
Induction of Suicidal Erythrocyte Death by Nelfinavir


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
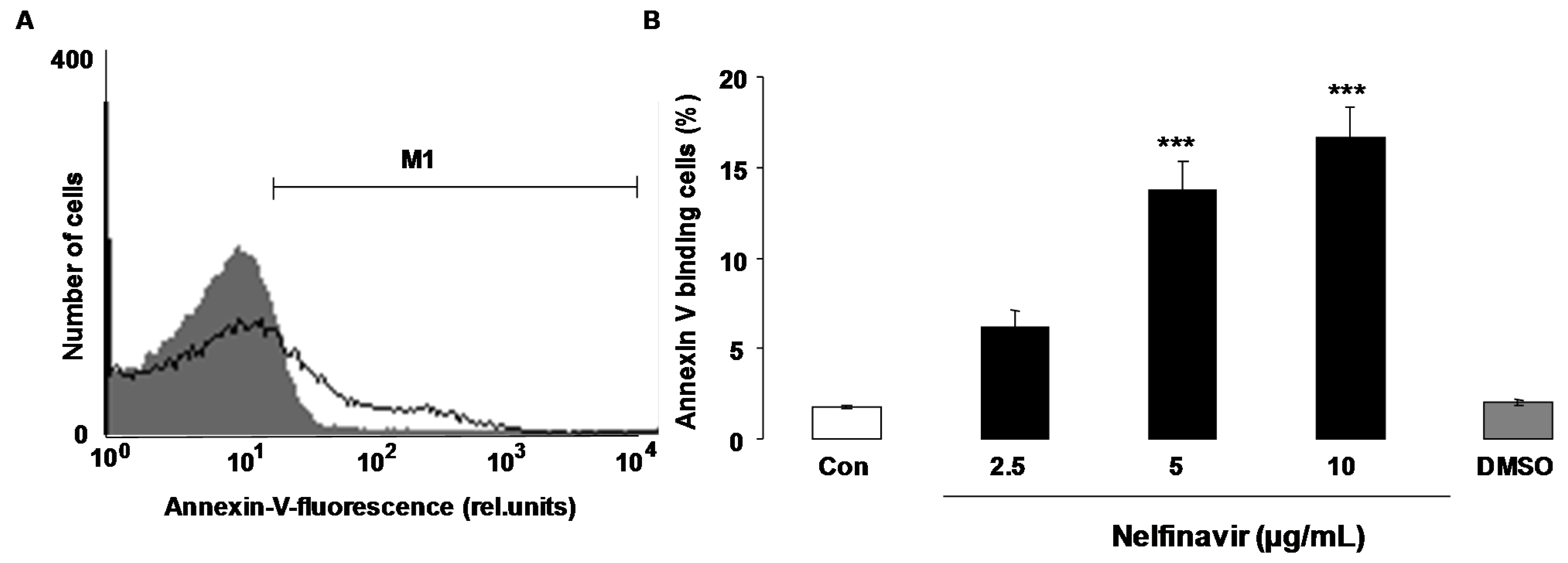
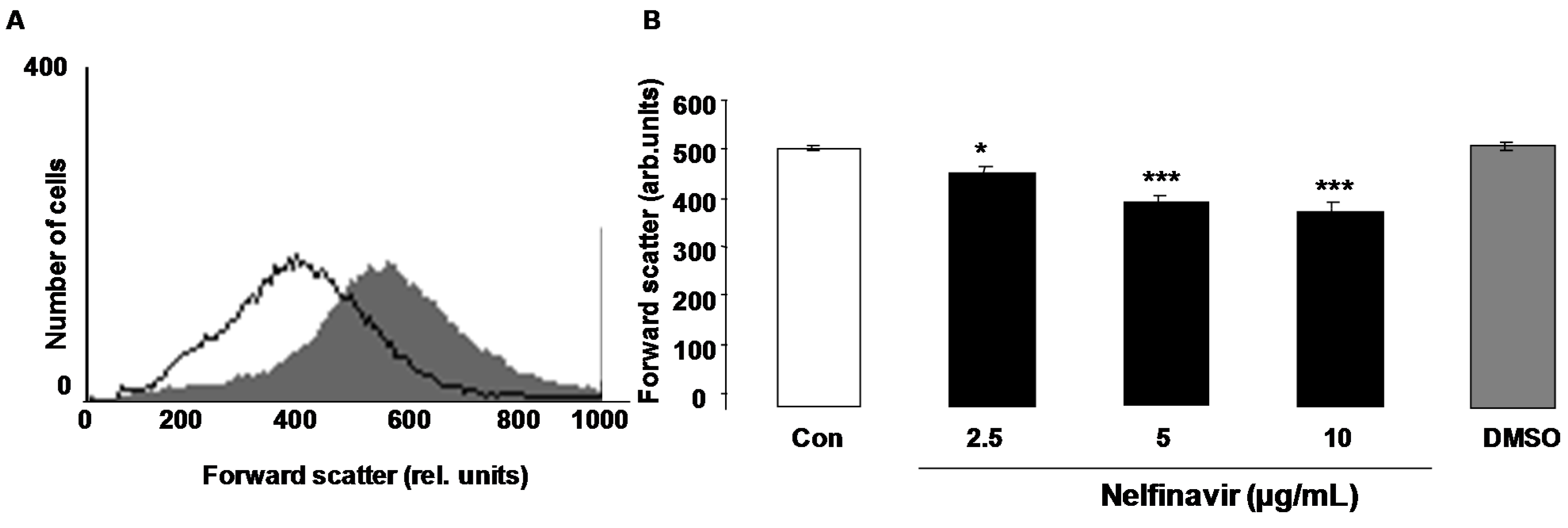
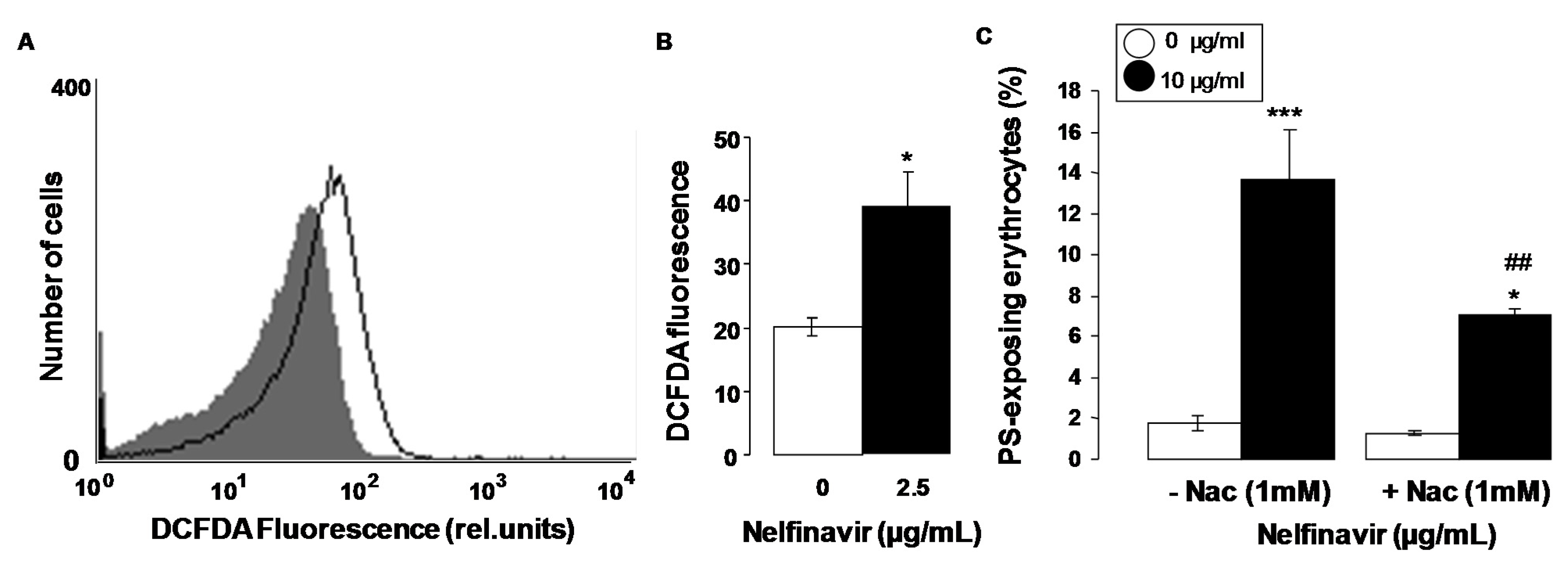
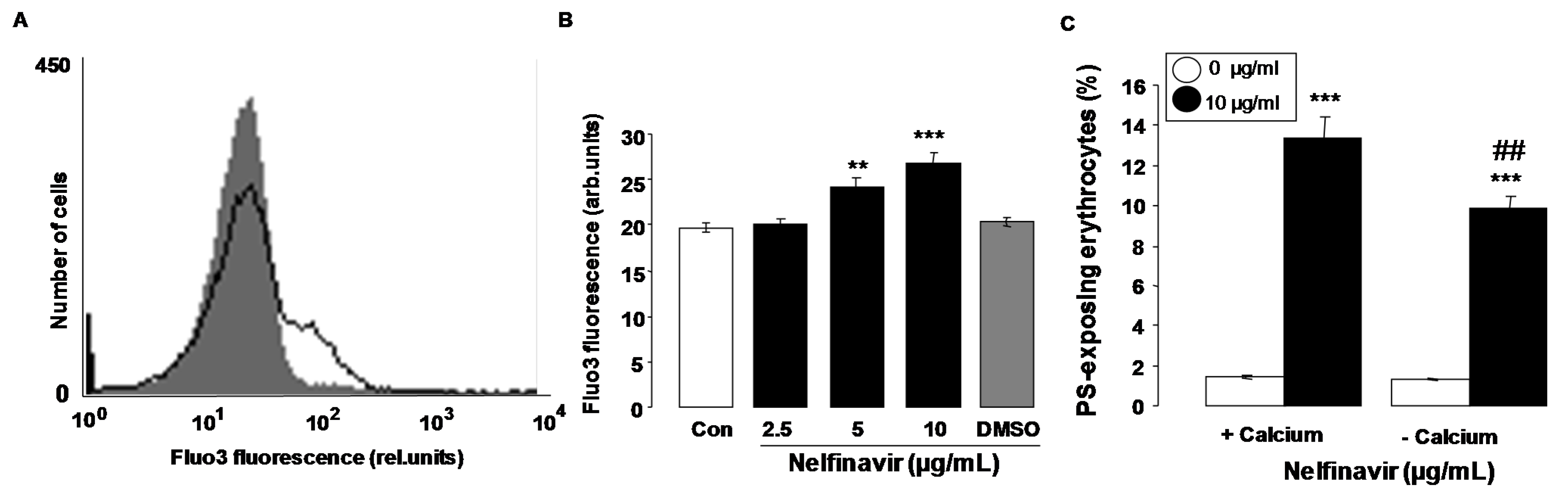
2. Results




3. Discussion
4. Experimental Section
4.1. Erythrocytes, Solutions and Chemicals
4.2. Annexin-V-Binding and Forward Scatter
4.3. Reactive Oxidant Species (ROS)
4.4. Intracellular Ca2+
4.5. Hemolysis
4.6. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baril, J.G.; Lefebvre, E.A.; Lalonde, R.G.; Shafran, S.D.; Conway, B. Nelfinavir and non-nucleoside reverse transcriptase inhibitor-based salvage regimens in heavily HIV pretreated patients. Can. J. Infect. Dis. 2003, 14, 201–205. [Google Scholar] [PubMed]
- Bruning, A.; Gingelmaier, A.; Friese, K.; Mylonas, I. New prospects for nelfinavir in non-HIV-related diseases. Curr. Mol. Pharmacol. 2010, 3, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Bruning, A.; Friese, K.; Burges, A.; Mylonas, I. Tamoxifen enhances the cytotoxic effects of nelfinavir in breast cancer cells. Breast Cancer Res. 2010, 12, R45. [Google Scholar] [CrossRef] [PubMed]
- Olmo, M.; Podzamczer, D. A review of nelfinavir for the treatment of HIV infection. Expert Opin. Drug Metab. Toxicol. 2006, 2, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M.; Frampton, J.E.; McCormack, P.L.; Siddiqui, M.A.; Cvetkovic, R.S. Nelfinavir: A review of its use in the management of HIV infection. Drugs 2005, 65, 2209–2244. [Google Scholar] [CrossRef] [PubMed]
- Gantt, S.; Casper, C.; Ambinder, R.F. Insights into the broad cellular effects of nelfinavir and the HIV protease inhibitors supporting their role in cancer treatment and prevention. Curr. Opin. Oncol. 2013, 25, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Halatsch, M.E. Matrix metalloproteinase-2 and -9 in glioblastoma: A trio of old drugs-captopril, disulfiram and nelfinavir-are inhibitors with potential as adjunctive treatments in glioblastoma. Arch. Med. Res. 2012, 43, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Wieder, T.; Lang, F.; Huber, S.M. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. Cell Physiol. 2003, 285, C1553–C1560. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly - suicidal erythrocyte death. Int J Biochem Cell Biol 2012, 44, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Towhid, S.T.; Mia, S.; Pakladok, T.; Alesutan, I.; Borst, O.; Gawaz, M.; Gulbins, E.; Lang, F. Sphingomyelinase-induced adhesion of eryptotic erythrocytes to endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 303, C991–C999. [Google Scholar] [CrossRef] [PubMed]
- Lau, I.P.; Chen, H.; Wang, J.; Ong, H.C.; Leung, K.C.; Ho, H.P.; Kong, S.K. In vitro effect of CTAB- and PEG-coated gold nanorods on the induction of eryptosis/erythroptosis in human erythrocytes. Nanotoxicology 2012, 6, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Maellaro, E.; Leoncini, S.; Moretti, D.; Del Bello, B.; Tanganelli, I.; De Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2013, 50, 489–495. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Stivison, E.A.; Hod, E.A.; Spitalnik, S.L.; Cowan, P.J.; Randis, T.M.; Ratner, A.J. Human-specific bacterial pore-forming toxins induce programmed necrosis in erythrocytes. MBio 2014, 5, e01251-14. [Google Scholar] [CrossRef] [PubMed]
- Bottger, E.; Multhoff, G.; Kun, J.F.; Esen, M. Plasmodium falciparum-infected erythrocytes induce granzyme B by NK cells through expression of host-Hsp70. PLoS One 2012, 7, e33774. [Google Scholar] [CrossRef] [PubMed]
- Firat, U.; Kaya, S.; Cim, A.; Buyukbayram, H.; Gokalp, O.; Dal, M.S.; Tamer, M.N. Increased caspase-3 immunoreactivity of erythrocytes in STZ diabetic rats. Exp. Diabetes Res. 2012, 2012, 316384. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Chaurasiya, N.D.; Sahu, R.; Walker, L.A.; Tekwani, B.L. Understanding the mechanisms for metabolism-linked hemolytic toxicity of primaquine against glucose 6-phosphate dehydrogenase deficient human erythrocytes: Evaluation of eryptotic pathway. Toxicology 2012, 294, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Cheung, K.L.; Lau, I.P.; Yu, W.S.; Fung, K.P.; Yu, B.; Loo, J.F.; Kong, S.K. Polyphyllin D induces apoptosis in human erythrocytes through Ca2+ rise and membrane permeabilization. Arch. Toxicol. 2012, 86, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Cluitmans, J.C.; Akel, A.; Dreischer, P.; Toulany, M.; Koberle, M.; Skabytska, Y.; Saki, M.; Biedermann, T.; Duszenko, M.; et al. The impact of erythrocyte age on eryptosis. Br. J. Haematol. 2012, 157, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Jilani, K.; Qadri, S.M.; Lang, F. Geldanamycin-induced phosphatidylserine translocation in the erythrocyte membrane. Cell. Physiol. Biochem. 2013, 32, 1600–1609. [Google Scholar] [PubMed]
- Polak-Jonkisz, D.; Purzyc, L. Ca influx versus efflux during eryptosis in uremic erythrocytes. Blood Purif. 2012, 34, 209–210. [Google Scholar] [CrossRef] [PubMed]
- Qian, E.W.; Ge, D.T.; Kong, S.K. Salidroside protects human erythrocytes against hydrogen peroxide-induced apoptosis. J. Nat. Prod. 2012, 75, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Vota, D.M.; Maltaneri, R.E.; Wenker, S.D.; Nesse, A.B.; Vittori, D.C. Differential erythropoietin action upon cells induced to eryptosis by different agents. Cell. Biochem. Biophys. 2013, 65, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.; Cytlak, U.M.; Rees, D.C.; Osei, A.; Gibson, J.S. Deoxygenation-induced and Ca2+ dependent phosphatidylserine externalisation in red blood cells from normal individuals and sickle cell patients. Cell Calcium 2012, 51, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Zappulla, D. Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: Adaptations to CO2 increases? J. Cardiometab. Syndr. 2008, 3, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Zbidah, M.; Lupescu, A.; Jilani, K.; Lang, F. Stimulation of suicidal erythrocyte death by fumagillin. Basic Clin. Pharmacol. Toxicol. 2013, 112, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Herrmann, T.; Alzoubi, K.; Pakladok, T.; Lang, F. Tannic Acid induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Langer, H.; Abed, M.; Voelkl, J.; Lang, F. The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press. Res 2013, 37, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Cluitmans, J.C.; Toulany, M.; Saki, M.; Koberle, M.; Lang, E.; Dreischer, P.; Biedermann, T.; Duszenko, M.; Lang, F.; et al. Age sensitivity of NFkappaB abundance and programmed cell death in erythrocytes induced by NFkappaB inhibitors. Cell. Physiol. Biochem. 2013, 32, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.; Honisch, S.; Abed, M.; Lang, F. Triggering of suicidal erythrocyte death by penta-o-galloyl-beta-d-glucose. Toxins 2014, 6, 54–65. [Google Scholar] [CrossRef]
- Jilani, K.; Lang, F. Carmustine-induced phosphatidylserine translocation in the erythrocyte membrane. Toxins 2013, 5, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Jilani, K.; Enkel, S.; Bissinger, R.; Almilaji, A.; Abed, M.; Lang, F. Fluoxetine induced suicidal erythrocyte death. Toxins 2013, 5, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Jilani, K.; Lang, F. Triggering of suicidal erythrocyte death by celecoxib. Toxins 2013, 5, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Patulin-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Zoubi, K.A.; Theurer, M.; Lang, F. Effect of dermaseptin on erythrocytes. Basic Clin. Pharmacol. Toxicol. 2013, 113, 347–352. [Google Scholar] [PubMed]
- Arnold, M.; Lang, E.; Modicano, P.; Bissinger, R.; Faggio, C.; Abed, M.; Lang, F. Effect of nitazoxanide on erythrocytes. Basic Clin. Pharmacol. Toxicol. 2014, 114, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Oswald, G.; Alzoubi, K.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by ribavirin. Basic Clin. Pharmacol. Toxicol. 2014, 114, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Malik, A.; Jilani, K.; Lang, F. Triggering of erythrocyte cell membrane scrambling by salinomycin. Basic Clin. Pharmacol. Toxicol. 2014, 115, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, J.; Lang, E.; Bissinger, R.; Frauenfeld, L.; Modicano, P.; Faggio, C.; Abed, M.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by mitotane. Cell. Physiol. Biochem. 2014, 33, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Herrmann, T.; Oswald, G.; Jilani, K.; Lang, F. Induction of suicidal erythrocyte death by novobiocin. Cell. Physiol. Biochem. 2014, 33, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Warsi, J.; Jilani, K.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by gedunin. Cell. Physiol. Biochem. 2014, 33, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Feger, M.; Alzoubi, K.; Pakladok, T.; Frauenfeld, L.; Geiger, C.; Towhid, S.T.; Lang, F. Sensitization of erythrocytes to suicidal erythrocyte death following water deprivation. Kidney Blood Press. Res. 2013, 37, 567–578. [Google Scholar] [PubMed]
- Alzoubi, K.; Calabro, S.; Bissinger, R.; Abed, M.; Faggio, C.; Lang, F. Stimulation of suicidal erythrocyte death by artesunate. Cell. Physiol. Biochem. 2014, 34, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Bissinger, R.; Lang, F. Mitoxantrone-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2014, 34, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Fischer, S.; Jilani, K.; Lang, F. Stimulation of erythrocyte death by phloretin. Cell. Physiol. Biochem. 2014, 34, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Lupescu, A.; Zelenak, C.; Jilani, K.; Lang, F. Stimulation of eryptosis by cryptotanshinone. Cell. Physiol. Biochem. 2014, 34, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Modicano, P.; Frauenfeld, L.; Lang, E.; Jacobi, J.; Faggio, C.; Lang, F. Estramustine-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Bissinger, R.; Calabro, S.; Faggio, C.; Jilani, K.; Lang, F. Aristolochic acid induced suicidal erythrocyte death. Kidney Blood Press. Res. 2014, 39, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Cilla, A.; Gentile, C.; Livrea, M.A. Oxysterol mixture in hypercholesterolemia-relevant proportion causes oxidative stress-dependent eryptosis. Cell. Physiol. Biochem. 2014, 34, 1075–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelkl, J.; Alzoubi, K.; Mamar, A.K.; Ahmed, M.S.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by increased extracellular phosphate concentrations. Kidney Blood Press. Res. 2013, 38, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xiang, Y.; Ran, Q.; Deng, X.; Xiao, Y.; Xiang, L.; Li, Z. Involvement of calcium, reactive oxygen species, and ATP in hexavalent chromium-induced damage in red blood cells. Cell. Physiol. Biochem. 2014, 34, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Foller, M.; Bobbala, D.; Koka, S.; Huber, S.M.; Gulbins, E.; Lang, F. Suicide for survival—Death of infected erythrocytes as a host mechanism to survive malaria. Cell. Physiol. Biochem. 2009, 24, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Sugioka, N.; Haraya, K.; Maeda, Y.; Fukushima, K.; Takada, K. Pharmacokinetics of human immunodeficiency virus protease inhibitor, nelfinavir, in poloxamer 407-induced hyperlipidemic model rats. Biol. Pharm. Bull. 2009, 32, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.L.; Brundage, R.C.; Bushman, L.; Kakuda, T.N.; Remmel, R.P.; Fletcher, C.V. Indinavir plasma protein binding in HIV-1-infected adults. AIDS 2000, 14, 2293–2297. [Google Scholar] [CrossRef] [PubMed]
- Ben-Romano, R.; Rudich, A.; Etzion, S.; Potashnik, R.; Kagan, E.; Greenbaum, U.; Bashan, N. Nelfinavir induces adipocyte insulin resistance through the induction of oxidative stress: Differential protective effect of antioxidant agents. Antivir. Ther. 2006, 11, 1051–1060. [Google Scholar] [PubMed]
- Chandra, S.; Mondal, D.; Agrawal, K.C. HIV-1 protease inhibitor induced oxidative stress suppresses glucose stimulated insulin release: Protection with thymoquinone. Exp. Biol. Med. 2009, 234, 442–453. [Google Scholar] [CrossRef]
- Kumar, P.; Lodge, R.; Trudel, N.; Ouellet, M.; Ouellette, M.; Tremblay, M.J. Nelfinavir, an HIV-1 protease inhibitor, induces oxidative stress-mediated, caspase-independent apoptosis in Leishmania amastigotes. PLoS Negl. Trop. Dis. 2010, 4, e642. [Google Scholar] [CrossRef] [PubMed]
- Kushchayeva, Y.; Jensen, K.; Recupero, A.; Costello, J.; Patel, A.; Klubo-Gwiezdzinska, J.; Boyle, L.; Burman, K.; Vasko, V. The HIV protease inhibitor nelfinavir down-regulates RET signaling and induces apoptosis in medullary thyroid cancer cells. J. Clin. Endocrinol. Metab. 2014, 99, E734–E745. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.; Kost, B.; Renner-Muller, I.; Wolf, E.; Mylonas, I.; Bruning, A. Efavirenz causes oxidative stress, endoplasmic reticulum stress, and autophagy in endothelial cells. Cardiovasc. Toxicol. 2015, in press. [Google Scholar]
- Harrison, H.E.; Bunting, H.; Ordway, N.K.; Albrink, W.S. The pathogenesis of the renal injury produced in the dog by hemoglobin or methemoglobin. J. Exp. Med. 1947, 86, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K. Membrane transport in the malaria-infected erythrocyte. Physiol. Rev. 2001, 81, 495–537. [Google Scholar] [PubMed]
- Ayi, K.; Giribaldi, G.; Skorokhod, A.; Schwarzer, E.; Prendergast, P.T.; Arese, P. 16alpha-bromoepiandrosterone, an antimalarial analogue of the hormone dehydroepiandrosterone, enhances phagocytosis of ring stage parasitized erythrocytes: A novel mechanism for antimalarial activity. Antimicrob. Agents Chemother. 2002, 46, 3180–3184. [Google Scholar] [CrossRef] [PubMed]
- Ayi, K.; Turrini, F.; Piga, A.; Arese, P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: A common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood 2004, 104, 3364–3371. [Google Scholar] [CrossRef] [PubMed]
- Cappadoro, M.; Giribaldi, G.; O’Brien, E.; Turrini, F.; Mannu, F.; Ulliers, D.; Simula, G.; Luzzatto, L.; Arese, P. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood 1998, 92, 2527–2534. [Google Scholar] [PubMed]
- Koka, S.; Huber, S.M.; Boini, K.M.; Lang, C.; Foller, M.; Lang, F. Lead decreases parasitemia and enhances survival of Plasmodium berghei-infected mice. Biochem. Biophys. Res. Commun. 2007, 363, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Koka, S.; Lang, C.; Niemoeller, O.M.; Boini, K.M.; Nicolay, J.P.; Huber, S.M.; Lang, F. Influence of NO synthase inhibitor L-NAME on parasitemia and survival of Plasmodium berghei infected mice. Cell. Physiol. Biochem. 2008, 21, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Foller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef] [PubMed]
- Andrews, D.A.; Low, P.S. Role of red blood cells in thrombosis. Curr. Opin. Hematol. 1999, 6, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.G.; Chang, S.H.; Rettig, M.P.; Neely, J.E.; Hillery, C.A.; Smith, B.D.; Low, P.S. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood 2003, 101, 4625–4627. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, A.; di Pietro, N.; Sirolli, V.; Giardinelli, A.; di Silvestre, S.; Amoroso, L.; di Tomo, P.; Capani, F.; Consoli, A.; Bonomini, M. Mechanisms of uremic erythrocyte-induced adhesion of human monocytes to cultured endothelial cells. J. Cell. Physiol. 2007, 213, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow-cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bissinger, R.; Waibel, S.; Lang, F. Induction of Suicidal Erythrocyte Death by Nelfinavir. Toxins 2015, 7, 1616-1628. https://doi.org/10.3390/toxins7051616
Bissinger R, Waibel S, Lang F. Induction of Suicidal Erythrocyte Death by Nelfinavir. Toxins. 2015; 7(5):1616-1628. https://doi.org/10.3390/toxins7051616
Chicago/Turabian StyleBissinger, Rosi, Sabrina Waibel, and Florian Lang. 2015. "Induction of Suicidal Erythrocyte Death by Nelfinavir" Toxins 7, no. 5: 1616-1628. https://doi.org/10.3390/toxins7051616
APA StyleBissinger, R., Waibel, S., & Lang, F. (2015). Induction of Suicidal Erythrocyte Death by Nelfinavir. Toxins, 7(5), 1616-1628. https://doi.org/10.3390/toxins7051616