
Induction of Suicidal Erythrocyte Death by Cantharidin


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
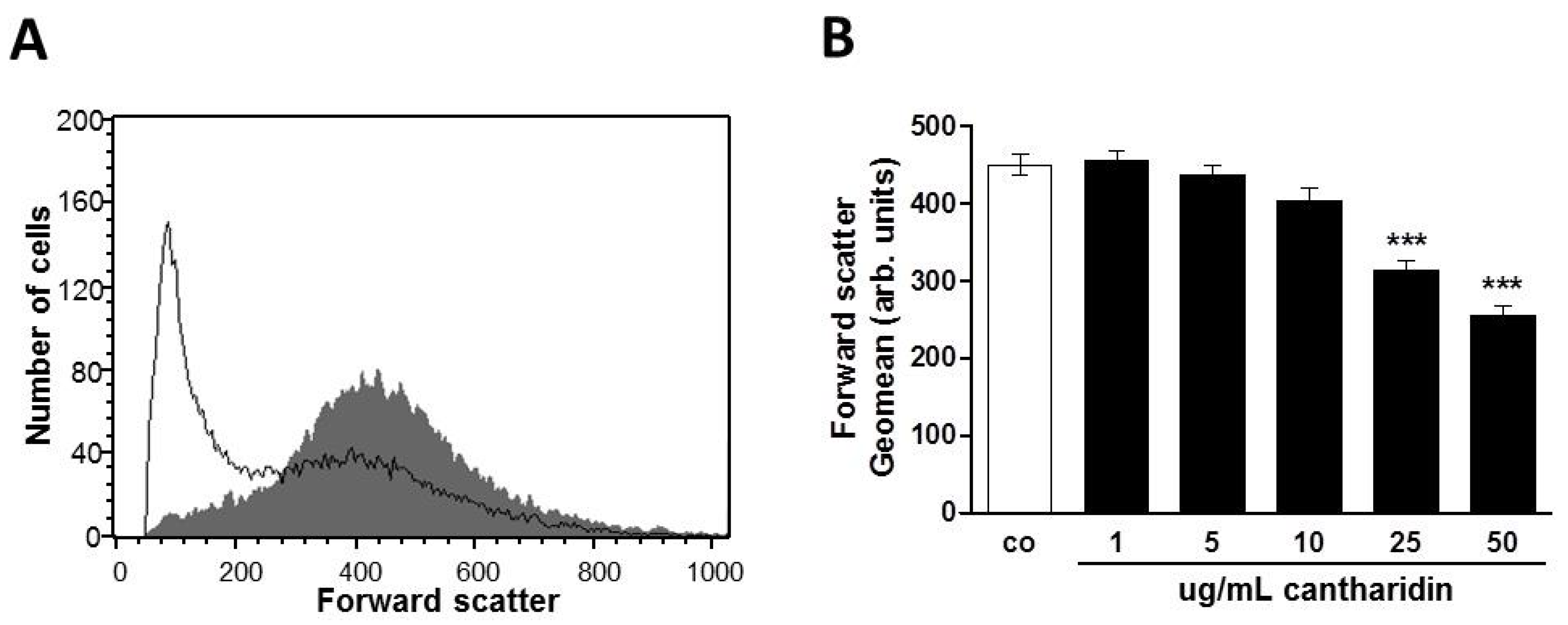
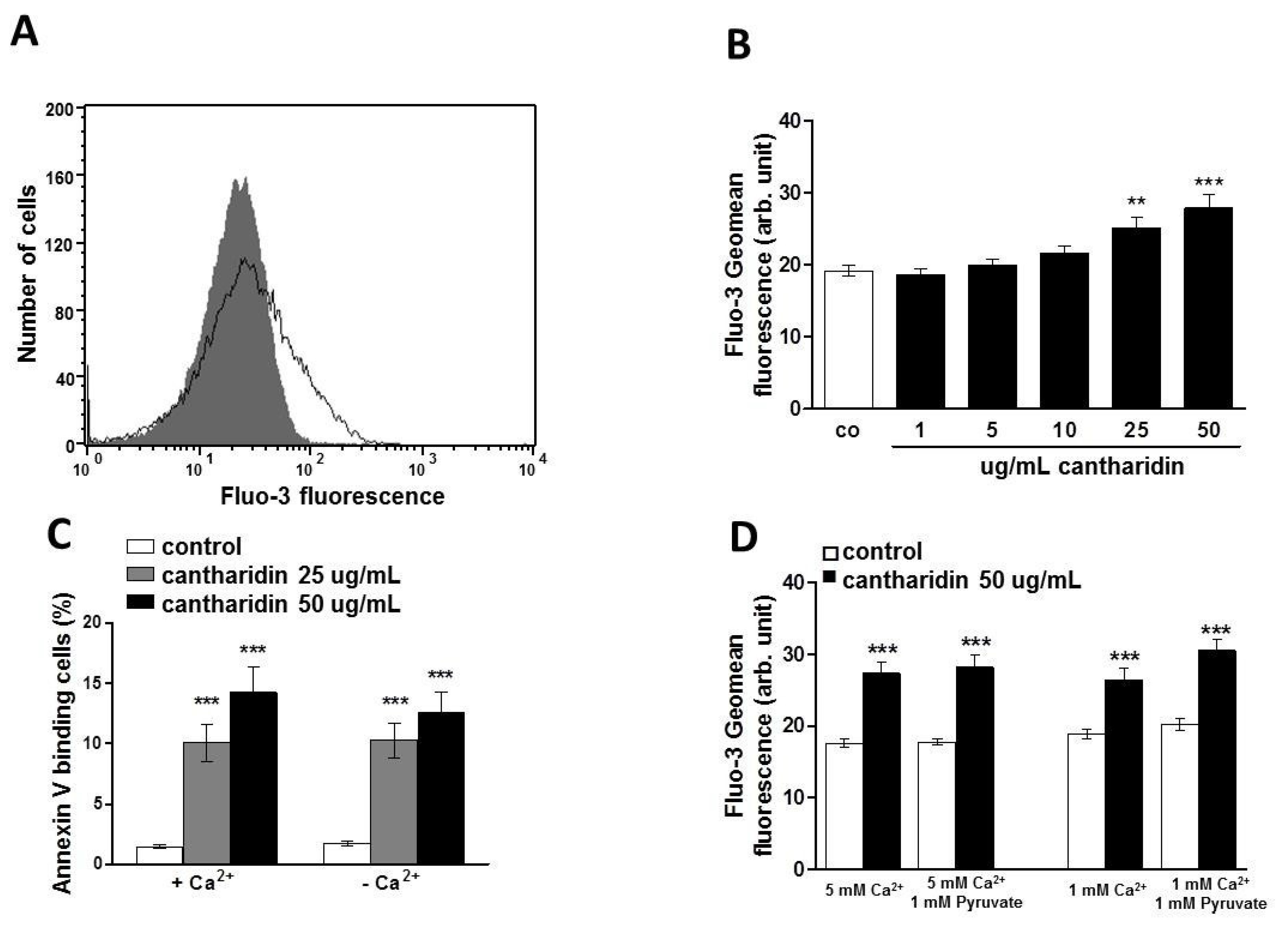
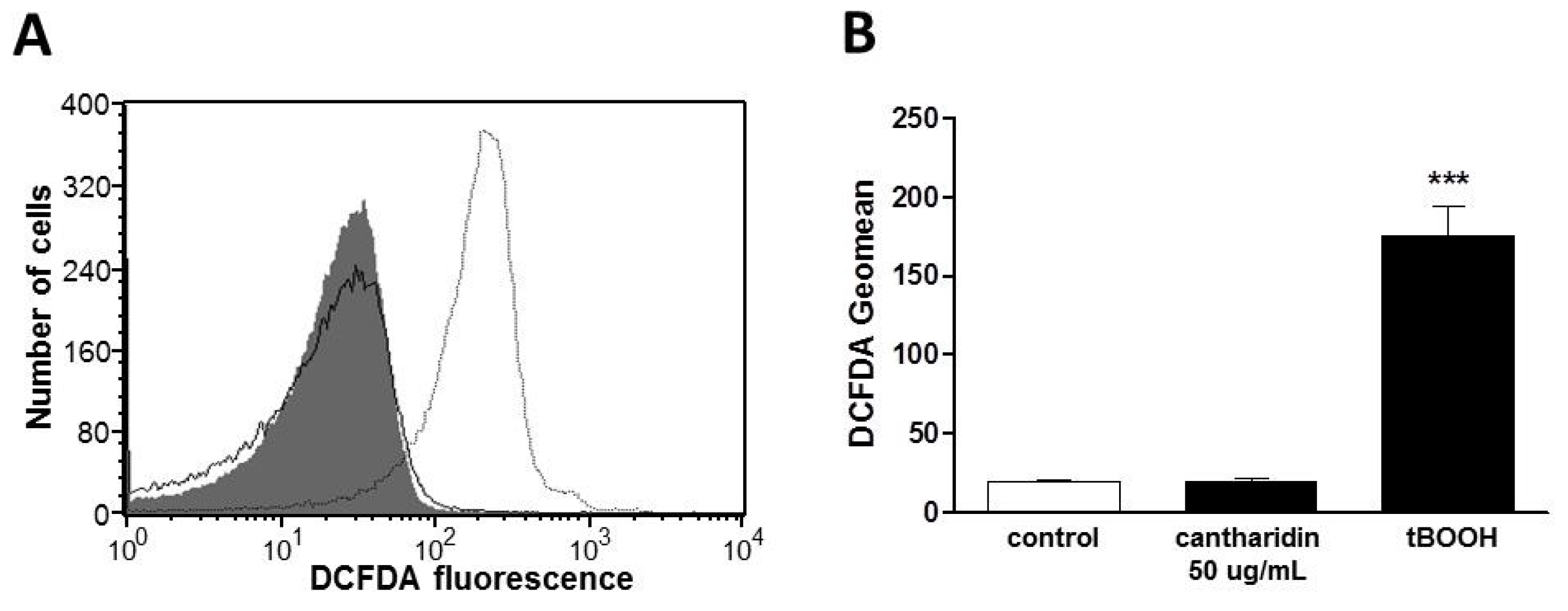
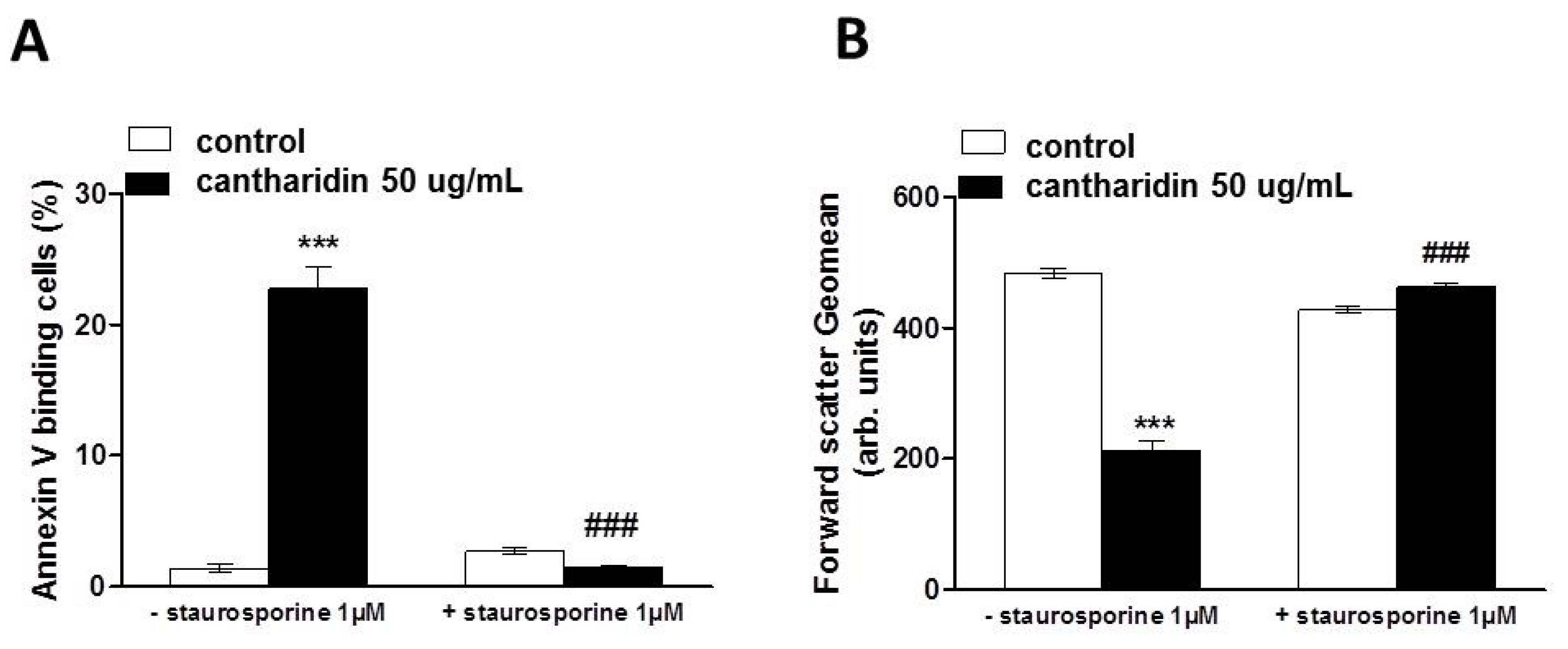
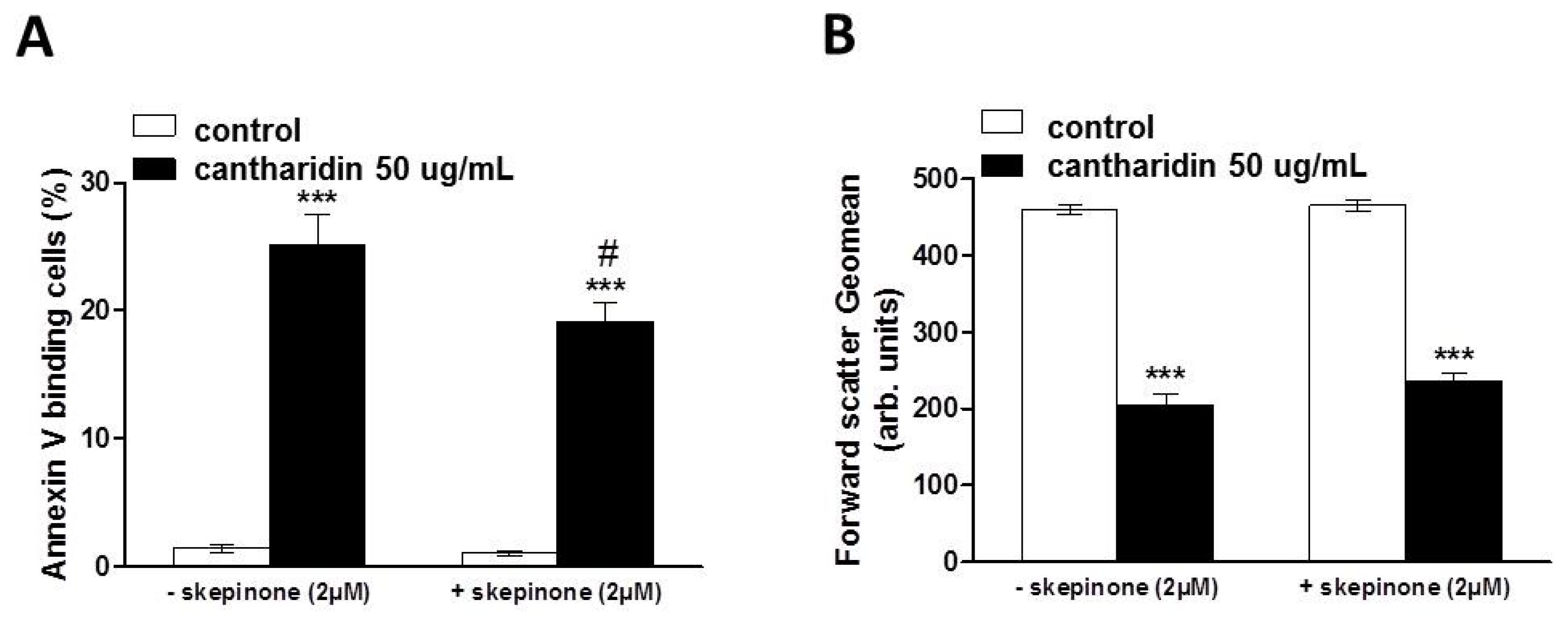
2. Results and Discussion






3. Experimental Section
3.1. Erythrocytes, Solutions and Chemicals
3.2. Annexin-V-Binding and Forward Scatter
3.3. Hemolysis
3.4. Intracellular Ca2+
3.5. Reactive Oxidant Species (ROS)
3.6. Ceramide Abundance
3.7. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Torbeck, R.; Pan, M.; DeMoll, E.; Levitt, J. Cantharidin: A comprehensive review of the clinical literature. Dermatol. Online J. 2014, 20. Available online: http://escholarship.org/uc/item/22845r22512w22860 (accessed on 27 July 2015). [Google Scholar]
- Deng, L.P.; Dong, J.; Cai, H.; Wang, W. Cantharidin as an antitumor agent: A retrospective review. Curr. Med. Chem. 2013, 20, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kok, S.H.; Cheng, S.J.; Hong, C.Y.; Lee, J.J.; Lin, S.K.; Kuo, Y.S.; Chiang, C.P.; Kuo, M.Y. Norcantharidin-induced apoptosis in oral cancer cells is associated with an increase of proapoptotic to antiapoptotic protein ratio. Cancer Lett. 2005, 217, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Puerto Galvis, C.E.; Vargas Mendez, L.Y.; Kouznetsov, V.V. Cantharidin-based small molecules as potential therapeutic agents. Chem. Biol. Drug Des. 2013, 82, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Dong, J.; Wang, W. Exploiting protein phosphatase inhibitors based on cantharidin analogues for cancer drug discovery. Mini Rev. Med. Chem. 2013, 13, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, R.E.; Golden, T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr. Med. Chem. 2002, 9, 2055–2075. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.W.; Ko, S.W.; Tsai, H.Y.; Chung, J.G.; Chiang, J.H.; Chen, K.T.; Chen, Y.C.; Chen, H.Y.; Chen, Y.F.; Yang, J.S. Cantharidin induces G2/M phase arrest and apoptosis in human colorectal cancer colo 205 cells through inhibition of CDK1 activity and caspase-dependent signaling pathways. Int. J. Oncol. 2011, 38, 1067–1073. [Google Scholar] [PubMed]
- Liu, D.; Chen, Z. The effects of cantharidin and cantharidin derivates on tumour cells. Anticancer Agents Med. Chem. 2009, 9, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.R.; Vasconcelos, V.M.; Antunes, A. The phosphoprotein phosphatase family of Ser/Thr phosphatases as principal targets of naturally occurring toxins. Crit. Rev. Toxicol. 2011, 41, 83–110. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Zhu, Y.Q.; Mei, J.J.; Liu, S.Q.; Luo, J. Involvement of mitochondrial pathway in NCTD-induced cytotoxicity in human hepG2 cells. J. Exp. Clin. Cancer Res. 2010, 29, 145. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.B.; Verma, A.K. Cantharidin-mediated ultrastructural and biochemical changes in mitochondria lead to apoptosis and necrosis in murine dalton’s lymphoma. Microsc. Microanal. 2013, 19, 1377–1394. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.N.; Chen, J.C.; Yin, S.C.; Wang, G.S.; Tsauer, W.; Hsu, S.F.; Hsu, S.L. Effector mechanisms of norcantharidin-induced mitotic arrest and apoptosis in human hepatoma cells. Int. J. Cancer 2002, 100, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Rauh, R.; Kahl, S.; Tomicic, M.; Bochzelt, H.; Tome, M.E.; Briehl, M.M.; Bauer, R.; Kaina, B. Molecular modes of action of cantharidin in tumor cells. Biochem. Pharmacol. 2005, 69, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.Y.; Huang, S.C.; Lin, S.K.; Lee, J.J.; Chueh, L.L.; Lee, C.H.; Lin, J.H.; Hsiao, M. Norcantharidin-induced post-G2/M apoptosis is dependent on wild-type p53 gene. Biochem. Biophys. Res. Commun. 2000, 276, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Wieder, T.; Lang, F.; Huber, S.M. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. Cell. Physiol. 2003, 285, C1553–C1560. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly—Suicidal erythrocyte death. Int. J. Biochem. Cell Biol. 2012, 44, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Towhid, S.T.; Mia, S.; Pakladok, T.; Alesutan, I.; Borst, O.; Gawaz, M.; Gulbins, E.; Lang, F. Sphingomyelinase-induced adhesion of eryptotic erythrocytes to endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 303, C991–C999. [Google Scholar] [CrossRef] [PubMed]
- Lau, I.P.; Chen, H.; Wang, J.; Ong, H.C.; Leung, K.C.; Ho, H.P.; Kong, S.K. In vitro effect of CTAB- and PEG-coated gold nanorods on the induction of eryptosis/erythroptosis in human erythrocytes. Nanotoxicology 2012, 6, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Maellaro, E.; Leoncini, S.; Moretti, D.; del Bello, B.; Tanganelli, I.; de Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2013, 50, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Vota, D.M.; Maltaneri, R.E.; Wenker, S.D.; Nesse, A.B.; Vittori, D.C. Differential erythropoietin action upon cells induced to eryptosis by different agents. Cell Biochem. Biophys. 2013, 65, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Zbidah, M.; Lupescu, A.; Jilani, K.; Lang, F. Stimulation of suicidal erythrocyte death by fumagillin. Basic Clin. Pharmacol. Toxicol. 2013, 112, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Langer, H.; Abed, M.; Voelkl, J.; Lang, F. The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press. Res. 2013, 37, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Cluitmans, J.C.; Toulany, M.; Saki, M.; Koberle, M.; Lang, E.; Dreischer, P.; Biedermann, T.; Duszenko, M.; Lang, F.; et al. Age sensitivity of nfkappab abundance and programmed cell death in erythrocytes induced by nfkappab inhibitors. Cell. Physiol. Biochem. 2013, 32, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.; Honisch, S.; Abed, M.; Lang, F. Triggering of suicidal erythrocyte death by penta-O-galloyl-β-d-glucose. Toxins (Basel) 2014, 6, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Jilani, K.; Enkel, S.; Bissinger, R.; Almilaji, A.; Abed, M.; Lang, F. Fluoxetine induced suicidal erythrocyte death. Toxins (Basel) 2013, 5, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Jilani, K.; Lang, F. Triggering of suicidal erythrocyte death by celecoxib. Toxins (Basel) 2013, 5, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Patulin-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Zoubi, K.A.; Theurer, M.; Lang, F. Effect of dermaseptin on erythrocytes. Basic Clin. Pharmacol. Toxicol. 2013, 113, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Lang, E.; Modicano, P.; Bissinger, R.; Faggio, C.; Abed, M.; Lang, F. Effect of nitazoxanide on erythrocytes. Basic Clin. Pharmacol. Toxicol. 2014, 114, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Oswald, G.; Alzoubi, K.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by ribavirin. Basic Clin. Pharmacol. Toxicol. 2014, 114, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, J.; Lang, E.; Bissinger, R.; Frauenfeld, L.; Modicano, P.; Faggio, C.; Abed, M.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by mitotane. Cell. Physiol. Biochem. 2014, 33, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Warsi, J.; Jilani, K.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by gedunin. Cell. Physiol. Biochem. 2014, 33, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Feger, M.; Alzoubi, K.; Pakladok, T.; Frauenfeld, L.; Geiger, C.; Towhid, S.T.; Lang, F. Sensitization of erythrocytes to suicidal erythrocyte death following water deprivation. Kidney Blood Press. Res. 2013, 37, 567–578. [Google Scholar] [PubMed]
- Alzoubi, K.; Calabro, S.; Bissinger, R.; Abed, M.; Faggio, C.; Lang, F. Stimulation of suicidal erythrocyte death by artesunate. Cell. Physiol. Biochem. 2014, 34, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Bissinger, R.; Lang, F. Mitoxantrone-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2014, 34, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Fischer, S.; Jilani, K.; Lang, F. Stimulation of erythrocyte death by phloretin. Cell. Physiol. Biochem. 2014, 34, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Lupescu, A.; Zelenak, C.; Jilani, K.; Lang, F. Stimulation of eryptosis by cryptotanshinone. Cell. Physiol. Biochem. 2014, 34, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Modicano, P.; Frauenfeld, L.; Lang, E.; Jacobi, J.; Faggio, C.; Lang, F. Estramustine-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Bissinger, R.; Calabro, S.; Faggio, C.; Jilani, K.; Lang, F. Aristolochic acid induced suicidal erythrocyte death. Kidney Blood Press. Res. 2014, 39, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Cilla, A.; Gentile, C.; Livrea, M.A. Oxysterol mixture in hypercholesterolemia-relevant proportion causes oxidative stress-dependent eryptosis. Cell. Physiol. Biochem. 2014, 34, 1075–1089. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, J.; Alzoubi, K.; Mamar, A.K.; Ahmed, M.S.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by increased extracellular phosphate concentrations. Kidney Blood Press. Res. 2013, 38, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xiang, Y.; Ran, Q.; Deng, X.; Xiao, Y.; Xiang, L.; Li, Z. Involvement of calcium, reactive oxygen species, and atp in hexavalent chromium-induced damage in red blood cells. Cell. Physiol. Biochem. 2014, 34, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sancho, J. Pyruvate prevents the atp depletion caused by formaldehyde or calcium-chelator esters in the human red cell. Biochim. Biophys. Acta 1985, 813, 148–150. [Google Scholar] [CrossRef]
- Tiffert, T.; Garcia-Sancho, J.; Lew, V.L. Irreversible atp depletion caused by low concentrations of formaldehyde and of calcium-chelator esters in intact human red cells. Biochim. Biophys. Acta 1984, 773, 143–156. [Google Scholar] [CrossRef]
- Dang, Y.J.; Zhu, C.Y. Oral bioavailability of cantharidin-loaded solid lipid nanoparticles. Chin. Med. 2013, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.J.; Zhu, C.Y. Determination of trace cantharidin in plasma and pharmacokinetic study in beagle dogs using gas chromatography-mass spectrometry. J. Anal. Toxicol. 2009, 33, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Cossins, A.R.; Gibson, J.S. Volume-sensitive transport systems and volume homeostasis in vertebrate red blood cells. J. Exp. Biol. 1997, 200, 343–352. [Google Scholar] [PubMed]
- De Los Heros, P.; Alessi, D.R.; Gourlay, R.; Campbell, D.G.; Deak, M.; Macartney, T.J.; Kahle, K.T.; Zhang, J. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl- co-transporters. Biochem. J. 2014, 458, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Merciris, P.; Claussen, W.J.; Joiner, C.H.; Giraud, F. Regulation of K-Cl cotransport by Syk and Src protein tyrosine kinases in deoxygenated sickle cells. Pflug. Arch. 2003, 446, 232–238. [Google Scholar]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Foller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef] [PubMed]
- Andrews, D.A.; Low, P.S. Role of red blood cells in thrombosis. Curr. Opin. Hematol. 1999, 6, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.G.; Chang, S.H.; Rettig, M.P.; Neely, J.E.; Hillery, C.A.; Smith, B.D.; Low, P.S. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood 2003, 101, 4625–4627. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, A.; di Pietro, N.; Sirolli, V.; Giardinelli, A.; di Silvestre, S.; Amoroso, L.; di Tomo, P.; Capani, F.; Consoli, A.; Bonomini, M. Mechanisms of uremic erythrocyte-induced adhesion of human monocytes to cultured endothelial cells. J. Cell. Physiol. 2007, 213, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow-cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzoubi, K.; Egler, J.; Briglia, M.; Fazio, A.; Faggio, C.; Lang, F. Induction of Suicidal Erythrocyte Death by Cantharidin. Toxins 2015, 7, 2822-2834. https://doi.org/10.3390/toxins7082822
Alzoubi K, Egler J, Briglia M, Fazio A, Faggio C, Lang F. Induction of Suicidal Erythrocyte Death by Cantharidin. Toxins. 2015; 7(8):2822-2834. https://doi.org/10.3390/toxins7082822
Chicago/Turabian StyleAlzoubi, Kousi, Jasmin Egler, Marilena Briglia, Antonella Fazio, Caterina Faggio, and Florian Lang. 2015. "Induction of Suicidal Erythrocyte Death by Cantharidin" Toxins 7, no. 8: 2822-2834. https://doi.org/10.3390/toxins7082822
APA StyleAlzoubi, K., Egler, J., Briglia, M., Fazio, A., Faggio, C., & Lang, F. (2015). Induction of Suicidal Erythrocyte Death by Cantharidin. Toxins, 7(8), 2822-2834. https://doi.org/10.3390/toxins7082822