
Effect of Fusarium-Derived Metabolites on the Barrier Integrity of Differentiated Intestinal Porcine Epithelial Cells (IPEC-J2)

 and
and 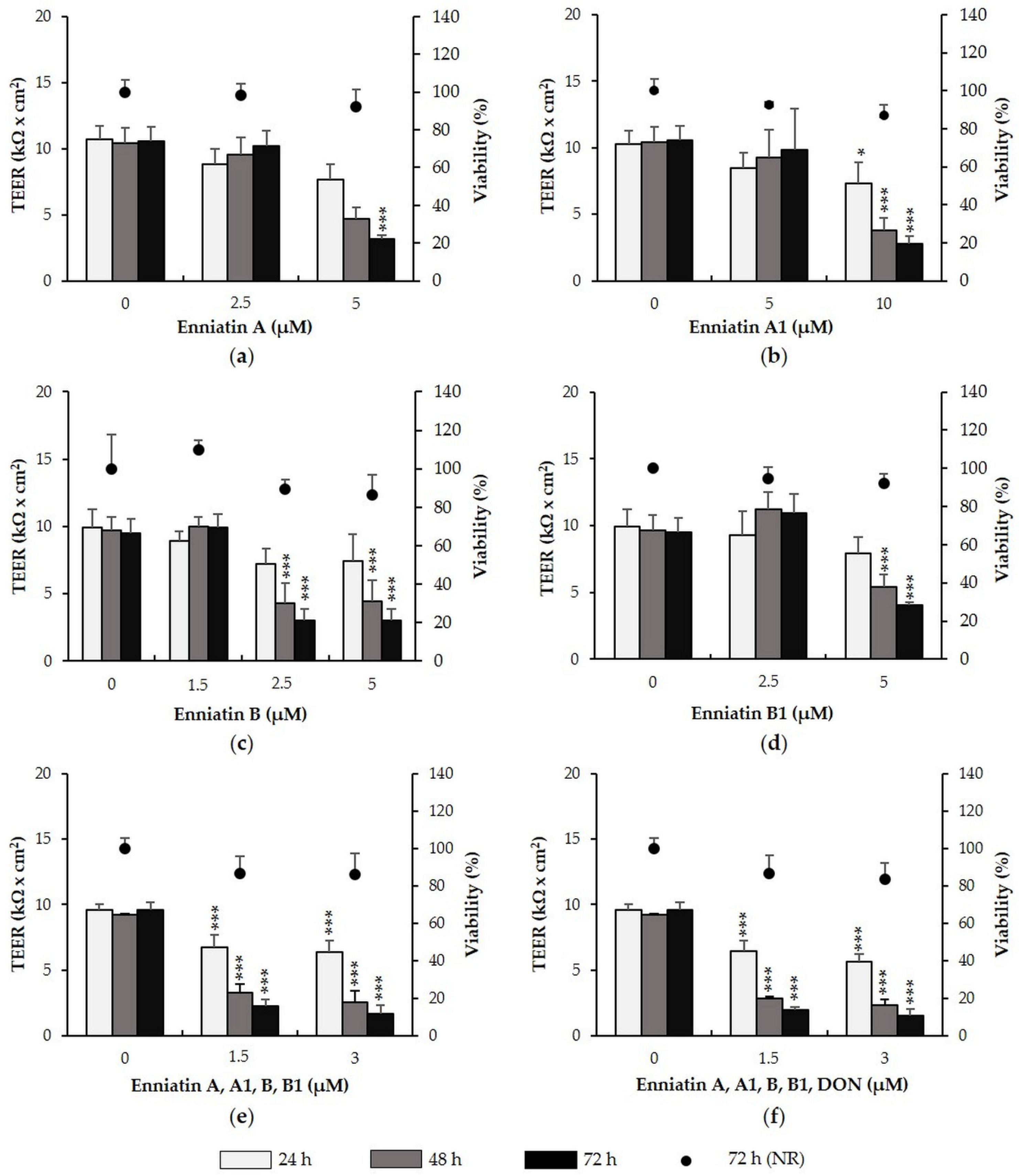{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Deoxynivalenol
2.2. Enniatins and Combinations of Enniatins
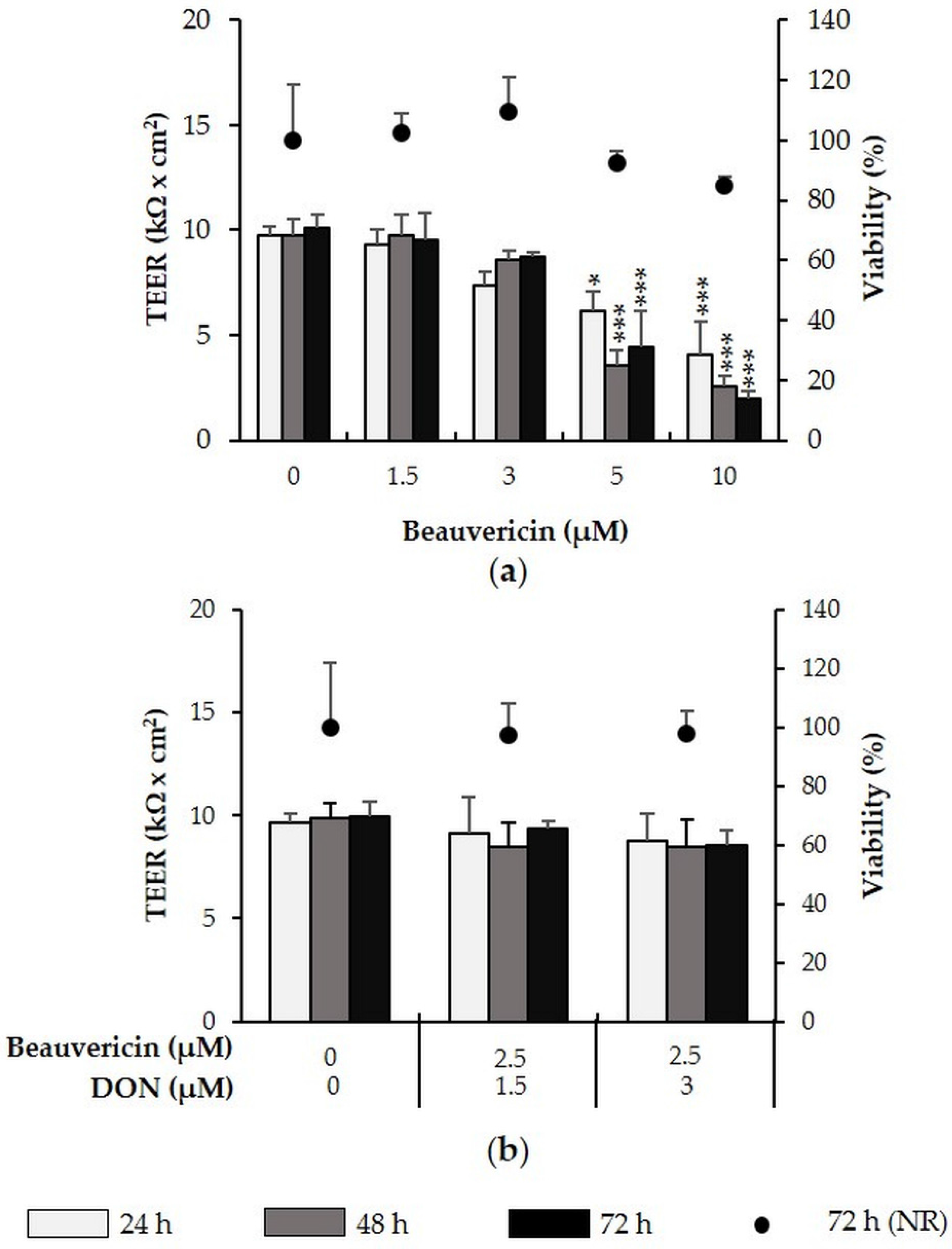
2.3. Beauvericin
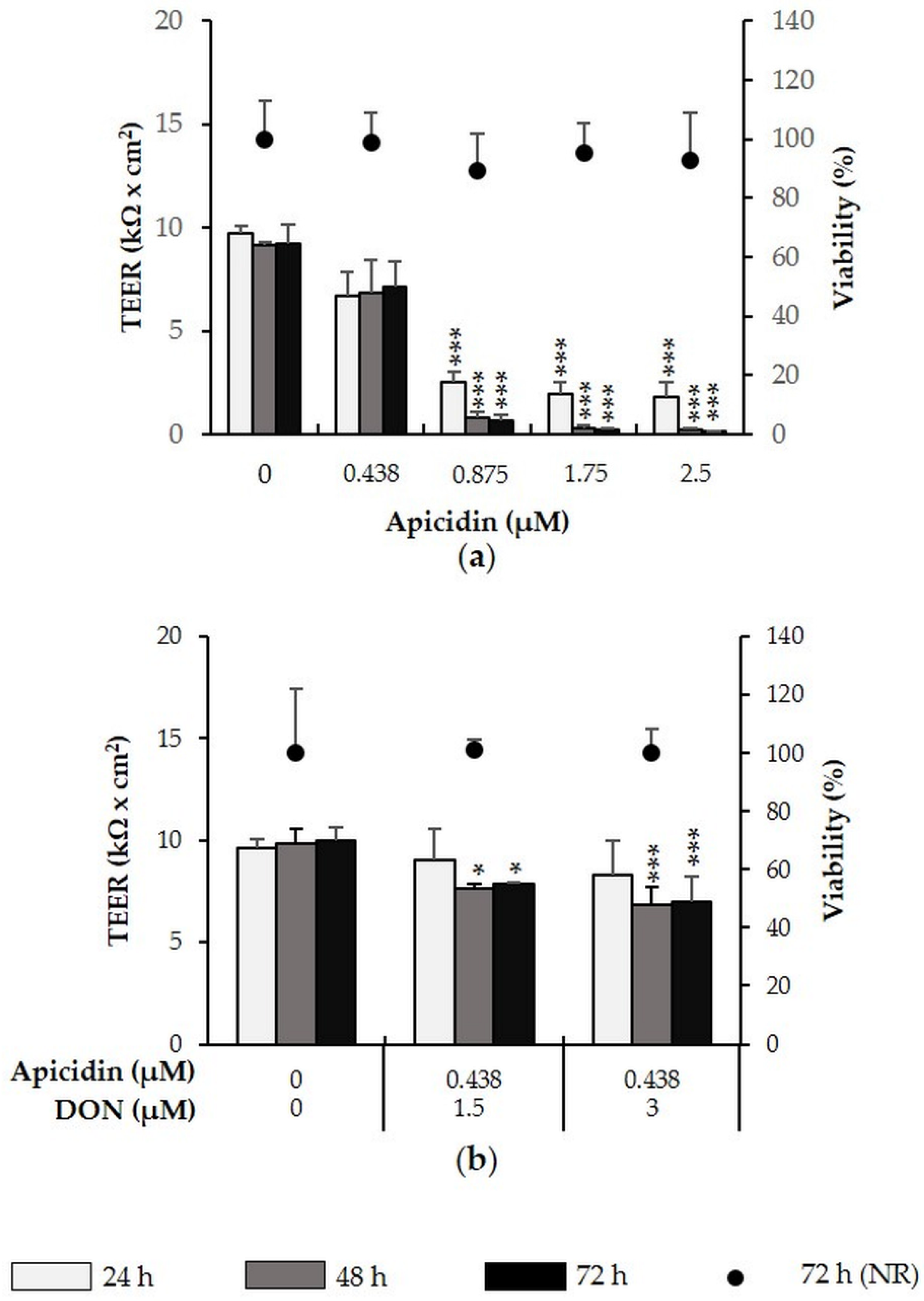
2.4. Apicidin
2.5. Aurofusarin
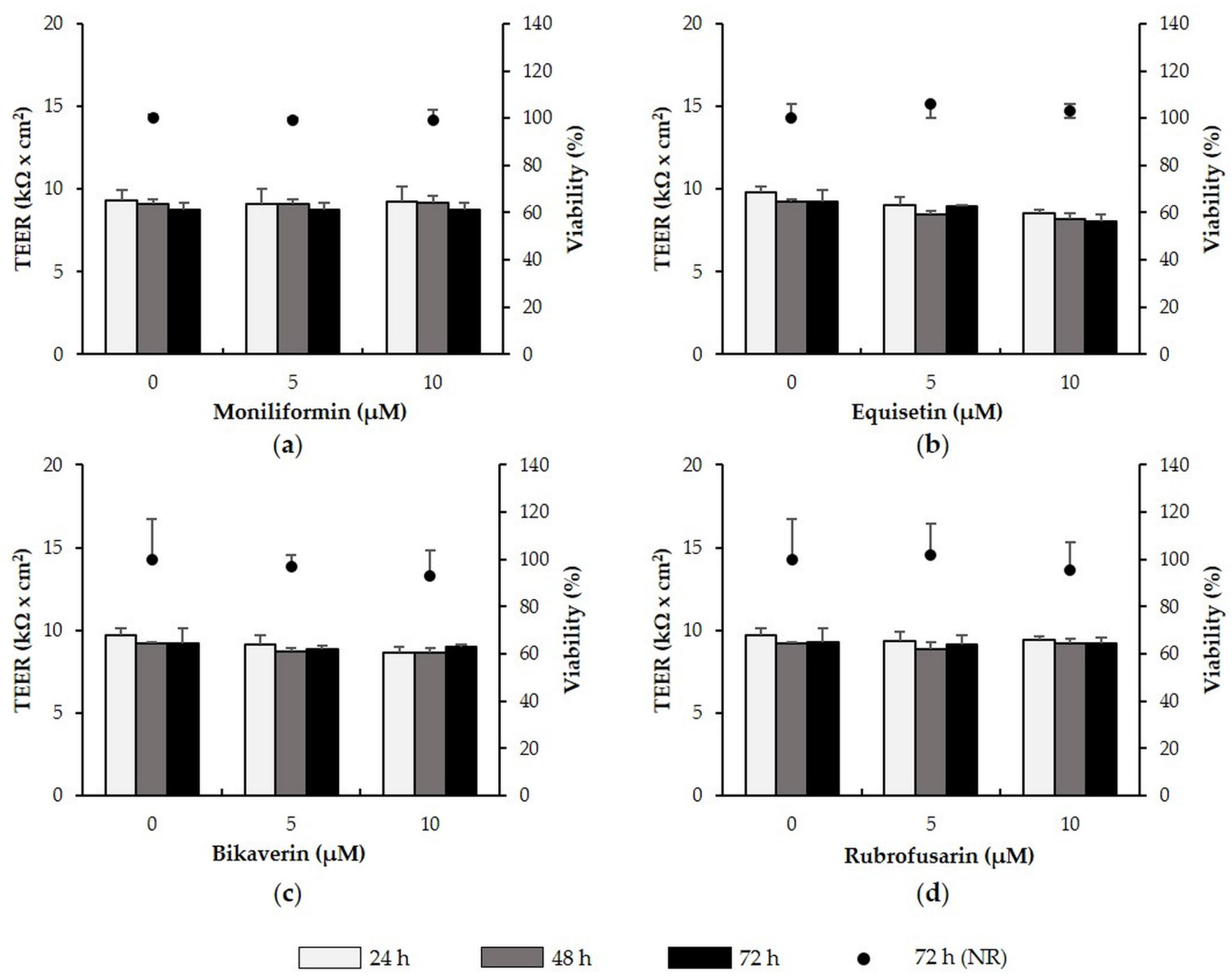
2.6. Metabolites with No Effect on TEER
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture
5.2. Chemicals
5.3. Measurement of Transepithelial Electrical Resistance (TEER)
5.4. Viability Assay
5.5. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACAT | acyl-CoA:cholesterol acyltransferase |
| BW | body weight |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | dimethyl sulfoxide |
| DON | deoxynivalenol |
| EFSA | European food safety authority |
| HDAC | histone deacetylase |
| i.p. | intraperitoneal |
| i.v. | intravenous |
| IPEC | intestinal porcine epithelial cells |
| LC-MS | liquid chromatography-mass spectrometry |
| MAPK | mitogen activated protein kinase |
| NR | neutral red |
| OD | optical density |
| TEER | transepithelial electrical resistance |
| TJ | tight junction |
References
- Frisvad, J.C.; Thrane, U.; Samson, R.A.; Pitt, J.I. Important mycotoxins and the fungi which produce them. Adv. Exp. Med. Biol. 2006, 571, 3–31. [Google Scholar] [PubMed]
- Streit, E.; Schatzmayr, G.; Tassis, P.; Tzika, E.; Marin, D.; Taranu, I.; Tabuc, C.; Nicolau, A.; Aprodu, I.; Puel, O.; et al. Current situation of mycotoxin contamination and co-occurrence in animal feed—Focus on Europe. Toxins (Basel) 2012, 4, 788–809. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Brera, C.; Crews, C.; Iha, M.H.; Krska, R.; Lattanzio, V.M.T.; MacDonald, S.; Malone, R.J.; Maragos, C.; Solfrizzo, M.; et al. Developments in mycotoxin analysis: An update for 2013–2014. World Mycotoxin J. 2015, 8, 5–35. [Google Scholar] [CrossRef]
- Berthiller, F.; Brera, C.; Crews, C.; Iha, M.H.; Krska, R.; Lattanzio, V.M.T.; MacDonald, S.; Malone, R.J.; Maragos, C.; Solfrizzo, M.; et al. Developments in mycotoxin analysis: An update for 2014–2015. World Mycotoxin J. 2016, 9, 5–30. [Google Scholar] [CrossRef]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schumacher, R. Application of an LC-MS/MS based multi-mycotoxin method for the semi-quantitative determination of mycotoxins occurring in different types of food infected by moulds. Food Chem. 2010, 119, 408–416. [Google Scholar] [CrossRef]
- Kovalsky, P.; Kos, G.; Nährer, K.; Schwab, C.; Jenkins, T.; Schatzmayr, G.; Sulyok, M.; Krska, R. Co-occurence of regulated, masked and emerging mycotoxins and secondary metabolites in finished feed and maize—A global review. Toxins 2016. Under review. [Google Scholar]
- Gruber-Dorninger, C.; Novak, B.; Nagl, V.; Berthiller, F. Emerging mycotoxins: Beyond traditionally determined food contaminants. J. Agric. Food Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Streit, E.; Schwab, C.; Sulyok, M.; Naehrer, K.; Krska, R.; Schatzmayr, G. Multi-mycotoxin screening reveals the occurrence of 139 different secondary metabolites in feed and feed ingredients. Toxins (Basel) 2013, 5, 504–523. [Google Scholar] [CrossRef] [PubMed]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Turner, J.R. Intestinal permeability defects: Is it time to treat? Clin. Gastroenterol. Hepatol. 2013, 11, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Akbari, P.; Braber, S.; Gremmels, H.; Koelink, P.J.; Verheijden, K.A.; Garssen, J.; Fink-Gremmels, J. Deoxynivalenol: A trigger for intestinal integrity breakdown. FASEB J. 2014, 28, 2414–2429. [Google Scholar] [CrossRef] [PubMed]
- Sergent, T.; Parys, M.; Garsou, S.; Pussemier, L.; Schneider, Y.J.; Larondelle, Y. Deoxynivalenol transport across human intestinal Caco-2 cells and its effects on cellular metabolism at realistic intestinal concentrations. Toxicol. Lett. 2006, 164, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Braicu, C.; Nougayrede, J.P.; Laffitte, J.; Taranu, I.; Oswald, I.P. Deoxynivalenol impairs porcine intestinal barrier function and decreases the protein expression of claudin-4 through a mitogen-activated protein kinase-dependent mechanism. J. Nutr. 2010, 140, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Nougayrede, J.P.; del Rio, J.C.; Moreno, C.; Marin, D.E.; Ferrier, L.; Bracarense, A.P.; Kolf-Clauw, M.; Oswald, I.P. The food contaminant deoxynivalenol, decreases intestinal barrier permeability and reduces claudin expression. Toxicol. Appl. Pharmacol. 2009, 237, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diesing, A.K.; Nossol, C.; Danicke, S.; Walk, N.; Post, A.; Kahlert, S.; Rothkotter, H.J.; Kluess, J. Vulnerability of polarised intestinal porcine epithelial cells to mycotoxin deoxynivalenol depends on the route of application. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Goossens, J.; Pasmans, F.; Verbrugghe, E.; Vandenbroucke, V.; De Baere, S.; Meyer, E.; Haesebrouck, F.; De Backer, P.; Croubels, S. Porcine intestinal epithelial barrier disruption by the Fusarium mycotoxins deoxynivalenol and T-2 toxin promotes transepithelial passage of doxycycline and paromomycin. BMC Vet. Res. 2012, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EFSA. Panel on Contaminants in the Food Chain (CONTAM). Scientific opinion on the risks to human and animal health related to the presence of beauvericin and enniatins in food and feed. EFSA J. 2014, 12, 3802. [Google Scholar]
- Kamyar, M.; Rawnduzi, P.; Studenik, C.R.; Kouri, K.; Lemmens-Gruber, R. Investigation of the electrophysiological properties of enniatins. Arch. Biochem. Biophys. 2004, 429, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Kouri, K.; Duchen, M.R.; Lemmens-Gruber, R. Effects of beauvericin on the metabolic state and ionic homeostasis of ventricular myocytes of the guinea pig. Chem. Res. Toxicol. 2005, 18, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Kouri, K.; Lemmens, M.; Lemmens-Gruber, R. Beauvericin-induced channels in ventricular myocytes and liposomes. Biochim. Biophys. Acta 2003, 1609, 203–210. [Google Scholar] [CrossRef]
- Meca, G.; Font, G.; Ruiz, M.J. Comparative cytotoxicity study of enniatins A, A(1), A(2), B, B(1), B(4) and J(3) on Caco-2 cells, Hep-G(2) and HT-29. Food Chem. Toxicol. 2011, 49, 2464–2469. [Google Scholar] [CrossRef] [PubMed]
- Dornetshuber, R.; Heffeter, P.; Kamyar, M.R.; Peterbauer, T.; Berger, W.; Lemmens-Gruber, R. Enniatin exerts p53-dependent cytostatic and p53-independent cytotoxic activities against human cancer cells. Chem. Res. Toxicol. 2007, 20, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Song, H.H.; Jeong, J.H.; Shin, C.G.; Choi, S.U.; Lee, C. Cytotoxicities of enniatins H, I, and MK1688 from Fusarium oxysporum KFCC 11363P. Toxicon 2008, 51, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Fornelli, F.; Minervini, F.; Logrieco, A. Cytotoxicity of fungal metabolites to lepidopteran (Spodoptera frugiperda) cell line (SF-9). J. Invertebr. Pathol. 2004, 85, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Skjerve, E.; Eriksen, G.S.; Uhlig, S. Cytotoxicity of enniatins A, A1, B, B1, B2 and B3 from Fusarium avenaceum. Toxicon 2006, 47, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Burns, A.M.; Liu, M.X.; Faeth, S.H.; Gunatilaka, A.A. Search for cell motility and angiogenesis inhibitors with potential anticancer activity: Beauvericin and other constituents of two endophytic strains of Fusarium oxysporum. J. Nat. Prod. 2007, 70, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Tonshin, A.A.; Teplova, V.V.; Andersson, M.A.; Salkinoja-Salonen, M.S. The Fusarium mycotoxins enniatins and beauvericin cause mitochondrial dysfunction by affecting the mitochondrial volume regulation, oxidative phosphorylation and ion homeostasis. Toxicology 2010, 276, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.I.; Lee, Y.J.; Chen, B.F.; Tsai, M.C.; Lu, J.L.; Chou, C.J.; Jow, G.M. Involvement of Bcl-2 family, cytochrome c and caspase 3 in induction of apoptosis by beauvericin in human non-small cell lung cancer cells. Cancer Lett. 2005, 230, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Gammelsrud, A.; Solhaug, A.; Dendele, B.; Sandberg, W.J.; Ivanova, L.; Kocbach Bolling, A.; Lagadic-Gossmann, D.; Refsnes, M.; Becher, R.; Eriksen, G.; et al. Enniatin B-induced cell death and inflammatory responses in RAW 267.4 murine macrophages. Toxicol. Appl. Pharmacol. 2012, 261, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Egge-Jacobsen, W.M.; Solhaug, A.; Thoen, E.; Faeste, C.K. Lysosomes as a possible target of enniatin B-induced toxicity in Caco-2 cells. Chem. Res. Toxicol. 2012, 25, 1662–1674. [Google Scholar] [CrossRef] [PubMed]
- Watjen, W.; Debbab, A.; Hohlfeld, A.; Chovolou, Y.; Proksch, P. The mycotoxin beauvericin induces apoptotic cell death in H4IIE hepatoma cells accompanied by an inhibition of NF-kappaB-activity and modulation of MAP-kinases. Toxicol. Lett. 2014, 231, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Prosperini, A.; Juan-Garcia, A.; Font, G.; Ruiz, M.J. Reactive oxygen species involvement in apoptosis and mitochondrial damage in Caco-2 cells induced by enniatins A, A(1), B and B(1). Toxicol. Lett. 2013, 222, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Dornetshuber, R.; Heffeter, P.; Lemmens-Gruber, R.; Elbling, L.; Marko, D.; Micksche, M.; Berger, W. Oxidative stress and DNA interactions are not involved in Enniatin- and Beauvericin-mediated apoptosis induction. Mol. Nutr. Food Res. 2009, 53, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Mallebrera, B.; Font, G.; Ruiz, M.J. Disturbance of antioxidant capacity produced by beauvericin in CHO-K1 cells. Toxicol. Lett. 2014, 226, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Tomoda, H.; Huang, X.H.; Cao, J.; Nishida, H.; Nagao, R.; Okuda, S.; Tanaka, H.; Omura, S.; Arai, H.; Inoue, K. Inhibition of acyl-CoA: Cholesterol acyltransferase activity by cyclodepsipeptide antibiotics. J. Antibiot. (Tokyo) 1992, 45, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Carrasco, Y.; Heilos, D.; Richter, L.; Sussmuth, R.D.; Heffeter, P.; Sulyok, M.; Kenner, L.; Berger, W.; Dornetshuber-Fleiss, R. Mouse tissue distribution and persistence of the food-born fusariotoxins Enniatin B and Beauvericin. Toxicol. Lett. 2016, 247, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.; Manyes, L.; Font, G.; Juan-Garcia, A. Evaluation of immunologic effect of Enniatin A and quantitative determination in feces, urine and serum on treated Wistar rats. Toxicon 2014, 87, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Manyes, L.; Escriva, L.; Serrano, A.B.; Rodriguez-Carrasco, Y.; Tolosa, J.; Meca, G.; Font, G. A preliminary study in Wistar rats with enniatin A contaminated feed. Toxicol. Mech. Methods 2014, 24, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Leitgeb, R.; Lew, H.; Khidr, R.; Böhm, J.; Zollitsch, W.; Wagner, E. Influence of Fusarium toxins on growth and carcass characteristics of turkeys. Die Bodenkult. 2000, 51, 171–178. [Google Scholar]
- Leitgeb, R.; Lew, H.; Wetscherek, W.; Böhm, J.; Quinz, A. Influence of fusariotoxins on growing and slaughtering performance of broilers. Die Bodenkult. 1999, 50, 57–66. [Google Scholar]
- Leitgeb, R.; Raffaseder, C.; Ruckenbauer, P.; Lemmens, M.; Bohm, J.; Wagner, E.; Krska, R.; Parich, A. Impact of fusarium toxins on growth and slaughter performance of broilers and turkeys. Mycotoxin Res. 2003, 19, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Zollitsch, W.; Raffaseder, C.; Böhm, J.; Wagner, W.; Leitgeb, R. Impact of the mycotoxins moniliformin and beauvericin on growh and carcass traits of broilers. Wien. Tierärztliche Mschr. 2003, 90, 238–243. [Google Scholar]
- McKee, T.C.; Bokesch, H.R.; McCormick, J.L.; Rashid, M.A.; Spielvogel, D.; Gustafson, K.R.; Alavanja, M.M.; Cardelline, J.H., 2nd; Boyd, M.R. Isolation and characterization of new anti-HIV and cytotoxic leads from plants, marine, and microbial organisms. J. Nat. Prod. 1997, 60, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Bauden, M.; Tassidis, H.; Ansari, D. In vitro cytotoxicity evaluation of HDAC inhibitor Apicidin in pancreatic carcinoma cells subsequent time and dose dependent treatment. Toxicol. Lett. 2015, 236, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Darkin-Rattray, S.J.; Gurnett, A.M.; Myers, R.W.; Dulski, P.M.; Crumley, T.M.; Allocco, J.J.; Cannova, C.; Meinke, P.T.; Colletti, S.L.; Bednarek, M.A.; et al. Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. USA 1996, 93, 13143–13147. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yan, H.; Zhuang, S. Histone deacetylases as targets for treatment of multiple diseases. Clin. Sci. (Lond.) 2013, 124, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Ahn, J.W.; Kim, H.S.; Lee, J.; Yoon, J.H. Apicidin inhibits cell growth by downregulating IGF-1R in salivary mucoepidermoid carcinoma cells. Oncol. Rep. 2015, 33, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Lee, J.; Na, Y.J.; Choi, W.S.; Lee, B.M.; Kang, K.W.; Kim, H.S. Mechanism of apicidin-induced cell cycle arrest and apoptosis in Ishikawa human endometrial cancer cells. Chem. Biol. Interact. 2009, 179, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Lee, K.R.; Kim, J.C.; Lim, S.H.; Seo, J.A.; Lee, Y.W. A hemorrhagic factor (Apicidin) produced by toxic Fusarium isolates from soybean seeds. Appl. Environ. Microbiol. 1999, 65, 126–130. [Google Scholar] [PubMed]
- Vejdovszky, K.; Warth, B.; Sulyok, M.; Marko, D. Non-synergistic cytotoxic effects of Fusarium and Alternaria toxin combinations in Caco-2 cells. Toxicol. Lett. 2015, 241, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dvorska, J.E.; Surai, P.F.; Speake, B.K.; Sparks, N.H. Effect of the mycotoxin aurofusarin on the antioxidant composition and fatty acid profile of quail eggs. Br. Poult. Sci. 2001, 42, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Dvorska, J.E.; Surai, P.F.; Speake, B.K.; Sparks, N.H. Antioxidant systems of the developing quail embryo are compromised by mycotoxin aurofusarin. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2002, 131, 197–205. [Google Scholar] [CrossRef]
- Dvorska, J.E. Effect of Dimeric Naphthoquinone Aurofusarin on Chicken Meat Quality. In Proceedings of the 21st World’s Poultry Congress, Montreal, QC, Canada, 20–24 August 2000; pp. 46–49.
- Branco, A.; Pinto, A.C.; Braz-Filho, R.; Silva, E.F.; Grynberg, N.F.; Echevarria, A. Rubrofusarin, a natural polyketide as new human topoisomerase II-α inhibitor. Rev. Bras. Farmacogn. 2008, 18, 703–708. [Google Scholar] [CrossRef]
- Desjardins, A.E.; Proctor, R.H. Molecular biology of Fusarium mycotoxins. Int. J. Food Microbiol. 2007, 119, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, D.; Kjaer, A.; Pedersen, C.; Bu’lock, J.D.; Smith, J.R. Bikaverin and norbikaverin, benzoxanthentrione pigments of Gibberella fujikuroi. J. Chem. Soc. Perkin 1 1971, 16, 2792–2797. [Google Scholar] [CrossRef] [PubMed]
- Linnemannstons, P.; Schulte, J.; del Mar Prado, M.; Proctor, R.H.; Avalos, J.; Tudzynski, B. The polyketide synthase gene pks4 from Gibberella fujikuroi encodes a key enzyme in the biosynthesis of the red pigment bikaverin. Fungal Genet. Biol. 2002, 37, 134–148. [Google Scholar] [CrossRef]
- Fuska, J.; Proksa, B.; Fuskova, A. New potential cytotoxic and antitumor substances I. In vitro effect of bikaverin and its derivatives on cells of certain tumors. Neoplasma 1975, 22, 335–338. [Google Scholar] [PubMed]
- Thiel, P.G. A molecular mechanism for the toxic action of moniliformin, a mycotoxin produced by Fusarium moniliforme. Biochem. Pharmacol. 1978, 27, 483–486. [Google Scholar] [CrossRef]
- Ficheux, A.S.; Sibiril, Y.; Le Garrec, R.; Parent-Massin, D. In vitro myelotoxicity assessment of the emerging mycotoxins Beauvericin, Enniatin b and Moniliformin on human hematopoietic progenitors. Toxicon 2012, 59, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Cetin, Y.; Bullerman, L.B. Cytotoxicity of Fusarium mycotoxins to mammalian cell cultures as determined by the MTT bioassay. Food Chem. Toxicol. 2005, 43, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Liu, T.; Vesonder, R.F. Comparative cytotoxicity of fumonisin B1 and moniliformin in chicken primary cell cultures. Mycopathologia 1995, 132, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Celik, M.; Yilmaz, S.; Aksoy, H.; Unal, F.; Yuzbasioglu, D.; Donbak, L. Evaluation of the genotoxicity of Fusarium mycotoxin moniliformin in human peripheral blood lymphocytes. Environ. Mol. Mutagen. 2009, 50, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Knasmuller, S.; Bresgen, N.; Kassie, F.; Mersch-Sundermann, V.; Gelderblom, W.; Zohrer, E.; Eckl, P.M. Genotoxic effects of three Fusarium mycotoxins, fumonisin B1, moniliformin and vomitoxin in bacteria and in primary cultures of rat hepatocytes. Mutat. Res. 1997, 391, 39–48. [Google Scholar] [CrossRef]
- Norred, W.P.; Plattner, R.D.; Vesonder, R.F.; Bacon, C.W.; Voss, K.A. Effects of selected secondary metabolites of Fusarium moniliforme on unscheduled synthesis of DNA by rat primary hepatocytes. Food Chem. Toxicol. 1992, 30, 233–237. [Google Scholar] [CrossRef]
- Cole, R.J.; Kirksey, J.W.; Cutler, H.G.; Doupnik, B.L.; Peckham, J.C. Toxin from Fusarium moniliforme: Effects on Plants and Animals. Science 1973, 179, 1324–1326. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.K.; Fitzgerald, S.D.; Rottinghaus, G.E.; Bursian, S.J.; Aulerich, R.J. Toxic effects to mink of moniliformin extracted from Fusarium fujikuroi culture material. Vet. Hum. Toxicol. 1999, 41, 1–5. [Google Scholar] [PubMed]
- Allen, N.K.; Burmeister, H.R.; Weaver, G.A.; Mirocha, C.J. Toxicity of dietary and intravenously administered moniliformin to broiler chickens. Poult. Sci. 1981, 60, 1415–1417. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, H.R.; Ciegler, A.; Vesonder, R.F. Moniliformin, a metabolite of Fusarium moniliforme NRRL 6322: Purification and toxicity. Appl. Environ. Microbiol. 1979, 37, 11–13. [Google Scholar] [PubMed]
- Kriek, N.P.; Marasas, W.F.; Steyn, P.S.; van Rensburg, S.J.; Steyn, M. Toxicity of a moniliformin-producing strain of Fusarium moniliforme var. subglutinans isolated from maize. Food Cosmet. Toxicol. 1977, 15, 579–587. [Google Scholar] [CrossRef]
- Jonsson, M.; Jestoi, M.; Nathanail, A.V.; Kokkonen, U.M.; Anttila, M.; Koivisto, P.; Karhunen, P.; Peltonen, K. Application of OECD Guideline 423 in assessing the acute oral toxicity of moniliformin. Food Chem. Toxicol. 2013, 53, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, A.J.; Ledoux, D.R.; Rottinghaus, G.E.; Bennett, G.A. The individual and combined effects of the Fusarium mycotoxins moniliformin and fumonisin B1 in turkeys. Avian Dis. 1997, 41, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Ledoux, D.R.; Bermudez, A.J.; Fritsche, K.L.; Rottinghaus, G.E. The individual and combined effects of fumonisin B1 and moniliformin on performance and selected immune parameters in turkey poults. Poult. Sci. 2000, 79, 871–878. [Google Scholar] [CrossRef] [PubMed]
- European Union. Commission Regulation (EC) No 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs; Food and Agriculture Organization of the United States: Washinghton, DC, USA, 2006. [Google Scholar]
- Jestoi, M. Emerging fusarium-mycotoxins fusaproliferin, beauvericin, enniatins, and moniliformin: A review. Crit. Rev. Food Sci. Nutr. 2008, 48, 21–49. [Google Scholar] [CrossRef] [PubMed]
- Bottalico, A.; Logrieco, A.; Ritieni, A.; Moretti, A.; Randazzo, G.; Corda, P. Beauvericin and fumonisin B1 in preharvest Fusarium moniliforme maize ear rot in Sardinia. Food Addit. Contam. 1995, 12, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Munkvold, G.; Stahr, H.M.; Logrieco, A.; Moretti, A.; Ritieni, A. Occurrence of fusaproliferin and beauvericin in Fusarium-contaminated livestock feed in Iowa. Appl. Environ. Microbiol. 1998, 64, 3923–3926. [Google Scholar] [PubMed]
- Jestoi, M.; Rokka, M.; Yli-Mattila, T.; Parikka, P.; Rizzo, A.; Peltonen, K. Presence and concentrations of the Fusarium-related mycotoxins beauvericin, enniatins and moniliformin in finnish grain samples. Food Addit. Contam. 2004, 21, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, S.; Torp, M.; Heier, B.T. Beauvericin and enniatins A, A1, B, and B1 in Norwegian grain: A survey. Food Chem. 2006, 94, 193–201. [Google Scholar] [CrossRef]
- Malachova, A.; Dzuman, Z.; Veprikova, Z.; Vaclavikova, M.; Zachariasova, M.; Hajslova, J. Deoxynivalenol, deoxynivalenol-3-glucoside, and enniatins: The major mycotoxins found in cereal-based products on the Czech market. J. Agric. Food Chem. 2011, 59, 12990–12997. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.; Ritieni, A.; Manes, J. Occurrence of Fusarium mycotoxins in Italian cereal and cereal products from organic farming. Food Chem. 2013, 141, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, M.; Gidlund, A.; Sulyok, M.; Borjesson, T.; Krska, R.; Olsen, M.; Fredlund, E. Deoxynivalenol and other selected Fusarium toxins in Swedish wheat—Occurrence and correlation to specific Fusarium species. Int. J. Food Microbiol. 2013, 167, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Mikusova, P.; Srobarova, A.; Sulyok, M.; Santini, A. Fusarium fungi and associated metabolites presence on grapes from Slovakia. Mycotoxin Res. 2013, 29, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Shimshoni, J.A.; Cuneah, O.; Sulyok, M.; Krska, R.; Galon, N.; Sharir, B.; Shlosberg, A. Mycotoxins in corn and wheat silage in Israel. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2013, 30, 1614–1625. [Google Scholar] [CrossRef] [PubMed]
- Springler, A.; Hessenberger, S.; Schatzmayr, G.; Mayer, E. Early Activation of MAPK p44/42 Is Partially Involved in DON-Induced Disruption of the Intestinal Barrier Function and Tight Junction Network. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Oswald, I.P. Effect of deoxynivalenol and other Type B trichothecenes on the intestine: A review. Toxins (Basel) 2014, 6, 1615–1643. [Google Scholar] [CrossRef] [PubMed]
- Maresca, M. From the gut to the brain: Journey and pathophysiological effects of the food-associated trichothecene mycotoxin deoxynivalenol. Toxins (Basel) 2013, 5, 784–820. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Ares, I.; Ramos, E.; Castellano, V.; Martinez, M.; Martinez-Larranaga, M.R.; Anadon, A.; Martinez, M.A. Mycotoxins modify the barrier function of Caco-2 cells through differential gene expression of specific claudin isoforms: Protective effect of illite mineral clay. Toxicology 2016, 353–354, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Naehrer, K.; Applegate, T.J. Interactive effects of dietary protein concentration and aflatoxin B1 on performance, nutrient digestibility, and gut health in broiler chicks. Poult. Sci. 2016, 95, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Marin, D.E.; Motiu, M.; Taranu, I. Food contaminant zearalenone and its metabolites affect cytokine synthesis and intestinal epithelial integrity of porcine cells. Toxins (Basel) 2015, 7, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gao, R.; Meng, Q.; Zhang, Y.; Bi, C.; Shan, A. Toxic effects of maternal zearalenone exposure on intestinal oxidative stress, barrier function, immunological and morphological changes in rats. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, J.; Padfield, P.J.; Burt, J.P.; O’Neill, C.A. Ochratoxin A increases permeability through tight junctions by removal of specific claudin isoforms. Am. J. Physiol. Cell Physiol. 2004, 287, C1412–C1417. [Google Scholar] [CrossRef] [PubMed]
- Maresca, M.; Mahfoud, R.; Pfohl-Leszkowicz, A.; Fantini, J. The mycotoxin ochratoxin A alters intestinal barrier and absorption functions but has no effect on chloride secretion. Toxicol. Appl. Pharmacol. 2001, 176, 54–63. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, J.; Lambert, D.; Padfield, P.J.; Burt, J.P.; O'Neill, C.A. The mycotoxin patulin, modulates tight junctions in caco-2 cells. Toxicol. In Vitro 2009, 23, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Mohan, H.M.; Collins, D.; Maher, S.; Walsh, E.G.; Winter, D.C.; O'Brien, P.J.; Brayden, D.J.; Baird, A.W. The mycotoxin patulin increases colonic epithelial permeability in vitro. Food Chem. Toxicol. 2012, 50, 4097–4102. [Google Scholar] [CrossRef] [PubMed]
- Bouhet, S.; Hourcade, E.; Loiseau, N.; Fikry, A.; Martinez, S.; Roselli, M.; Galtier, P.; Mengheri, E.; Oswald, I.P. The mycotoxin fumonisin B1 alters the proliferation and the barrier function of porcine intestinal epithelial cells. Toxicol. Sci. 2004, 77, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Bouhet, S.; Oswald, I.P. The intestine as a possible target for fumonisin toxicity. Mol. Nutr. Food Res. 2007, 51, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Bracarense, A.P.; Lucioli, J.; Grenier, B.; Drociunas Pacheco, G.; Moll, W.D.; Schatzmayr, G.; Oswald, I.P. Chronic ingestion of deoxynivalenol and fumonisin, alone or in interaction, induces morphological and immunological changes in the intestine of piglets. Br. J. Nutr. 2012, 107, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Verbrugghe, E.; Vandenbroucke, V.; Dhaenens, M.; Shearer, N.; Goossens, J.; De Saeger, S.; Eeckhout, M.; D’Herde, K.; Thompson, A.; Deforce, D.; et al. T-2 toxin induced Salmonella Typhimurium intoxication results in decreased Salmonella numbers in the cecum contents of pigs, despite marked effects on Salmonella-host cell interactions. Vet. Res. 2012, 43. [Google Scholar] [CrossRef] [PubMed]
- Schierack, P.; Nordhoff, M.; Pollmann, M.; Weyrauch, K.D.; Amasheh, S.; Lodemann, U.; Jores, J.; Tachu, B.; Kleta, S.; Blikslager, A.; et al. Characterization of a porcine intestinal epithelial cell line for in vitro studies of microbial pathogenesis in swine. Histochem. Cell Biol. 2006, 125, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Berschneider, H.M. Development of normal cultured small intestinal epithelial cell lines which transport Na and Cl. Gastroenterology 1989, 96, A41. [Google Scholar]
- Nossol, C.; Barta-Boszormenyi, A.; Kahlert, S.; Zuschratter, W.; Faber-Zuschratter, H.; Reinhardt, N.; Ponsuksili, S.; Wimmers, K.; Diesing, A.K.; Rothkotter, H.J. Comparing Two Intestinal Porcine Epithelial Cell Lines (IPECs): Morphological Differentiation, Function and Metabolism. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Meca, G.; Zinedine, A.; Blesa, J.; Font, G.; Manes, J. Further data on the presence of Fusarium emerging mycotoxins enniatins, fusaproliferin and beauvericin in cereals available on the Spanish markets. Food Chem. Toxicol. 2010, 48, 1412–1416. [Google Scholar] [CrossRef] [PubMed]
- Prosperini, A.; Font, G.; Ruiz, M.J. Interaction effects of Fusarium enniatins (A, A1, B and B1) combinations on in vitro cytotoxicity of Caco-2 cells. Toxicol. In Vitro 2014, 28, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.H.; Flick, J.; Levine, S.A.; Madara, J.L.; Sharp, G.W.; Donowitz, M. Regulation of tight junction resistance in T84 monolayers by elevation in intracellular Ca2+: A protein kinase C effect. J. Membr. Biol. 1996, 149, 71–79. [Google Scholar] [PubMed]
- Kan, K.S.; Coleman, R. The calcium ionophore A23187 increases the tight-junctional permeability in rat liver. Biochem. J. 1988, 256, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.L.; Lin, H.I.; Chen, B.F.; Jow, G.M. Beauvericin-induced cell apoptosis through the mitogen-activated protein kinase pathway in human nonsmall cell lung cancer A549 cells. J. Toxicol. Sci. 2016, 41, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; Ahn, S.H.; Park, S.H.; Wang, S.Y.; Bae, G.U.; Seo, D.W.; Kwon, H.K.; Hong, S.; Lee, H.Y.; Lee, Y.W.; et al. Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Res 2000, 60, 6068–6074. [Google Scholar] [PubMed]
- Krishnan, M.; Singh, A.B.; Smith, J.J.; Sharma, A.; Chen, X.; Eschrich, S.; Yeatman, T.J.; Beauchamp, R.D.; Dhawan, P. HDAC inhibitors regulate claudin-1 expression in colon cancer cells through modulation of mRNA stability. Oncogene 2010, 29, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Dvorska, J.E.; Surai, P.F.; Speake, B.K.; Sparks, N.H. Protective effect of modified glucomannans against aurofusarin-induced changes in quail egg and embryo. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2003, 135C, 337–343. [Google Scholar] [CrossRef]
- Ficheux, A.S.; Sibiril, Y.; Parent-Massin, D. Effects of beauvericin, enniatin b and moniliformin on human dendritic cells and macrophages: An in vitro study. Toxicon 2013, 71, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Speijers, G.J.; Speijers, M.H. Combined toxic effects of mycotoxins. Toxicol. Lett. 2004, 153, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kouadio, J.H.; Dano, S.D.; Moukha, S.; Mobio, T.A.; Creppy, E.E. Effects of combinations of Fusarium mycotoxins on the inhibition of macromolecular synthesis, malondialdehyde levels, DNA methylation and fragmentation, and viability in Caco-2 cells. Toxicon 2007, 49, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.J.; Macakova, P.; Juan-Garcia, A.; Font, G. Cytotoxic effects of mycotoxin combinations in mammalian kidney cells. Food Chem. Toxicol. 2011, 49, 2718–2724. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.J.; Franzova, P.; Juan-Garcia, A.; Font, G. Toxicological interactions between the mycotoxins beauvericin, deoxynivalenol and T-2 toxin in CHO-K1 cells in vitro. Toxicon 2011, 58, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Visconti, A.; Minervini, F.; Lucivero, G.; Gambatesa, V. Cytotoxic and immunotoxic effects of Fusarium mycotoxins using a rapid colorimetric bioassay. Mycopathologia 1991, 113, 181–186. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Springler, A.; Vrubel, G.-J.; Mayer, E.; Schatzmayr, G.; Novak, B. Effect of Fusarium-Derived Metabolites on the Barrier Integrity of Differentiated Intestinal Porcine Epithelial Cells (IPEC-J2). Toxins 2016, 8, 345. https://doi.org/10.3390/toxins8110345
Springler A, Vrubel G-J, Mayer E, Schatzmayr G, Novak B. Effect of Fusarium-Derived Metabolites on the Barrier Integrity of Differentiated Intestinal Porcine Epithelial Cells (IPEC-J2). Toxins. 2016; 8(11):345. https://doi.org/10.3390/toxins8110345
Chicago/Turabian StyleSpringler, Alexandra, Galina-Jacqueline Vrubel, Elisabeth Mayer, Gerd Schatzmayr, and Barbara Novak. 2016. "Effect of Fusarium-Derived Metabolites on the Barrier Integrity of Differentiated Intestinal Porcine Epithelial Cells (IPEC-J2)" Toxins 8, no. 11: 345. https://doi.org/10.3390/toxins8110345
APA StyleSpringler, A., Vrubel, G. -J., Mayer, E., Schatzmayr, G., & Novak, B. (2016). Effect of Fusarium-Derived Metabolites on the Barrier Integrity of Differentiated Intestinal Porcine Epithelial Cells (IPEC-J2). Toxins, 8(11), 345. https://doi.org/10.3390/toxins8110345