
Maternal-Fetal Cancer Risk Assessment of Ochratoxin A during Pregnancy


Abstract
:
1. Introduction
2. Results
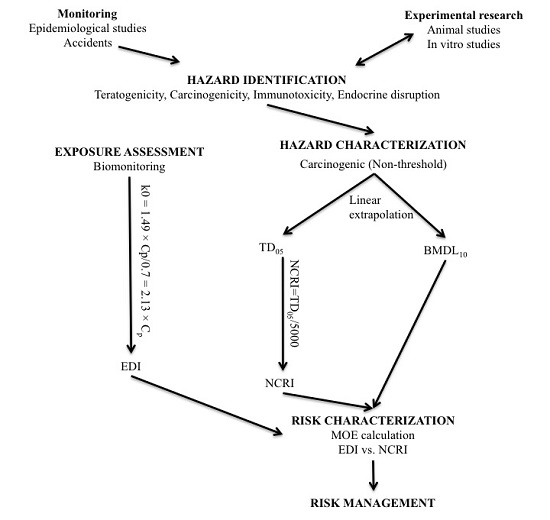
2.1. Hazard Characterization
2.1.1. Derivation of the Benchmark Dose Lower Limit (BMDL10)
2.1.2. Derivation of the Tumorigenic Dose (TD05) and Negligible Cancer Risk Intake (NCRI)
2.2. Exposure Assessment
2.3. Risk Characterization
2.3.1. Pregnant Women
2.3.2. Fetus
3. Discussion
3.1. Hazard Characterization
3.2. Maternal Risk Assessment of OTA Based on Serum OTA Level
3.3. Fetal Risk Assessment of OTA from Maternal Exposure
3.3.1. Fetal Exposure to OTA during Early Pregnancy
3.3.2. Chronic and Early-Life Exposure Poses Extra Risk to the Individual
3.4. Challenges and the Need of Fetal OTA Risk Assessment from Maternal Exposure
4. Conclusions
5. Materials and Methods
5.1. Chemicals
5.2. Sample Collection
5.3. Extraction of Serum OTA
5.4. Analysis of OTA by High Performance Liquid Chromatography
5.5. Data Management and Statistical Treatment of the Data
5.6. Risk Assessment
5.6.1. Hazard Identification
5.6.2. Hazard Characterization
5.6.3. Exposure Assessment
Pregnant Women
Fetus
5.6.4. Risk Characterization
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AIC | Akaike information criterion |
| AOAC | Association of analytical communities |
| BEN | Balkan Endemic Nephropathy |
| BMD | Benchmark dose |
| BMDL | Benchmark dose lower limit |
| BMR | Benchmark response |
| EDI | Estimated daily intake |
| EFSA | European food safety authority |
| GOF | Goodness-of-fit |
| ILSI | International life sciences institute |
| LOAEL | Lowest observed adverse effect level |
| LOD | Limit of detection |
| MOA | Mode of action |
| MOE | Margin of exposure |
| NCRI | Negligible cancer risk intake |
| NOAEL | No observed adverse effect level |
| NTP | National toxicology program |
| OTA | Ochratoxin A |
| PoD | Point of departure |
| SPE | Solid-phase extraction |
| TD | Tumorigenic dose |
| TDI | Tolerable daily intake |
| UF | Uncertainty factor |
| UTT | Urinary tract tumor |
References
- Bhat, R.V.; Miller, J.D. Mycotoxins and food supply. In Food, Nutrition and Agriculture Review 01: Food for the Future; Albert, J.L., Tucker, R., Roland, N., Gigli, H., Eds.; Food and Agriculture Organization (FAO): Rome, Italy, 1991; Volume 1. [Google Scholar]
- Kuiper-Goodman, T. Food safety: Mycotoxins and phycotoxins in perspective. In Mycotoxins and Phycotoxins: Developments in Chemistry, Toxicology, and Food Safety: Proceeding of the IX International IUPAC Symposium on Mycotoxins and Phycotoxins; Miraglia, M., van Egmond, H.P., Brera, C., Gilbert, J., Eds.; Alaken, Inc.: Fort Collins, CO, USA, 1998; pp. 25–48. [Google Scholar]
- Anderson, L.M.; Diwan, B.A.; Fear, N.T.; Roman, E. Critical windows of exposure for children’s health: Cancer in human epidemiological studies and neoplasms in experimental animal models. Environ. Health Perspect. 2000, 108, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Godschalk, R.W.; Kleinjans, J.C. Characterization of the exposure-disease continuum in neonates of mothers exposed to carcinogens during pregnancy. Basic Clin. Pharmacol. Toxicol. 2008, 102, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.P.; Jedrychowski, W.; Rauh, V.; Whyatt, R.M. Molecular epidemiologic research on the effects of environmental pollutants on the fetus. Environ. Health Perspect. 1999, 107, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Vanhees, K.; de Bock, L.; Godschalk, R.W.L.; van Schooten, F.J.; van Waalwijk van Doorn-Khosrovani, S.B. Prenatal exposure to flavonoids: Implication for cancer risk. Toxicol. Sci. 2011, 120, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Soffritti, M.; Belpoggi, F.; Tibaldi, E.; Esposti, D.D.; Lauriola, M. Life-span exposure to low doses of aspartame beginning during prenatal life increases cancer effects in rats. Environ. Health Perspect. 2007, 115, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Wangikar, P.B.; Dwivedi, P.; Sinha, N.; Sharma, A.K.; Telang, A.G. Teratogenic effects in rabbits of simultaneous exposure to Ochratoxin A and Aflatoxin B1 with special reference to microscopic effects. Toxicology 2005, 215, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Kuiper-Goodman, T.; Hilts, C.; Billiard, S.M.; Kiparissis, Y.; Richard, I.D.; Hayward, S. Health risk assessment of Ochratoxin A for all age-sex strata in a market economy. Food Addit. Contam. A 2010, 27, 212–240. [Google Scholar] [CrossRef] [PubMed]
- Boon, P.E.; Bakker, M.I.; van Klaveren, J.D.; van Rossum, C.T.M. Risk Assessment of the Dietary Exposure to Contaminants and Pesticide Residues in Young Children in the Netherlands; National Institute for Public Health and the Environment: Bilthoven, The Netherlands, 2009. [Google Scholar]
- Hallen, I.P.; Breitholtz-Emanuelsson, A.; Hult, K.; Olsen, M.; Oskarsson, A. Placental and lactational transfer of Ochratoxin A in rats. Nat. Toxins 1998, 6, 43–49. [Google Scholar] [CrossRef]
- Fukui, Y.; Hoshino, K.; Kameyama, Y.; Yasui, T.; Toda, C.; Nagano, H. Placental transfer of Ochratoxin A and its cytotoxic effect on the mouse embryonic brain. Food Chem. Toxicol. 1987, 25, 17–24. [Google Scholar] [CrossRef]
- Appelgren, L.E.; Arora, R.G. Distribution of 14C-labelled Ochratoxin A in pregnant mice. Food Chem. Toxicol. 1983, 21, 563–568. [Google Scholar] [CrossRef]
- Ballinger, M.B.; Phillips, T.D.; Kubena, L.F. Assessment of the distribution and elimination of Ochratoxin A in the pregnant rat. J. Food Saf. 1986, 8, 11–24. [Google Scholar] [CrossRef]
- Petkova-Bocharova, T.; Stoichev, I.I.; Chernozemsky, I.N.; Castegnaro, M.; Pfohl-Leszkowicz, A. Formation of DNA adducts in tissues of mouse progeny through transplacental contamination and/or lactation after administration of a single dose of ochratoxin a to the pregnant mother. Environ. Mol. Mutagen. 1998, 32, 155–162. [Google Scholar] [CrossRef]
- Zimmerli, B.; Dick, R. Determination of Ochratoxin A at the PPT level in human blood, serum, milk and some foodstuffs by high-performance liquid chromatography with enhanced fluorescence detection and immunoaffinity column cleanup: Methodology and swiss data. J. Chromatogr. B Biomed. Appl. 1995, 666, 85–99. [Google Scholar] [CrossRef]
- Postupolski, J.; Karlowski, K.; Kubik, P. Ochratoxin A in maternal and foetal blood and in maternal milk. Rocz. Panstw. Zakl. Hig. 2006, 57, 23–30. [Google Scholar] [PubMed]
- Malir, F.; Ostry, V.; Pfohl-Leszkowicz, A.; Novotna, E. Ochratoxin A: Developmental and reproductive toxicity—An overview. Birth Defects Res. B Dev. Reprod. Toxicol. 2013, 98, 493–502. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program (NTP). Toxicology and Carcinogenesis Studies of Ochratoxin A (CAS No. 303-47-9) in F344/N Rats (Gavage Studies); Department of Health and Human Services: Durham, NC, USA, 1989.
- Knaap, A.; Anderson, C.; Brantom, P.; Bridges, J.; Crebelli, R.; Greim, H.; Larsen, J.C.; McGregor, D.; Renwick, A.; Schlatter, J. Opinion of the scientific committee on a request from efsa related to a harmonised approach for risk assessment of substances which are both genotoxic and carcinogenic. EFSA J. 2005, 282, 1–31. [Google Scholar]
- Krogh, P.; Gyrd-Hansen, N.; Hald, B.; Larsen, S.; Nielsen, J.P.; Smith, M.; Ivanoff, C.; Meisner, H. Renal enzyme activities in experimental Ochratoxin A-induced porcine nephropathy: Diagnostic potential of phosphoenolpyruvate carboxykinase and gamma-glutamyl transpeptidase activity. J. Toxicol. Environ. Health 1988, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Krogh, P.; Elling, F.; Friis, C.; Hald, B.; Larsen, A.E.; Lillehoj, E.B.; Madsen, A.; Mortensen, H.P.; Rasmussen, F.; Ravnskov, U. Porcine nephropathy induced by long-term ingestion of Ochratoxin A. Vet. Pathol. 1979, 16, 466–475. [Google Scholar] [PubMed]
- Elling, F. Ochratoxin A-induced mycotoxic porcine nephropathy: Alterations in enzyme activity in tubular cells. Acta Pathol. Microbiol. Scand. A 1979, 87A, 237–243. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Bartsch, H.; Azémar, B.; Mohr, U.; Estève, J.; Castegnaro, M. Mesna protects rats against nephrotoxicity but carcinogenicity induced by Ochratoxin A, implicating two separate pathways. FU Med. Biol. 2002, 9, 57–63. [Google Scholar]
- Dourson, M.L.; Felter, S.P.; Robinson, D. Evolution of science-based uncertainty factors in noncancer risk assessment. Regul. Toxicol. Pharmacol. 1996, 24, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Kuiper-Goodman, T. Risk assessment and risk management of mycotoxins in food. In Mycotoxins in Food. Detection and Control; Magan, N., Olsen, M., Eds.; CRC Press LLC: Boca Raton, FL, USA, 2004; pp. 3–31. [Google Scholar]
- Benford, D.; Bolger, P.M.; Carthew, P.; Coulet, M.; DiNovi, M.; Leblanc, J.-C.; Renwick, A.G.; Setzer, W.; Schlatter, J.; Smith, B.; et al. Application of the margin of exposure (MOE) approach to substances in food that are genotoxic and carcinogenic. Food Chem. Toxicol. 2010, 48, S2–S24. [Google Scholar] [CrossRef] [PubMed]
- Thuvander, A.; Paulsen, J.E.; Axberg, K.; Johansson, N.; Vidnes, A.; Enghardt-Barbieri, H.; Trygg, K.; Lund-Larsen, K.; Jahrl, S.; Widenfalk, A.; et al. Levels of Ochratoxin A in blood from norwegian and swedish blood donors and their possible correlation with food consumption. Food Chem. Toxicol. 2001, 39, 1145–1151. [Google Scholar] [CrossRef]
- Biasucci, G.; Calabrese, G.; Di Giuseppe, R.; Carrara, G.; Colombo, F.; Mandelli, B.; Maj, M.; Bertuzzi, T.; Pietri, A.; Rossi, F. The presence of Ochratoxin A in cord serum and in human milk and its correspondence with maternal dietary habits. Eur. J. Nutr. 2010, 50, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, K.; Vega, M.; Rios, G.; Muñoz, S.; Madariaga, R. Preliminary study of Ochratoxin A in human plasma in agricultural zones of chile and its relation to food consumption. Food Chem. Toxicol. 2006, 44, 1884–1889. [Google Scholar] [CrossRef] [PubMed]
- Miraglia, M.; Brera, C.; Colatosti, M. Application of biomarkers to assessment of risk to human health from exposure to mycotoxins. Microchem. J. 1996, 54, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Brereton, P.; MacDonald, S. Assessment of dietary exposure to Ochratoxin A in the UK using a duplicate diet approach and analysis of urine and plasma samples. Food Addit. Contam. 2001, 18, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Breitholtz, A.; Olsen, M.; Dahlback, A.; Hult, K. Plasma Ochratoxin A levels in three swedish populations surveyed using an ion-pair HPLC technique. Food Addit. Contam. 1991, 8, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Merlo, D.F.; Wild, C.P.; Kogevinas, M.; Kyrtopoulos, S.; Kleinjans, J. Newgeneris: A european study on maternal diet during pregnancy and child health. Cancer Epidemiol. Biomark. Prev. 2009, 18, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Vähäkangas, K.; Myllynen, P. Experimental methods to study human transplacental exposure to genotoxic agents. Mutat. Res. 2006, 608, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.S.; Partanen, H.; Myllynen, P.; Vahakangas, K.; El-Nezami, H. Fate of the teratogenic and carcinogenic Ochratoxin A in human perfused placenta. Toxicol. Lett. 2012, 208, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Appelgren, L.E.; Arora, R.G. Distribution studies of 14C-labelled aflatoxin B1 and Ochratoxin A in pregnant mice. Vet. Res. Commun. 1983, 7, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A.; Cooper, C.; Thornburg, K.L. Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 2008, 359, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Pallapies, D. Trends in childhood disease. Mutat. Res. 2006, 608, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Loebstein, R.; Lalkin, A.; Koren, G. Pharmacokinetic changes during pregnancy and their clinical relevance. Clin. Pharmacokinet. 1997, 33, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.J. Drug disposition in mother and foetus. Clin. Exp. Pharmacol Physiol. 1997, 24, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Steyn, P.S. Mycotoxins, general view, chemistry and structure. Toxicol. Lett. 1995, 82–83, 843–851. [Google Scholar] [CrossRef]
- Miraglia, M.; Brera, C. Task 3.2.7 Assessment of Dietary Intake of Ochratoxin A by the Population of EU Member States; Istituto Superiore di Sanità: Rome, Italy, 2002; pp. 1–153. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.M. Biomarkers of human exposure to Ochratoxin A. Food Addit. Contam. 2005, 22, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.C.; Pena, A.; Lino, C.M. Human Ochratoxin A biomarkers—From exposure to effect. Crit. Rev. Toxicol. 2011, 41, 187–212. [Google Scholar] [CrossRef] [PubMed]
- Ghali, R.; Hmaissia-Khlifa, K.; Mezigh, C.; Ghorberl, H.; Maaroufi, K.; Maschgoul, S.; Hedilli, A. HPLC determination of Ochratoxin A in a low volume of human blood serum. Anal. Lett. 2008, 41, 757–766. [Google Scholar] [CrossRef]
- World Health Organization (WHO). IPCS Risk Assessment Terminology; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Lock, E.A.; Hard, G.C. Chemically induced renal tubule tumors in the laboratory rat and mouse: Review of the NCI/NTP database and categorization of renal carcinogens based on mechanistic information. Crit. Rev. Toxicol. 2004, 34, 211–299. [Google Scholar] [CrossRef] [PubMed]
- Pfohl-Leszkowicz, A.; Petkova-Bocharova, T.; Chernozemsky, I.N.; Castegnaro, M. Balkan endemic nephropathy and associated urinary tract tumours: A review on aetiological causes and the potential role of mycotoxins. Food Addit. Contam. 2002, 19, 282–302. [Google Scholar] [CrossRef] [PubMed]
- Pfohl-Leszkowicz, A.; Tozlovanu, M.; Manderville, R.; Peraica, M.; Castegnaro, M.; Stefanovic, V. New molecular and field evidences for the implication of mycotoxins but not aristolochic acid in human nephropathy and urinary tract tumor. Mol. Nutr. Food Res. 2007, 51, 1131–1146. [Google Scholar] [CrossRef] [PubMed]
- Pfohl-Leszkowicz, A. Ochratoxin A and aristolochic acid involvement in nephropathies and associated urothelial tract tumours. Arh. Hig. Rada Toksikol. 2009, 60, 465–483. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G. Hypothesis: Does Ochratoxin A cause testicular cancer? Cancer Causes Control 2002, 13, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Jennings-Gee, J.E.; Tozlovanu, M.; Manderville, R.; Miller, M.S.; Pfohl-Leszkowicz, A.; Schwartz, G.G. Ochratoxin A: In utero exposure in mice induces adducts in testicular DNA. Toxins 2010, 2, 1428–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Kuan, M.; Cavin, C.; Delatour, T.; Schilter, B. Ochratoxin A carcinogenicity involves a complex network of epigenetic mechanisms. Toxicon 2008, 52, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Cavin, C.; Delatour, T.; Marin-Kuan, M.; Fenaille, F.; Holzhäuser, D.; Guignard, G.; Bezençon, C.; Piguet, D.; Parisod, V.; Richoz-Payot, J.; et al. Ochratoxin A-mediated DNA and protein damage: Roles of nitrosative and oxidative stresses. Toxicol. Sci. 2009, 110, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Mally, A. Ochratoxin A and mitotic disruption: Mode of action analysis of renal tumor formation by Ochratoxin A. Toxicol. Sci. 2012, 127, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Manderville, R.A. A case for the genotoxicity of Ochratoxin A by bioactivation and covalent DNA adduction. Chem. Res. Toxicol. 2005, 18, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Manderville, R.A.; Pfohl-Leszkowicz, A. Bioactivation and DNA adduction as a rationale for Ochratoxin A carcinogenesis. World Mycotoxin J. 2008, 1, 357–367. [Google Scholar] [CrossRef]
- Malaveille, C.; Brun, G.; Bartsch, H. Genotoxicity of Ochratoxin A and structurally related compounds in escherichia coli strains: Studies on their mode of action. IARC Sci. Publ. 1991, 261–266. [Google Scholar]
- Gillman, I.G.; Clark, T.N.; Manderville, R.A. Oxidation of Ochratoxin A by an FE-porphyrin system: Model for enzymatic activation and DNA cleavage. Chem. Res. Toxicol. 1999, 12, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.S.; Monks, T.J.; Everitt, J.I.; Kleymenova, E.; Walker, C.L. Carcinogenicity of a nephrotoxic metabolite of the “nongenotoxic” carcinogen hydroquinone. Chem. Res. Toxicol. 2001, 14, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Mally, A.; Dekant, W. DNA adduct formation by Ochratoxin A: Review of the available evidence. Food Addit. Contam. 2005, 22, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Mantle, P.G.; Faucet-Marquis, V.; Manderville, R.A.; Squillaci, B.; Pfohl-Leszkowicz, A. Structures of covalent adducts between DNA and Ochratoxin A: A new factor in debate about genotoxicity and human risk assessment. Chem. Res. Toxicol. 2010, 23, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Stoev, S.D. Studies on carcinogenic and toxic effects of ochratoxin a in chicks. Toxins 2010, 2, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Follmann, W.; Lucas, S. Effects of the mycotoxin Ochratoxin A in a bacterial and a mammalian in vitro mutagenicity test system. Arch. Toxicol. 2003, 77, 298–304. [Google Scholar] [PubMed]
- Pfohl-Leszkowicz, A.; Castegnaro, M. Further arguments in favour of direct covalent binding of Ochratoxin A (OTA) after metabolic biotransformation. Food Addit. Contam. 2005, 22, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Turesky, R.J. Perspective: Ochratoxin A is not a genotoxic carcinogen. Chem. Res. Toxicol. 2005, 18, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Tozlovanu, M.; Faucet-Marquis, V.; Pfohl-Leszkowicz, A.; Manderville, R.A. Genotoxicity of the hydroquinone metabolite of Ochratoxin A: Structure-activity relationships for covalent DNA adduction. Chem. Res. Toxicol. 2006, 19, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Pfohl-Leszkowicz, A.; Gabryelski, W.; Manderville, R.A. Formation of 2′-deoxyguanosine-carbon 8-bound ochratoxin a adduct in rat kidney DNA. Mol. Nutr. Food Res. 2009, 53, 154–155. [Google Scholar] [CrossRef] [PubMed]
- Hadjeba-Medjdoub, K.; Tozlovanu, M.; Pfohl-Leszkowicz, A.; Frenette, C.; Paugh, R.J.; Manderville, R.A. Structure-activity relationships imply different mechanisms of action for ochratoxin A-mediated cytotoxicity and genotoxicity. Chem. Res. Toxicol. 2012, 25, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as a molecular mechanism of ochratoxin A carcinogenicity. Chem. Res. Toxicol. 2012, 25, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Bolt, H.M.; Foth, H.; Hengstler, J.G.; Degen, G.H. Carcinogenicity categorization of chemicals—New aspects to be considered in a european perspective. Toxicol. Lett. 2004, 151, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Lima, B.S.; van der Laan, J.W. Mechanisms of nongenotoxic carcinogenesis and assessment of the human hazard. Regul. Toxicol. Pharmacol. 2000, 32, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Bendele, A.M.; Carlton, W.W.; Krogh, P.; Lillehoj, E.B. Ochratoxin A carcinogenesis in the (C57BL/6J × C3H)F1 mouse. J. Natl. Cancer Inst. 1985, 75, 733–742. [Google Scholar] [PubMed]
- Boorman, G.A.; McDonald, M.R.; Imoto, S.; Persing, R. Renal lesions induced by Ochratoxin A exposure in the F344 rat. Toxicol. Pathol. 1992, 20, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Piekkola, S.; Turner, P.C.; Abdel-Hamid, M.; Ezzat, S.; El-Daly, M.; El-Kafrawy, S.; Savchenko, E.; Poussa, T.; Woo, J.C.S.; Mykkanen, H.; et al. Characterisation of aflatoxin and deoxynivalenol exposure among pregnant egyptian women. Food Addit. Contam. Part A 2012, 29, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Polychronaki, N.; Turner, C.P.; Mykkanen, H.; Gong, Y.; Amra, H.; Abdel-Wahhab, M.; El-Nezami, H. Determinants of aflatoxin M1 in breast milk in a selected group of egyptian mothers. Food Addit. Contam. 2006, 23, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Zohri, A.A.; Abdel-Gawad, K.M. Survey of mycoflora and mycotoxins of some dried fruits in egypt. J. Basic Microbiol. 1993, 33, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Abd Alla, E.-S. Natural occurrence of Ochratoxin A and citrinin in food stuffs in Egypt. Mycotoxin Res. 1996, 12, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Hagelberg, S.; Hult, K.; Fuchs, R. Toxicokinetics of Ochratoxin A in several species and its plasma-binding properties. J. Appl. Toxicol. 1989, 9, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Schlatter, C.; Studer-Rohr, J.; Rasonyi, T. Carcinogenicity and kinetic aspects of Ochratoxin A. Food Addit. Contam. 1996, 13, 43–44. [Google Scholar] [PubMed]
- Frederiksen, M.C. Physiologic changes in pregnancy and their effect on drug disposition. Semin. Perinatol. 2001, 25, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Parry, E.; Shields, R.; Turnbull, A.C. Transit time in the small intestine in pregnancy. J. Obstet. Gynaecol. Br. Commonw. 1970, 77, 900–901. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.N.; Murray, F.A. Gastric function in pregnancy. J. Obstet. Gynaecol. Br. Commonw. 1958, 65, 78–83. [Google Scholar] [CrossRef]
- Mattison, D.R.; Blann, E.; Malek, A. Physiological alterations during pregnancy: Impact on toxicokinetics. Fundam. Appl. Toxicol. 1991, 16, 215–218. [Google Scholar] [CrossRef]
- Anger, G.J.; Costantine, M.M.; Piquette-Miller, M. Chapter 4—Pharmacokinetics in pregnancy. In Reproductive and Developmental Toxicology; Ramesh, C.G., Ed.; Academic Press: San Diego, CA, USA, 2011; pp. 39–45. [Google Scholar]
- Dunlop, W.; Davison, J.M. 1 Renal haemodynamics and tubular function in human pregnancy. Baillieres Clin. Obstet. Gynaecol. 1987, 1, 769–787. [Google Scholar] [CrossRef]
- Loebstein, R.; Koren, G. Clinical relevance of therapeutic drug monitoring during pregnancy. Ther. Drug Monit. 2002, 24, 15–22. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Dose (μg OTA/kg bw/Day) | Number of Animals in the Dose Group | Number of Animals with Renal Carcinomas | Number of Animals with Renal Adenomas | Number of Animals with Both Renal Carcinomas and Adenomas |
|---|---|---|---|---|
| 0 | 50 | 0 | 1 (2%) | 1 (2%) |
| 21 | 51 | 0 | 1 (2%) | 1 (2%) |
| 70 | 51 | 16 (31%) | 6 (12%) | 20 (39%) |
| 210 | 50 | 30 (60%) | 10 (20%) | 36 (72%) |
| Model | Number of Parameters | Likelihood Ratio Test | Akaike Information Criterion (AIC) | Goodness of Fit (GOF) Test | BMD10 (μg/kg bw/Day) | BMDL10 (μg/kg bw/Day) | ||
|---|---|---|---|---|---|---|---|---|
| Log-Likelihood | p-Value | χ2 | p-Value | |||||
| Null | 1 | −121.11 | - | - | - | - | - | - |
| Logistic | 2 | −82.22 | <0.01 | 168.4 | 16.33 | <0.01 | 52.4 | 42.9 |
| Probit | 2 | −81.32 | <0.01 | 166.6 | 15.02 | <0.01 | 49.2 | 40.8 |
| Quantal-linear | 2 | −77.88 | 0.01 | 159.8 | 6.14 | 0.05 | 18.6 | 14.8 |
| Gamma multi-hit | 3 | −76.43 | 0.02 | 158.9 | 5.01 | 0.03 | 31.0 | 18.2 |
| Multistage | 3 | −77.39 | 0.01 | 160.8 | 6.10 | 0.01 | 24.4 | 15.5 |
| LogLogistic | 3 | −75.64 | 0.05 | 157.3 | 3.55 | 0.06 | 31.9 | 20.7 |
| LogProbit | 3 | −75.10 | 0.09 | 156.2 | 2.70 | 0.10 | 33.2 | 22.5 |
| Weibull | 3 | −76.76 | 0.01 | 159.5 | 5.38 | 0.02 | 28.7 | 16.9 |
| Full | 4 | −73.63 | - | - | - | - | - | - |
| Exposure | High | Median | Low |
|---|---|---|---|
| Pregnant women | |||
| Serum Level | 1.53 ng/mL | 0.26 ng/mL | 0.20 ng/mL |
| Estimated Daily Intake (EDI) | 3.26 ng/kg bw/day | 0.55 ng/kg bw/day | 0.43 ng/kg bw/day |
| Fetus | |||
| Serum Level | 3.06 ng/mL | 0.52 ng/mL | 0.40 ng/mL |
| EDI | 6.52 ng/kg bw/day | 1.10 ng/kg bw/day | 0.86 ng/kg bw/day |
| Exposure | High | Median | Low |
|---|---|---|---|
| Pregnant women | |||
| MOE | 4.9 × 103 | 2.9 × 104 | 3.7 × 104 |
| NCRI comparison | <NCRI | <NCRI | <NCRI |
| Fetus | |||
| MOE | 2.5 × 103 | 1.5 × 104 | 1.9 × 104 |
| NCRI comparison | >NCRI | <NCRI | <NCRI |
| Uncertainty Factor | Reason (References) |
|---|---|
| 10 | Intraspecies difference [25]. |
| 25 | Interspecies difference based on half-life difference between pigs and human with the same route of exposure (oral) [9]. |
| 10 | Lowest Observed Adverse Effect Level (LOAEL) → No Observed Adverse Effect Level (NOAEL) |
| In the absence of NOAEL, UF of 3 should be applied if the LOAEL is of sufficient quality. Because of the small number of animals per group, a more conservative UF up to 10 is reasonably applied [25]. | |
| 1–10 | Highly susceptible life-stages such as childhood and pregnancy [26]. |
| >2500 | If all of the above taken on account. |
| Characteristics | Threshold | Non-Threshold | OTA | References |
|---|---|---|---|---|
| Species | Often only in single species | Not restricted to single species | Shown in two species | [19,75] |
| Sex | Single | Both | Both sexes | [19] |
| Site | Single | Multiple | Liver and kidney (both sex) Breast (female). | [19] |
| Tumorigenic potency | Low | High | Unusual high incidence of renal cell carcinomas (60%) a | [49,76] |
| Ratio of carcinomas to adenomas | Low | High | High | [19] |
| Aggressiveness | Mutation frequency similar to spontaneous tumors | Rapid progression | High degree of atypia, rapid progression, large size (2–6.5 cm) b, and invasive | [19,76] |
| Often bilateral and multiple | ||||
| High cytoplasmic atypia; invasive | ||||
| Metastases | Rare | More common | Common c | [19] |
| Lifespan | Tumors do not reduce lifespan | Tumors reduce lifespan | Decreased survival rates in the mid- and high-dose groups d | [76] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woo, C.S.J.; El-Nezami, H. Maternal-Fetal Cancer Risk Assessment of Ochratoxin A during Pregnancy. Toxins 2016, 8, 87. https://doi.org/10.3390/toxins8040087
Woo CSJ, El-Nezami H. Maternal-Fetal Cancer Risk Assessment of Ochratoxin A during Pregnancy. Toxins. 2016; 8(4):87. https://doi.org/10.3390/toxins8040087
Chicago/Turabian StyleWoo, Chit Shing Jackson, and Hani El-Nezami. 2016. "Maternal-Fetal Cancer Risk Assessment of Ochratoxin A during Pregnancy" Toxins 8, no. 4: 87. https://doi.org/10.3390/toxins8040087
APA StyleWoo, C. S. J., & El-Nezami, H. (2016). Maternal-Fetal Cancer Risk Assessment of Ochratoxin A during Pregnancy. Toxins, 8(4), 87. https://doi.org/10.3390/toxins8040087