
Acute Cardiotoxicity Evaluation of the Marine Biotoxins OA, DTX-1 and YTX

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
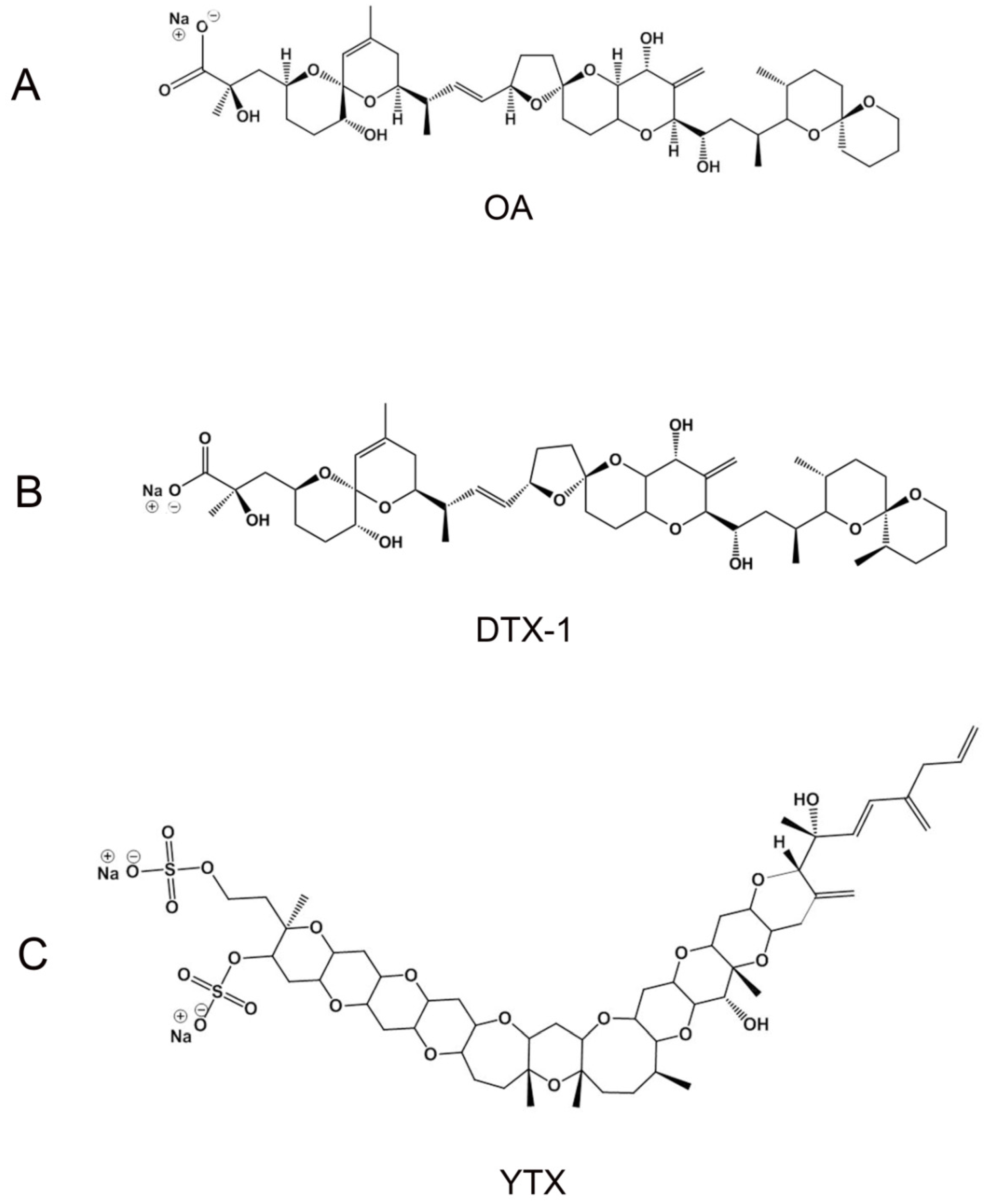
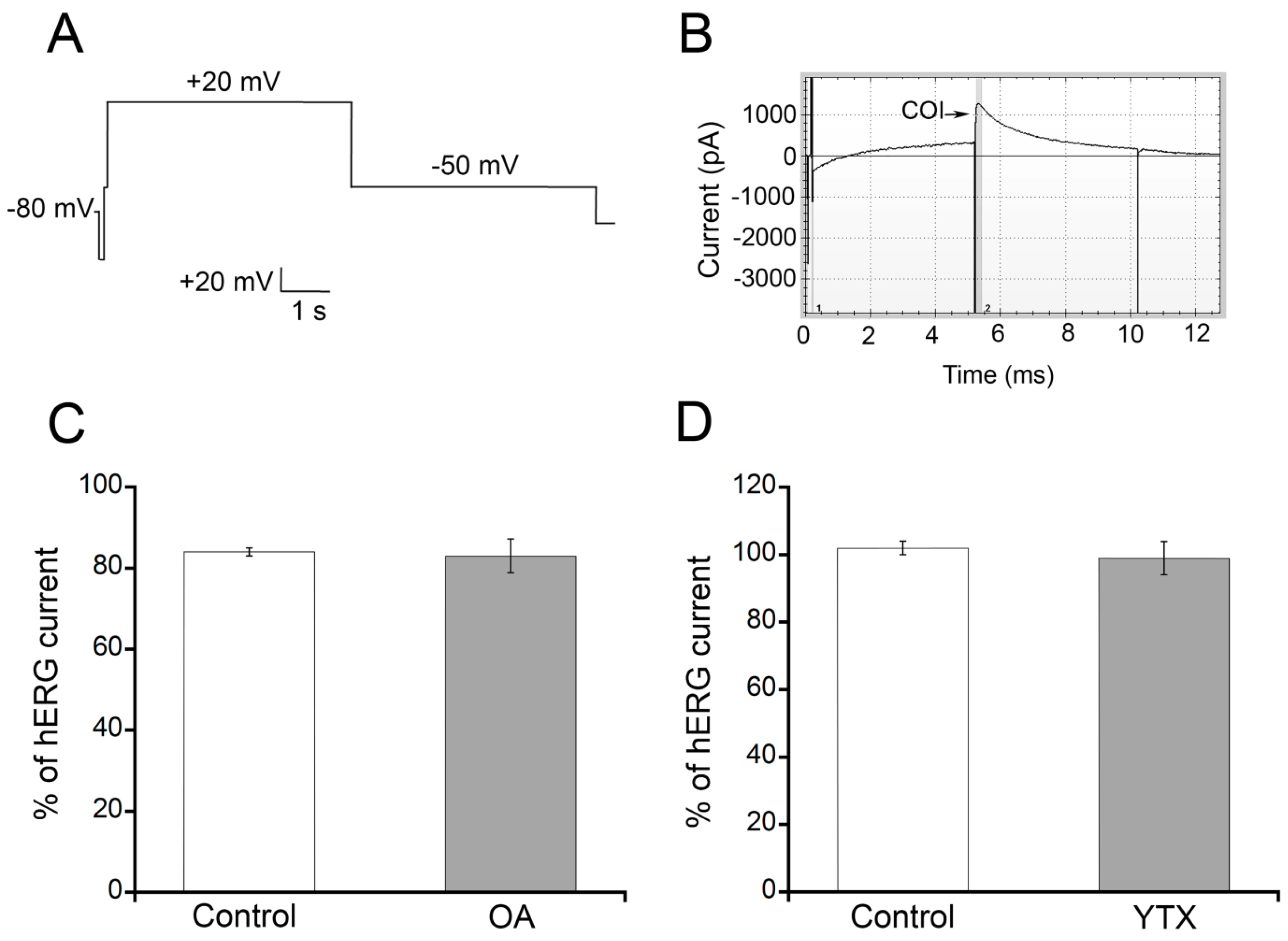
2.1. OA and YTX Effects on hERG Channel Activity


2.2. Effects of OA, DTX-1 and YTX on Rat ECG


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rat | Type | Time of Appearance | Duration (s) | Total N° | ECG Total Time (min) | Death before 240 min | |
|---|---|---|---|---|---|---|---|
| Control | 1 | 0 | 265 | no | |||
| 2 | 0 | 265 | no | ||||
| 3 | 0 | 265 | no | ||||
| 4 | 0 | 265 | no | ||||
| 5 | 0 | 265 | no | ||||
| 6 | VES | t68:45 | 3 | 2 | 265 | no | |
| 7 | VES | t52:31 | 1 | 265 | no | ||
| 8 | 0 | 265 | no | ||||
| 9 | 0 | 265 | no | ||||
| OA | 10 | 0 | 265 | no | |||
| 11 | 0 | 265 | no | ||||
| 12 | VES | t110: 30 | 66 | 3 | 265 | no | |
| VES | t174:45 | 1 | 265 | no | |||
| VES | t196:49 | 1 | 265 | no | |||
| 13 | 0 | 130 | yes | ||||
| 14 | 0 | 265 | no | ||||
| DTX-1 | 15 | 0 | 265 | no | |||
| 16 | 0 | 265 | no | ||||
| 17 | 0 | 265 | no | ||||
| 18 | 0 | 265 | no | ||||
| 19 | 0 | 265 | no | ||||
| YTX | 20 | 0 | 160 | yes | |||
| 21 | 0 | 265 | no | ||||
| 22 | 0 | 120 | yes | ||||
| 23 | VES | t236:08 | 1 | 1 | 265 | no | |
| 24 | VES | t77: 35 | 5 | 3 | 140 | yes | |
| 25 | 0 | 265 | no | ||||
| 26 | 0 | 265 | no | ||||
2.3. Effects of OA, DTX-1 and YTX on the Levels of Cardiac Biomarkers

2.4. Effects on Biochemistry Parameters
3. Experimental Section
3.1. Chemicals and Solutions
| Time | Before | After | Physiologic Range | |
|---|---|---|---|---|
| Biochemical Parameter | ||||
| Control (n = 9) | BUN (mg/dL) | 17 ± 1.0 | 23.7 ± 1.1 ** | 20.3–25.5 |
| CREA (mg/dL) | 0.4 ± 0.0 | 0.4 ± 0.0 | 0.5–0.92 | |
| PHOS (mg(dL) | 6.2 ± 0.2 | 6.4 ± 0.9 | 4.2–8.33 | |
| Ca (mg/dL) | 10.2 ± 0.1 | 10.1 ± 0.1 | 9.6–11.86 | |
| TP (g/dL) | 5.2 ± 0.1 | 3.9 ± 0.1 ** | 5.00–7.7 | |
| ALB (g/dL) | 2.8 ± 0.1 | 1.9 ± 0.1 ** | 2.9–4.6 | |
| GLOB (g/dL) | 2.4 ± 0.1 | 2.0 ± 0.1 ** | 2.1–3.1 | |
| ALT (U/L) | 49.1 ± 10.9 | 52.2 ± 23.8 | 32.7–84.1 | |
| ALKP (U/L) | 130.4 ± 10.1 | 76.3 ± 6.5 ** | 82.8–297.3 | |
| CHOL (mg/dL) | 59.1 ± 15.6 | 56 ± 2.2 * | 41.1–59.1 | |
| CK (U/L) | 131.4 ± 7.4 | 248.9 ± 28.0 ** | 494–4132 | |
| OA (n = 5) | BUN (mg/dL) | 18.3 ± 1.5 | 28.0 ± 0.9 ** | 20.3–25.5 |
| CREA (mg/dL) | 0.5 ± 0.1 | 0.6 ± 0.0 | 0.5–0.92 | |
| PHOS (mg(dL) | 6.9 ± 1.0 | 8.0 ± 0.7 | 4.2–8.33 | |
| Ca (mg/dL) | 10.2 ± 0.1 | 9.9 ± 0.1 * | 9.6–11.86 | |
| TP (g/dL) | 5.5 ± 0.2 | 4.4 ± 0.1 ** | 5.00–7.7 | |
| ALB (g/dL) | 2.8 ± 0.1 | 1.9 ± 0.1 ** | 2.9–4.6 | |
| GLOB (g/dL) | 2.7 ± 0.1 | 2.5 ± 0.1 | 2.1–3.1 | |
| ALT (U/L) | 51.5 ± 8.6 | 95.6 ± 24.6 | 32.7–84.1 | |
| ALKP (U/L) | 110.7 ± 11.8 | 83 ± 8.3 * | 82.8–297.3 | |
| CHOL (mg/dL) | 91.3 ± 7.3 | 69.6 ± 2.9 | 41.1–59.1 | |
| CK (U/L) | 98.5 ± 12.6 | 251.7 ± 33.3 ** | 494–4132 | |
| DTX-1 (n = 5) | BUN (mg/dL) | 18 ± 1.1 | 25.0 ± 2.1 ** | 20.3–25.5 |
| CREA (mg/dL) | 0.3 ± 0.0 | 0.3 ± 0.0 | 0.5–0.92 | |
| PHOS (mg(dL) | 7.5 ± 0.4 | 7.5 ± 0.6 | 4.2–8.33 | |
| Ca (mg/dL) | 10.1 ± 0.1 | 9.0 ± 0.6 | 9.6–11.86 | |
| TP (g/dL) | 5.5 ± 0.1 | 4.2 ± 0.1 ** | 5.00–7.7 | |
| ALB (g/dL) | 3.0 ± 0.1 | 2.0 ± 0.1 ** | 2.9–4.6 | |
| GLOB (g/dL) | 2.5 ± 0.1 | 2.1 ± 0.1 * | 2.1–3.1 | |
| ALT (U/L) | 34.5 ± 3.5 | 21.2 ± 6.2 | 32.7–84.1 | |
| ALKP (U/L) | 123.2 ± 13.7 | 78.8 ± 8.3 ** | 82.8–297.3 | |
| CHOL (mg/dL) | 74.2 ± 4.2 | 58.2 ± 8.7 | 41.1–59.1 | |
| CK (U/L) | 113.6 ± 16.6 | 162.4 ± 26.4 | 494–4132 | |
| YTX (n = 7) | BUN (mg/dL) | 17.7 ± 1.3 | 29.8 ± 1.5 ** | 20.3–25.5 |
| CREA (mg/dL) | 0.5 ± 0.1 | 0.6 ± 0.2 | 0.5–0.92 | |
| PHOS (mg(dL) | 7.1 ± 1.4 | 7.0 ± 1.3 | 4.2–8.33 | |
| Ca (mg/dL) | 10.1 ± 0.0 | 9.5 ± 0.1 ** | 9.6–11.86 | |
| TP (g/dL) | 5.4 ± 0.2 | 4.1 ± 0.1 ** | 5.00–7.7 | |
| ALB (g/dL) | 2.6 ± 0.1 | 1.8 ± 0.1 ** | 2.9–4.6 | |
| GLOB (g/dL) | 2.7 ± 0.1 | 2.5 ± 0.1 | 2.1–3.1 | |
| ALT (U/L) | 23.8 ± 4.5 | 30.8 ± 8.2 | 32.7–84.1 | |
| ALKP (U/L) | 113.8 ± 8.6 | 80 ± 6.4 ** | 82.8–297.3 | |
| CHOL (mg/dL) | 82 ± 6.1 | 64.5 ± 2.9 ** | 41.1–59.1 | |
| CK (U/L) | 151.3 ± 16.7 | 296 ± 119.0 | 494–4132 | |
3.2. Cell Line
3.3. Automated Patch Clamp
3.4. Animals and in vivo Experimental Design
3.5. Electrocardiography
3.6. Cardiac Biomarkers
3.7. Biochemistry Analysis
3.8. Data Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- EFSA. Opinion of the Scientific Panel on Contaminants in the Food chain on a request from the European Comission on marine biotoxins in shellfish-okadaic acid and analogues. EFSA J. 2008, 589, 1–62. [Google Scholar]
- Valdiglesias, V.; Prego-Faraldo, M.V.; Pasaro, E.; Mendez, J.; Laffon, B. Okadaic acid: More than a diarrheic toxin. Mar. Drugs 2013, 11, 4328–4349. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Regulation (EC) No. 853/2004 of the European Parliament and of the Council of 29 April 2004 laying down specific hygiene rules for food animal origin. Off. J. Eur. Union 2004, 139, 22–82. [Google Scholar]
- Takai, A.; Bialojan, C.; Troschka, M.; Ruegg, J.C. Smooth muscle myosin phosphatase inhibition and force enhancement by black sponge toxin. FEBS Lett. 1987, 217, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, R.E.; Codispoti, B.A.; Tse, K.; Boynton, A.L.; Honkanan, R.E. Characterization of natural toxins with inhibitory activity against serine/threonine protein phosphatases. Toxicon 1994, 32, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Tubaro, A.; Dell’ovo, V.; Sosa, S.; Florio, C. Yessotoxins: A toxicological overview. Toxicon 2010, 56, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Terao, K.; Ito, E.; Oarada, M.; Murata, M.; Yasumoto, T. Histopathological studies on experimental marine toxin poisoning--5. The effects in mice of yessotoxin isolated from Patinopecten yessoensis and of a desulfated derivative. Toxicon 1990, 28, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Aune, T.; Sorby, R.; Yasumoto, T.; Ramstad, H.; Landsverk, T. Comparison of oral and intraperitoneal toxicity of yessotoxin towards mice. Toxicon 2002, 40, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Tubaro, A.; Sosa, S.; Carbonatto, M.; Altinier, G.; Vita, F.; Melato, M.; Satake, M.; Yasumoto, T. Oral and intraperitoneal acute toxicity studies of yessotoxin and homoyessotoxins in mice. Toxicon 2003, 41, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Tubaro, A.; Giangaspero, A.; Ardizzone, M.; Soranzo, M.R.; Vita, F.; Yasumoto, T.; Maucher, J.M.; Ramsdell, J.S.; Sosa, S. Ultrastructural damage to heart tissue from repeated oral exposure to yessotoxin resolves in 3 months. Toxicon 2008, 51, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- De la Rosa, L.A.; Alfonso, A.; Vilarino, N.; Vieytes, M.R.; Botana, L.M. Modulation of cytosolic calcium levels of human lymphocytes by yessotoxin, a novel marine phycotoxin. Biochem. Pharmacol. 2001, 61, 827–833. [Google Scholar]
- Alfonso, A.; de la Rosa, L.; Vieytes, M.R.; Yasumoto, T.; Botana, L.M. Yessotoxin, a novel phycotoxin, activates phosphodiesterase activity. Effect of yessotoxin on cAMP levels in human lymphocytes. Biochem. Pharmacol. 2003, 65, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Paz, B.; Daranas, A.H.; Norte, M.; Riobo, P.; Franco, J.M.; Fernandez, J.J. Yessotoxins, a group of marine polyether toxins: An overview. Mar. Drugs 2008, 6, 73–102. [Google Scholar] [CrossRef] [PubMed]
- Tobio, A.; Fernandez-Araujo, A.; Alfonso, A.; Botana, L.M. Role of yessotoxin in calcium and cAMP-crosstalks in primary and K-562 human lymphocytes: The effect is mediated by anchor kinase A mitochondrial proteins. J. Cell Biochem. 2012, 113, 3752–3761. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Araujo, A.; Tobio, A.; Alfonso, A.; Botana, L.M. Role of AKAP 149-PKA-PDE4A complex in cell survival and cell differentiation processes. Int. J. Biochem. Cell Biol. 2014, 53, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; Lopez-Alonso, H.; Martinez, P.; Millan, A.; Cagide, E.; Vieytes, M.R.; Vega, F.V.; Botana, L.M. Yessotoxin induces ER-stress followed by autophagic cell death in glioma cells mediated by mTOR and BNIP3. Cell Signal 2014, 26, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Tubaro, A.; Sosa, S.; Altinier, G.; Soranzo, M.R.; Satake, M.; Della Loggia, R.; Yasumoto, T. Short-term oral toxicity of homoyessotoxins, yessotoxin and okadaic acid in mice. Toxicon 2004, 43, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Sosa, S.; Ardizzone, M.; Beltramo, D.; Vita, F.; Dell’Ovo, V.; Barreras, A.; Yasumoto, T.; Tubaro, A. Repeated oral co-exposure to yessotoxin and okadaic acid: A short term toxicity study in mice. Toxicon 2013, 76, 94–102. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Opinion of the Scientific Panel on Contaminants in the Food chain on a request from the European Comission on marine biotoxins in shellfish-yessotoxin group. EFSA J. 2008, 907, 1–62. [Google Scholar]
- EMA. International conference on harmonisation; guidance on s7b nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals; availability. Notice. Fed. Regist. 2005, 70, 61133–61134. [Google Scholar]
- Guth, B.D. Preclinical cardiovascular risk assessment in modern drug development. Toxicol. Sci. 2007, 97, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Stummann, T.C.; Beilmann, M.; Duker, G.; Dumotier, B.; Fredriksson, J.M.; Jones, R.L.; Hasiwa, M.; Kang, Y.J.; Mandenius, C.F.; Meyer, T.; et al. Report and recommendations of the workshop of the European centre for the validation of alternative methods for drug-induced cardiotoxicity. Cardiovasc. Toxicol. 2009, 9, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Priest, B.T.; Bell, I.M.; Garcia, M.L. Role of hERG potassium channel assays in drug development. Channels 2008, 2, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Gintant, G.A.; Su, Z.; Martin, R.L.; Cox, B.F. Utility of hERG assays as surrogate markers of delayed cardiac repolarization and QT safety. Toxicol. Pathol. 2006, 34, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Van der Heyden, M.A.; Smits, M.E.; Vos, M.A. Drugs and trafficking of ion channels: A new pro-arrhythmic threat on the horizon? Br. J. Pharmacol. 2008, 153, 406–409. [Google Scholar]
- Farraj, A.K.; Hazari, M.S.; Cascio, W.E. The utility of the small rodent electrocardiogram in toxicology. Toxicol. Sci. 2011, 121, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Sandri, M.T. Role of biomarkers in chemotherapy-induced cardiotoxicity. Prog. Cardiovasc. Dis. 2010, 53, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Kettenhofen, R.; Bohlen, H. Preclinical assessment of cardiac toxicity. Drug Discov. Today 2008, 13, 702–707. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J. Cardiac troponin is the most effective translational safety biomarker for myocardial injury in cardiotoxicity. Toxicology 2008, 245, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Garibo, D.; de la Iglesia, P.; Diogene, J.; Campas, M. Inhibition equivalency factors for dinophysistoxin-1 and dinophysistoxin-2 in protein phosphatase assays: Applicability to the analysis of shellfish samples and comparison with LC-MS/MS. J. Agric. Food Chem. 2013, 61, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- Tubaro, A.; Sosa, S.; Bornancin, A.; Hungerford, J. Seafood and freshwater toxins. Pharmacology, Physiology and Detection. Pharmacology and Toxicology of Diarrheic Shellfish Toxins; Botana, L.M., Ed.; CRC Press: Boca Raton, FL, USA, 2008; pp. 229–254. [Google Scholar]
- Katchman, A.N.; Koerner, J.; Tosaka, T.; Woosley, R.L.; Ebert, S.N. Comparative evaluation of HERG currents and QT intervals following challenge with suspected torsadogenic and nontorsadogenic drugs. J. Pharmacol. Exp. Ther. 2006, 316, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Zhou, Z.; Gong, Q.; January, C.T. Blockage of the HERG human cardiac K+ channel by the gastrointestinal prokinetic agent cisapride. Am. J. Physiol. 1997, 273, H2534–H2538. [Google Scholar] [PubMed]
- Ohtani, H.; Taninaka, C.; Hanada, E.; Kotaki, H.; Sato, H.; Sawada, Y.; Iga, T. Comparative pharmacodynamic analysis of Q-T interval prolongation induced by the macrolides clarithromycin, roxithromycin, and azithromycin in rats. Antimicrob. Agents Chemother. 2000, 44, 2630–2637. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, S.F.; Vilarino, N.; Carrera, C.; Louzao, M.C.; Santamarina, G.; Cantalapiedra, A.G.; Rodriguez, L.P.; Cifuentes, J.M.; Vieira, A.C.; Nicolaou, K.C.; et al. In vivo arrhythmogenicity of the marine biotoxin azaspiracid-2 in rats. Arch. Toxicol. 2014, 88, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Ogino, H.; Kumagai, M.; Yasumoto, T. Toxicologic evaluation of yessotoxin. Nat. Toxins 1997, 5, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Moulin, M.; Piquereau, J.; Mateo, P.; Fortin, D.; Rucker-Martin, C.; Gressette, M.; Lefebvre, F.; Gresikova, M.; Solgadi, A.; Veksler, V.; et al. Sexual dimorphism of doxorubicin-mediated cardiotoxicity: Potential role of energy metabolism remodeling. Circ. Heart Fail 2015, 8, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodzadeh, S.; Fliegner, D.; Dworatzek, E. Sex differences in animal models for cardiovascular diseases and the role of estrogen. Handb. Exp. Pharmacol. 2012, 23–48. [Google Scholar]
- Fernandez, D.A.; Louzao, M.C.; Fraga, M.; Vilarino, N.; Vieytes, M.R.; Botana, L.M. Experimental basis for the high oral toxicity of dinophysistoxin 1: A comparative study of DSP. Toxins 2014, 6, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Kemp, C.D.; Conte, J.V. The pathophysiology of heart failure. Cardiovasc. Pathol. 2012, 21, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.; York, M.; Scudamore, C.; Williams, T.; Griffiths, W.; Turton, J. Cardiac troponin I in isoproterenol-induced cardiac injury in the Hanover Wistar rat: Studies on low dose levels and routes of administration. Toxicol. Pathol. 2010, 38, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.B. Serum chemical biomarkers of cardiac injury for nonclinical safety testing. Toxicol. Pathol. 2006, 34, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Ozer, J.; Ratner, M.; Shaw, M.; Bailey, W.; Schomaker, S. The current state of serum biomarkers of hepatotoxicity. Toxicology 2008, 245, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Berven, G.; Saetre, F.; Halvorsen, K.; Seglen, P.O. Effects of the diarrhetic shellfish toxin, okadaic acid, on cytoskeletal elements, viability and functionality of rat liver and intestinal cells. Toxicon 2001, 39, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Tonomura, Y.; Tsuchiya, N.; Torii, M.; Uehara, T. Evaluation of the usefulness of urinary biomarkers for nephrotoxicity in rats. Toxicology 2010, 273, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Vaidya, V.S.; Schmouder, R.; Feig, P.; Dieterle, F. Next-generation biomarkers for detecting kidney toxicity. Nat. Biotechnol. 2010, 28, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Comission regulation (EU). Amending Annex III to Regulation (EC) No 853/2004 of the European Parliament and of the Council as Regards the Permitted Limits of Yessotoxins in Live Bivalve Molluscs. No 786/2013, 16 August 2013. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreiro, S.F.; Carrera, C.; Vilariño, N.; Louzao, M.C.; Santamarina, G.; Cantalapiedra, A.G.; Botana, L.M. Acute Cardiotoxicity Evaluation of the Marine Biotoxins OA, DTX-1 and YTX. Toxins 2015, 7, 1030-1047. https://doi.org/10.3390/toxins7041030
Ferreiro SF, Carrera C, Vilariño N, Louzao MC, Santamarina G, Cantalapiedra AG, Botana LM. Acute Cardiotoxicity Evaluation of the Marine Biotoxins OA, DTX-1 and YTX. Toxins. 2015; 7(4):1030-1047. https://doi.org/10.3390/toxins7041030
Chicago/Turabian StyleFerreiro, Sara F., Cristina Carrera, Natalia Vilariño, M. Carmen Louzao, Germán Santamarina, Antonio G. Cantalapiedra, and Luis M. Botana. 2015. "Acute Cardiotoxicity Evaluation of the Marine Biotoxins OA, DTX-1 and YTX" Toxins 7, no. 4: 1030-1047. https://doi.org/10.3390/toxins7041030
APA StyleFerreiro, S. F., Carrera, C., Vilariño, N., Louzao, M. C., Santamarina, G., Cantalapiedra, A. G., & Botana, L. M. (2015). Acute Cardiotoxicity Evaluation of the Marine Biotoxins OA, DTX-1 and YTX. Toxins, 7(4), 1030-1047. https://doi.org/10.3390/toxins7041030