglou, D. Refined structure of the pore-forming domain of colicin A at 2.4 Å resolution. J. Biol. Chem. 1992, 224, 639–657. [Google Scholar]
- Padmavathi, P.V.L.; Steinhoff, H.J. Conformation of the closed channel state of colicin A in proteoliposomes: An umbrella model. J. Mol. Biol. 2008, 378, 204–214. [Google Scholar] [CrossRef]
- Lakey, J.H.; van der Goot, F.G.; Pattus, F. All in the family: The toxic activity of pore-forming colicins. Toxicology 1994, 87, 85–108. [Google Scholar] [CrossRef]
- Braun, V.; Pilsl, H.; Gross, P. Colicins: Structures, modes of action, transfer through membranes, and evolution. Arch. Microbiol. 1994, 161, 199–206. [Google Scholar] [CrossRef]
- Kanagawa, M.; Satoh, T.; Ikeda, A.; Nakano, Y.; Yagi, H.; Kato, K.; Kojima-Aikawa, K.; Yamaguchi, Y. Crystal structures of human secretory proteins ZG16p and ZG16b reveal a Jacalin-related beta-prism fold. Biochem. Biophys. Res. Commun. 2011, 404, 201–205. [Google Scholar] [CrossRef]
- Holm, L.; Rosenstrom, R. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef]
- Kumazawa-Inoue, K.; Mimura, T.; Hosokawa-Tamiya, S.; Nakano, Y.; Dohmae, N.; Kinoshita-Toyoda, A.; Toyoda, H.; Kojima-Aikawa, K. ZG16p, an animal homolog of beta-prism fold plant lectins, interacts with heparan sulfate proteoglycans in pancreatic zymogen granules. Glycobiology 2012, 22, 258–266. [Google Scholar] [CrossRef]
- Kleene, R.; Dartsch, H.; Kern, H.F. The secretory lectin ZG16p mediates sorting of enzyme proteins to the zymogen granule membrane in pancreatic acinar cells. Eur. J. Cell. Biol. 1999, 18, 79–90. [Google Scholar] [CrossRef]
- Schmidt, K.; Schrader, M.; Kern, H.F.; Kleene, R. Regulated apical secretion of zymogens in rat pancreas involvement of the glycosylophophatidylinositol-anchored glycoprotein GP-2, the lectin ZG16p, and cholesterol-glycophingolipid-enriched microdomains. J. Biol. Chem. 2001, 276, 14314–14323. [Google Scholar]
- Sankaranarayanan, R.; Sekar, K.; Banerjee, R.; Sharma, V.; Surolia, A.; Vijayan, M. A novel mode of carbohydrate recognition in jacalin, a Moraceae plant lectin with a β-prism fold. Nat. Struct. Mol. Biol. 1996, 3, 596–603. [Google Scholar] [CrossRef]
- Michel, G.; Barbeyron, T.; Kloareg, B.; Czjzek, M. The family 6 carbohydrate-binding modules have coevolved with their appended catalytic modules toward similar substrate specificity. Glycobiology 2009, 19, 615–623. [Google Scholar] [CrossRef]
- Guillén, D.; Sánchez, S.; Rodríguez-Sanoja, R. Carbohydrate-binding domains: Multiplicity of biological roles. Appl. Microbiol. Biotechnol 2010, 85, 1241–1249. [Google Scholar] [CrossRef]
- Pires, V.M.R.; Henshaw, J.L.; Prates, J.A.M.; Bolam, D.N.; Ferreira, L.M.A.; Fontes, C.M.G.A.; Henrissat, B.; Planas, A.; Gilbert, H.J.; et al. The crystal structure of the family 6 carbohydrate binding module from Cellvibrio mixtus endoglucanase 5A in complex with oligosaccharides reveals two distinct binding sites with different ligand specificities. J. Biol. Chem. 2004, 279, 21560–21568. [Google Scholar] [CrossRef]
- Bravo, A.; Likitvivatanavong, S.; Gill, S.S.; Soberón, M. Bacillus thuringiensis: A story of a successful bioinsecticide. Insect Biochem. Mol. Biol. 2011, 41, 423–431. [Google Scholar] [CrossRef]
- Gómez, I.; Sanchez, J.; Miranda, R.; Bravo, A.; Soberon, M. Cadherin-like receptor binding facilitates proteolytic cleavage of helix α1 in domain I and oligomer pre-pore formation of Bacillus thuringiensis Cry1Ab toxin. FEBS Lett. 2002, 513, 242–246. [Google Scholar] [CrossRef]
- Soberón, M.; Gill, S.S.; Bravo, A. Signaling versus punching hole: How do Bacillus thuringiensis toxins kill insect midgut cells? Cell. Mol. Life Sci. 2009, 66, 1337–1349. [Google Scholar] [CrossRef]
- Greig, S.L.; Radjainia, M.; Mitra, A.K. Oligomeric structure of Colicin Ia channel in lipid bilayer membranes. J. Biol. Chem. 2009, 284, 16126–16134. [Google Scholar] [CrossRef]
- Groulx, N.; McGuire, H.; Laprade, R.; Schwartz, J.-L.; Blunck, R. Single molecule fluorescence study of the Bacillus thuringiensis toxin Cry1Aa reveals tetramerization. J. Biol. Chem. 2011, 286, 42274–42282. [Google Scholar]
- Pigott, C.R.; Ellar, D.J. Role of receptors in Bacillus thuringiensis crystal toxin activity. Microbiol. Mol. Biol. Rev. 2007, 71, 255–281. [Google Scholar] [CrossRef]
- Zhang, X.B.; Candas, M.; Griko, N.B.; Taussig, R.; Bulla, L.A. A mechanism of cell death involving an adenylyl cyclase/PKA signaling pathway is induced by the Cry1Ab toxin of Bacillus thuringiensis. Proc. Natl. Acad. Sci. USA 2006, 103, 9897–9902. [Google Scholar] [CrossRef]
- Vachon, V.; Laprade, R.; Schwartz, J.L. Current models of the mode of action of Bacillus thuringiensis insecticidal crystal proteins: A critical review. J. Invertebr. Pathol. 2012, 111, 1–12. [Google Scholar] [CrossRef]
- Schwartz, J.L.; Garneau, L.; Masson, L.; Brousseau, R. Early response of cultured lepidopteran cells to exposure to δ-endotoxin from Bacillus thuringiensis: Involvement of calcium and anionic channels. Biochim. Biophys. Acta. 1991, 1065, 250–260. [Google Scholar] [CrossRef]
- Jurat-Fuentes, J.L.; Adang, M.J. Cry toxin mode of action in susceptible and resistant Heliothis virescens larvae. J. Invertebr. Pathol. 2006, 92, 166–171. [Google Scholar] [CrossRef]
- Thomas, W.E.; Ellar, D.J. Bacillus thuringiensis var. israelensis crystal δ-endotoxin: Effects on insect and mammalian cells in vitro and in vivo. J. Cell. Sci. 1983, 60, 181–197. [Google Scholar]
- Koni, P.A.; Ellar, D.J. Biochemical characterization of Bacillus thuringiensis cytolytic δ-endotoxins. Microbiology 1994, 140, 1869–1880. [Google Scholar]
- Berry, C.; O’Neil, S.; Ben-Dov, E.; Jones, A.F.; Murphy, L.; Quail, M.A.; Holden, M.T.G.; Harris, D.; Zaritsky, A.; Parkhill, J. Complete sequence and organization of pBtoxis, the toxin-coding plasmid of Bacillus thuringiensis subsp. israelensis. Appl. Environ. Microbiol. 2002, 68, 5082–5095. [Google Scholar] [CrossRef]
- Cohen, S.; Albeck, S.; Ben-Dov, E.; Cahan, R.; Firer, M.; Zaritsky, A.; Dym, O. Cyt1Aa toxin: Crystal structure reveals implications for its membrane-perforating function. J. Mol. Biol. 2011, 413, 804–814. [Google Scholar] [CrossRef]
- Cohen, S.; Dym, O.; Albeck, S.; Ben-Dov, E.; Cahan, R.; Firer, M.; Zaritsky, A. High-resolution crystal structure of activated Cyt2Ba monomer from Bacillus thuringiensis subsp. israelensis. J. Mol. Biol. 2008, 380, 820–827. [Google Scholar] [CrossRef]
- Li, J.; Koni, P.A.; Ellar, D.J. Structure of the mosquitocidal δ-endotoxin CytB from Bacillus thuringiensis sp. kyushuensis and implications for membrane pore formation. J. Mol. Biol. 1996, 257, 129–152. [Google Scholar] [CrossRef]
- Butko, P. Cytolytic toxin Cyt1A and tts mechanism of membrane damage: Data and hypotheses. Appl. Environ. Microbiol. 2003, 69, 2415–2422. [Google Scholar] [CrossRef]
- Lin, S.C.; Lo, Y.C.; Lin, J.Y.; Liaw, Y.C. Crystal structures and electron micrographs of fungal volvatoxin A2. J. Mol. Biol. 2004, 343, 477–491. [Google Scholar] [CrossRef]
- Lin, S.C.; Lin, J.Y.; Liaw, Y.C. Crystallization and preliminary X-ray analysis of volvatoxin A2 from Volvariella volvacea. Proteins 1996, 24, 141–142. [Google Scholar] [CrossRef]
- Lin, J.F.; Jeng, T.W.; Chen, C.C.; Shi, G.Y.; Tung, T.C. Isolation of a new cardiotoxic protein from the edible mushroom, Volvariella volvacea. Nature 1973, 246, 524–525. [Google Scholar] [CrossRef]
- Wu, P.T.; Lin, S.C.; Hsu, C.I.; Liaw, Y.C.; Lin, J.Y. Inhibitory effects of nontoxic protein volvatoxin A1 on pore-forming cardiotoxic protein volvatoxin A2 by interaction with amphipathic alpha-helix. FEBS J. 2006, 273, 3160–3171. [Google Scholar] [CrossRef]
- Weng, Y.P.; Lin, Y.P.; Hsu, C.I.; Lin, J.Y. Functional domains of a pore-forming cardiotoxic protein, volvatoxin A2. J. Biol. Chem. 2004, 279, 6805–6814. [Google Scholar] [CrossRef]
- Rodriguez-Almazan, C.; de Escudero, I.R.; Canton, P.E.; Munoz-Garay, C.; Perez, C.; Gill, S.S.; Soberón, M.; Bravo, A. The amino- and carboxyl-terminal fragments of the Bacillus thuringensis Cyt1Aa toxin have differential roles in toxin oligomerization and pore formation. Biochemistry 2011, 50, 388–396. [Google Scholar] [CrossRef]
- Du, J.P.; Knowles, B.H.; Li, J.; Ellar, D.J. Biochemical characterization of Bacillus thuringiensis cytolytic toxins in association with a phospholipid bilayer. Biochem. J. 1999, 338, 185–193. [Google Scholar] [CrossRef]
- Gazit, E.; Burshtein, N.; Ellar, D.J.; Sawyer, T.; Shai, Y. Bacillus thuringiensis cytolytic toxin associates specifically with its synthetic helices A and C in the membrane bound state. Implications for the assembly of oligomeric transmembrane pores. Biochemistry 1997, 36, 15546–15554. [Google Scholar] [CrossRef]
- Promdonkoy, B.; Rungrod, A.; Promdonkoy, P.; Pathaichindachote, W.; Krittanai, C.; Panyim, S. Amino acid substitutions in αA and αC of Cyt2Aa2 alter hemolytic activity and mosquito-larvicidal specificity. J. Biotech. 2008, 133, 287–293. [Google Scholar] [CrossRef]
- Porter, A.G.; Davidson, E.W.; Liu, J.W. Mosquitocidal toxins of Bacilli and their genetic manipulation for effective bological control of mosquitos. Microbiol. Rev. 1993, 57, 838–861. [Google Scholar]
- Pérez, C.; Fernandez, L.E.; Sun, J.G.; Folch, J.L.; Gill, S.S.; Soberon, M.; Bravo, A. Bacillus thuringiensis subsp. israelensis Cyt1Aa synergizes Cry11Aa toxin by functioning as a membrane-bound receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 18303–18308. [Google Scholar] [CrossRef]
- Cantón, P.E.; Reyes, E.Z.; de Escudero, I.R.; Bravo, A.; Soberón, M. Binding of Bacillus thuringiensis subsp. israelensis Cry4Ba to Cyt1Aa has an important role in synergism. Peptides 2011, 32, 595–600. [Google Scholar] [CrossRef]
- Wirth, M.C.; Georghiou, G.P.; Federici, B.A. CytA enables CryIV endotoxins of Bacillus thuringiensis to overcome high levels of CryIV resistance in the mosquito, Culex quinquefasciatus. Proc. Natl. Acad. Sci. USA 1997, 94, 10536–10540. [Google Scholar] [CrossRef]
- Pérez, C.; Munoz-Garay, C.; Portugal, L.C.; Sanchez, J.; Gill, S.S.; Soberon, M.; Bravo, A. Bacillus thuringiensis ssp. israelensis Cyt1Aa enhances activity of Cry11Aa toxin by facilitating the formation of a pre-pore oligomeric structure. Cell. Microbiol. 2007, 9, 2931–2937. [Google Scholar] [CrossRef]
- Hua, G.; Jurat-Fuentes, J.L.; Adang, M.J. Fluorescent-based assays establish Manduca sexta Bt-R1a cadherin as a receptor for multiple Bacillus thuringiensis Cry1A toxins in Drosophila S2 cells. Insect Biochem. Mol. Biol. 2004, 34, 193–202. [Google Scholar] [CrossRef]
- Bravo, A.; Soberón, M. How to cope with insect resistance to Bt toxins? Trends Biotechnol. 2008, 26, 573–579. [Google Scholar] [CrossRef]
- Bravo, A.; Gill, S.S.; Soberón, M. Mode of action of Bacillus thuringiensis Cry and Cyt toxins and their potential for insect control. Toxicon 2007, 49, 423–435. [Google Scholar] [CrossRef]
- Mizuki, E.; Ohba, M.; Akao, T.; Yamashita, S.; Saitoh, H.; Park, Y.S. Unique activity associated with non-insecticidal Bacillus thuringiensis parasporal inclusions: In vitro cell-killing action on human cancer cells. J. Appl. Microbiol 1999, 86, 477–486. [Google Scholar] [CrossRef]
- Ohba, M.; Mizuki, E.; Uemori, A. Parasporin, a newanticancer protein group from Bacillus thuringiensis. Anticancer Res. 2009, 29, 427–433. [Google Scholar]
- Kitada, S.; Abe, Y.; Shimada, H.; Kusaka, Y.; Matsuo, Y.; Katayama, H.; Okumura, S.; Akao, T.; Mizuki, E.; Kuge, O.; et al. Cytocidal actions of parasporin-2, an anti-tumor crystal toxin from Bacillus thuringiensis. J. Biol. Chem. 2006, 281, 26350–26360. [Google Scholar] [CrossRef]
- Katayama, H.; Yokota, H.; Akao, T.; Nakamura, O.; Ohba, M.; Mekada, E.; Mizuki, E. Parasporin-1, a novel cytotoxic protein to human cells from non-insecticidal parasporal inclusions of Bacillus thuringiensis. J. Biochem. 2005, 137, 17–25. [Google Scholar] [CrossRef]
- Katayama, H.; Kusaka, Y.; Mizuk, E. Parasproin-1 Receptor and Use Thereof. U.S. Patent 20110038880; filed 30 March 200, and issued 17 February 2011,
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar]
- Wirawan, E.; Lippens, S.; Vanden Berghe, T.; Romagnoli, A.; Fimia, G.M.; Piacentini, M.; Vandenabeele, P. Beclin1: A role in membrane dynamics and beyond. Autophagy 2012, 8, 6–17. [Google Scholar] [CrossRef]
- Kim, H.S.; Yamashita, S.; Akao, T.; Saitoh, H.; Higuchi, K.; Park, Y.S.; Mizuki, E.; Ohba, M. In vitro cytotoxicity of non-Cyt inclusion proteins of a Bacillus thuringiensis isolate against human cells, including cancer cells. J. Appl. Microbiol. 2000, 89, 16–23. [Google Scholar] [CrossRef]
- Ito, A.; Sasaguri, Y.; Kitada, S.; Kusaka, Y.; Kuwano, K.; Masutomi, K.; Mizuki, E.; Akao, T.; Ohba, M. A Bacillus thuringiensis crystal protein with selective cytocidal action to human cells. J. Biol. Chem. 2004, 279, 21282–21286. [Google Scholar] [CrossRef]
- Akiba, T.; Abe, Y.; Kitada, S.; Kusaka, Y.; Ito, A.; Ichimatsu, T.; Katayama, H.; Akao, T.; Higuchi, K.; Mizuki, E.; et al. Crystal structure of the parasporin-2 Bacillus thuringiensis toxin that recognizes cancer cells. J. Mol. Biol. 2009, 386, 121–133. [Google Scholar]
- Abe, Y.; Shimada, H.; Kitada, S. Raft-targeting and oligomerization of parasporin-2, a Bacillus thuringiensis crystal protein with anti-tumour activity. J. Biochem. 2008, 143, 269–275. [Google Scholar]
- Legler, D.F.; Doucey, M.A.; Schneider, P.; Chapatte, L.; Bender, F.C.; Bron, C. Differential insertion of GPI-anchored GFPs into lipid rafts of live cells. FASEB J. 2004, 18, 73–75. [Google Scholar]
- Lingwood, D.; Kaiser, H.J.; Levental, I.; Simons, K. Lipid rafts as functional heterogeneity in cell membranes. Biochem. Soc. Trans. 2009, 37, 955–960. [Google Scholar] [CrossRef]
- Kitada, S.; Abe, Y.; Maeda, T.; Shimada, H. Parasporin-2 requires GPI-anchored proteins for the efficient cytocidal action to human hepatoma cells. Toxicology 2009, 264, 80–88. [Google Scholar] [CrossRef]
- Yamashita, S.; Akao, T.; Mizuki, E.; Saitoh, H.; Higuchi, K.; Park, Y.S.; Kim, H.S.; Ohba, M. Characterization of the anti-cancer-cell parasporal proteins of a Bacillus thuringiensis isolate. Can. J. Microbiol. 2000, 46, 913–919. [Google Scholar] [CrossRef]
- Lee, D.W.; Akao, T.; Yamashita, S.; Katayama, H.; Maeda, M.; Saitoh, H.; Mizuki, E.; Ohba, M. Noninsecticidal parasporal proteins of a Bacillus thuringiensis serovar shandongiensis isolate exhibit a preferential cytotoxicity against human leukemic T cells. Biochem. Biophys. Res. Commun. 2000, 272, 218–223. [Google Scholar] [CrossRef]
- Lee, D.W.; Katayama, H.; Akao, T.; Maeda, M.; Tanaka, R.; Yamashita, S.; Saitoh, H.; Mizuki, E.; Ohba, M. A 28 kDa protein of the Bacillus thuringiensis serovar shandongiensis isolate 89-T-34-22 induces a human leukemic cell-specific cytotoxicity. Biochim. Biophys. Acta 2001, 1547, 57–63. [Google Scholar]
- Okumura, S.; Akao, T.; Higuchi, K.; Saitoh, H.; Mizuki, E.; Ohba, M.; Inouye, K. Bacillus thuringiensis serovar shandongiensis strain 89-T-34-22 produces multiple cytotoxic proteins with similar molecular masses against human cancer cells. Lett. Appl. Microbiol. 2004, 39, 89–92. [Google Scholar] [CrossRef]
- Okumura, S.; Saitoh, H.; Ishikawa, T.; Wasano, N.; Yamashita, S.; Kusumoto, K.; Akao, T.; Mizuki, E.; Ohba, M.; Inouye, K. Identification of a novel cytotoxic protein, Cry45Aa, from Bacillus thuringiensis A1470 and its selective cytotoxic activity against various mammalian cell lines. J. Agric. Food. Chem. 2005, 53, 6313–6318. [Google Scholar] [CrossRef]
- Saitoh, H.; Okumura, S.; Ishikawa, T.; Akao, T.; Mizuki, E.; Ohba, M. Investigation of a novel Bacillus thuringiensis gene encoding a parasporal protein, parasporin-4, that preferentially kills human leukemic T cells. Biosci. Biotechnol. Biochem. 2006, 70, 2935–2941. [Google Scholar] [CrossRef]
- Okumura, S.; Saitoh, H.; Ishikawa, T.; Mizuki, E.; Inouye, K. Identification and characterization of a novel cytotoxic protein, parasporin-4, produced by Bacillus thuringiensis A1470 strain. Biotechnol. Annu. Rev. 2008, 14, 225–252. [Google Scholar] [CrossRef]
- Okumura, S.; Saitoh, H.; Ishikawa, T.; Inouye, K.; Mizuki, E. Mode of action of parasporin-4, a cytocidal protein from Bacillus thuringiensis. Biochim. Biophys. Acta. 2011, 1808, 1476–1482. [Google Scholar] [CrossRef]
- Parker, M.W.; Buckley, J.T.; Postma, J.P.M.; Tucker, A.D.; Leonard, K.; Pattus, F.; Tsernoglou, D. Structure of the Aeromonas toxin proaerolysin in its water-soluble and membrane-channel states. Nature 1994, 367, 292–295. [Google Scholar] [CrossRef]
- Szczesny, P.; Iacovache, I.; Muszewska, A.; Ginalski, K.; van der Goot, F.G.; Grynberg, M. Extending the aerolysin family: From bacteria to vertebrates. PLoS One 2011, 6, e20349. [Google Scholar]
- Knapp, O.; Stiles, B.G.; Popoff, M.R. The aerolysin-like toxin family of cytolytic, pore-forming toxins. Open Toxicol. J. 2010, 3, 53–68. [Google Scholar] [CrossRef]
- Briggs, D.C.; Naylor, C.E.; Smedley, J.G.; Lukoyanova, N.; Robertson, S.; Moss, D.S.; McClane, B.A.; Basak, A.K. Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J. Mol. Biol. 2011, 413, 138–149. [Google Scholar] [CrossRef]
- Akiba, T.; Higuchi, K.; Mizuki, E.; Ekino, K.; Shin, T.; Ohba, M.; Kanai, R.; Harata, K. Nontoxic crystal protein from Bacillus thuringiensis demonstrates a remarkable structural similarity to pore-forming toxins. Proteins: Struct. Funct. Bioinf. 2006, 63, 243–248. [Google Scholar] [CrossRef]
- Akiba, T.; Abe, Y.; Kitada, S.; Kusaka, Y.; Ito, A.; Ichimatsu, T.; Katayama, H.; Akao, T.; Higuchi, K.; Mizuki, E.; et al. Crystallization of parasporin-2, a Bacillus thuringiensis crystal protein with selective cytocidal activity against human cells. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2004, 60, 2355–2357. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, 162–173. [Google Scholar]
- Cole, A.R.; Gibert, M.; Popoff, M.; Moss, D.S.; Titball, R.W.; Basak, A.K. Clostridium perfringens epsilon-toxin shows structural similarity to the pore-forming toxin aerolysin. Nat. Struct. Mol. Biol. 2004, 11, 797–798. [Google Scholar]
- Payne, C.M.; Bomble, Y.J.; Taylor, C.B.; McCabe, C.; Himmel, M.E.; Crowley, M.F.; Beckham, G.T. Multiple functions of aromatic-carbohydrate interactions in a processive cellulase examined with molecular simulation. J. Biol. Chem. 2011, 286, 41028–42035. [Google Scholar]
- Schwartz, R.; Istrail, S.; King, J. Frequencies of amino acid strings in globular protein sequences indicate suppression of blocks of consecutive hydrophobic residues. Protein Sci. 2001, 10, 1023–1031. [Google Scholar]
- Melton, J.A.; Parker, M.W.; Rossjohn, J.; Buckley, J.T.; Tweten, R.K. The identification and structure of the membrane-spanning domain of the Clostridium septicum α-toxin. J. Biol. Chem. 2004, 279, 14315–14322. [Google Scholar]
- Knapp, O.; Maier, E.; Benz, R.; Geny, B.; Popoff, M.R. Identification of the channel-forming domain of Clostridium perfringens epsilon-toxin (ETX). Biochim. Biophys. Acta-Biomembr. 2009, 1788, 2584–2593. [Google Scholar]
- Mancheño, J.M.; Tateno, H.; Goldstein, I.J.; Martínez-Ripoll, M.; Hermoso, J.A. Structural analysis of the Laetiporus sulphureus hemolytic pore-forming lectin in complex with sugars. J. Biol. Chem. 2005, 280, 17251–17259. [Google Scholar]
- Kitadokoro, K.; Nishimura, K.; Kamitani, S.; Fukui-Miyazaki, A.; Toshima, H.; Abe, H.; Kamata, Y.; Sugita-Konishi, Y.; Yamamoto, S.; Karatani, H.; et al. Crystal structure of Clostridium perfringens enterotoxin dsplays features of β-pore-forming toxins. J. Biol. Chem. 2011, 286, 19549–19555. [Google Scholar]
- Hong, Y.J.; Ohishi, K.; Inoue, N.; Kang, J.Y.; Shime, H.; Horiguchi, Y.; van der Goot, F.G.; Sugimoto, N.; Kinoshita, T. Requirement of N-glycan on GPI-anchored proteins for efficient binding of aerolysin but not Clostridium septicum alpha-toxin. EMBO J. 2002, 21, 5047–5056. [Google Scholar]
- Simpson, P.J.; Xie, H.F.; Bolam, D.N.; Gilbert, H.J.; Williamson, M.P. The structural basis for the ligand specificity of family 2 carbohydrate-binding modules. J. Biol. Chem. 2000, 275, 41137–41142. [Google Scholar]
- Hashimoto, H. Recent structural studies of carbohydrate-binding modules. Cell. Mol. Life. Sci. 2006, 63, 2954–2967. [Google Scholar]
- Mancheño, J.M.; Tateno, H.; Sher, D.; Goldstein, I.J. Laetiporus sulphureus lectin and aerolysin protein family. In Proteins Membrane Binding and Pore Formation; Springer New York: New York, NY, USA, 2010; Volume 677, pp. 67–80. [Google Scholar]
- Rossjohn, J.; Feil, S.C.; McKinstry, W.J.; Tsernoglou, D.; van der Goot, G.; Buckley, J.T.; Parker, M.W. Aerolysin-a paradigm for membrane insertion of beta-sheet protein toxins? J. Struct. Biol. 1998, 121, 92–100. [Google Scholar] [CrossRef]
- Vandergoot, F.G.; Hardie, K.R.; Parker, M.W.; Buckley, J.T. The C-terminal peptide produced upon proteolytic activation of the cytolytic toxin aerolysin is not involved in channel formation. J. Biol. Chem. 1994, 269, 30496–30501. [Google Scholar]
- Degiacomi, M.T.; Lacovache, I.; Pernot, L.; Chami, M.; Kudryashev, M.; Stahlberg, H.; van der Goot, F.G.; dal Peraro, M. Molecular assembly of the aerolysin pore reveals a swirling membrane-insertion mechanism. Nat. Chem. Biol. 2013, 9, 623–629. [Google Scholar] [CrossRef]
- Wilmsen, H.U.; Leonard, K.R.; Tichelaar, W.; Buckley, J.T.; Pattus, F. The aerolysin membrane channel is formed by heptamerization of the monomer. EMBO J. 1992, 11, 2457–2463. [Google Scholar]
- Tsitrin, Y.; Morton, C.J.; El Bez, C.; Paumard, P.; Velluz, M.C.; Adrian, M.; Dubochet, J.; Parker, M.W.; Lanzavecchia, S.; van der Goot, F.G. Conversion of a transmembrane to a watersoluble protein complex by a single point mutation. Nat. Struct. Biol. 2002, 9, 729–733. [Google Scholar]
- Iacovache, I.; Paumard, P.; Scheib, H.; Lesieur, C.; Sakai, N.; Matile, S.; Parker, M.W.; van der Goot, F.G. A rivet model for channel formation by aerolysin-like pore-forming toxins. EMBO J. 2006, 25, 457–466. [Google Scholar] [CrossRef]
- Whisstock, J.C.; Dunstone, M.A. Torqueing about pores. Nat. Chem. Biol. 2013, 9, 605–606. [Google Scholar] [CrossRef]
- Yu, Z.; Luo, H.; Xiong, J.; Zhou, Q.; Xia, L.; Sun, M.; Li, L. Bacillus thuringiensis Cry6A exhibits nematicidal activity to Caenorhabditis elegans bre mutants and synergistic activity with Cry5B to C. elegans. Lett. Appl. Microbiol. 2014, 58, 511–519. [Google Scholar]
- Naimov, S.; Boncheva, R.; Karlova, R.; Dukiandjiev, S.; Minkov, I.; de Maagd, R.A. Solubilization, activation, and insecticidal activity of Bacillus thuringiensis serovar thompsoni HD542 crystal proteins. Appl. Environ. Microbiol. 2008, 74, 7145–7151. [Google Scholar] [CrossRef]
- Masson, L.; Schwab, G.; Mazza, A.; Brousseau, R.; Potvin, L.; Schwartz, J.L. A novel Bacillus thuringiensis (PS149B1) containing a Cry34Abl/Cry35Abl binary toxin specific for the western corn rootworm Diabrotica virgifera virgifera LeConte forms ion channels in lipid membranes. Biochemistry 2004, 43, 12349–12357. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, C.; Wang, B.-C.; Yu, Z.; Sun, M. Structural Insights into Bacillus thuringiensis Cry, Cyt and Parasporin Toxins. Toxins 2014, 6, 2732-2770. https://doi.org/10.3390/toxins6092732
Xu C, Wang B-C, Yu Z, Sun M. Structural Insights into Bacillus thuringiensis Cry, Cyt and Parasporin Toxins. Toxins. 2014; 6(9):2732-2770. https://doi.org/10.3390/toxins6092732
Chicago/Turabian StyleXu, Chengchen, Bi-Cheng Wang, Ziniu Yu, and Ming Sun. 2014. "Structural Insights into Bacillus thuringiensis Cry, Cyt and Parasporin Toxins" Toxins 6, no. 9: 2732-2770. https://doi.org/10.3390/toxins6092732
APA StyleXu, C., Wang, B. -C., Yu, Z., & Sun, M. (2014). Structural Insights into Bacillus thuringiensis Cry, Cyt and Parasporin Toxins. Toxins, 6(9), 2732-2770. https://doi.org/10.3390/toxins6092732