
Docking Simulation of the Binding Interactions of Saxitoxin Analogs Produced by the Marine Dinoflagellate Gymnodinium catenatum to the Voltage-Gated Sodium Channel Nav1.4

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
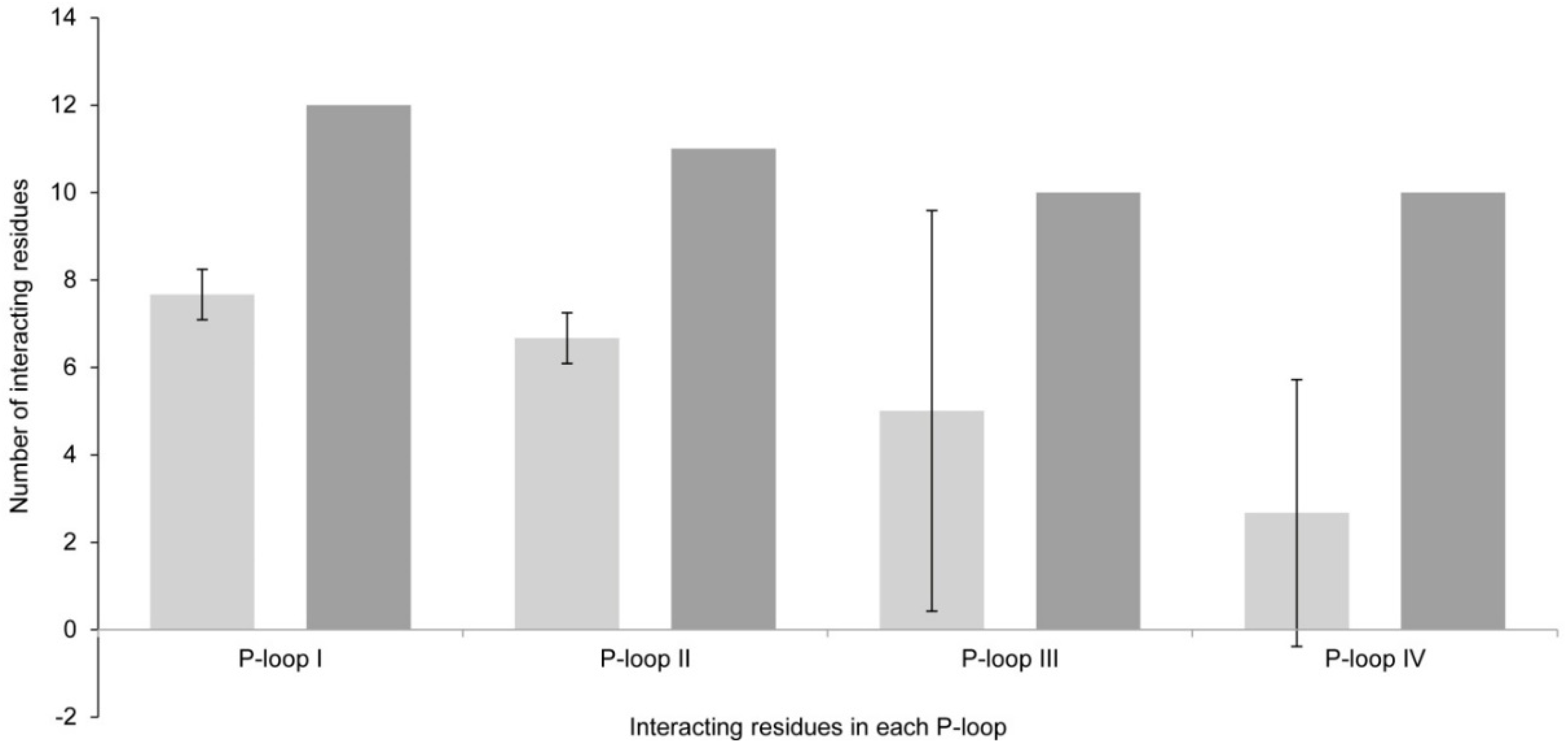
2.1. Interaction of STX and Analogs with Nav1.4 Amino Acid Residues
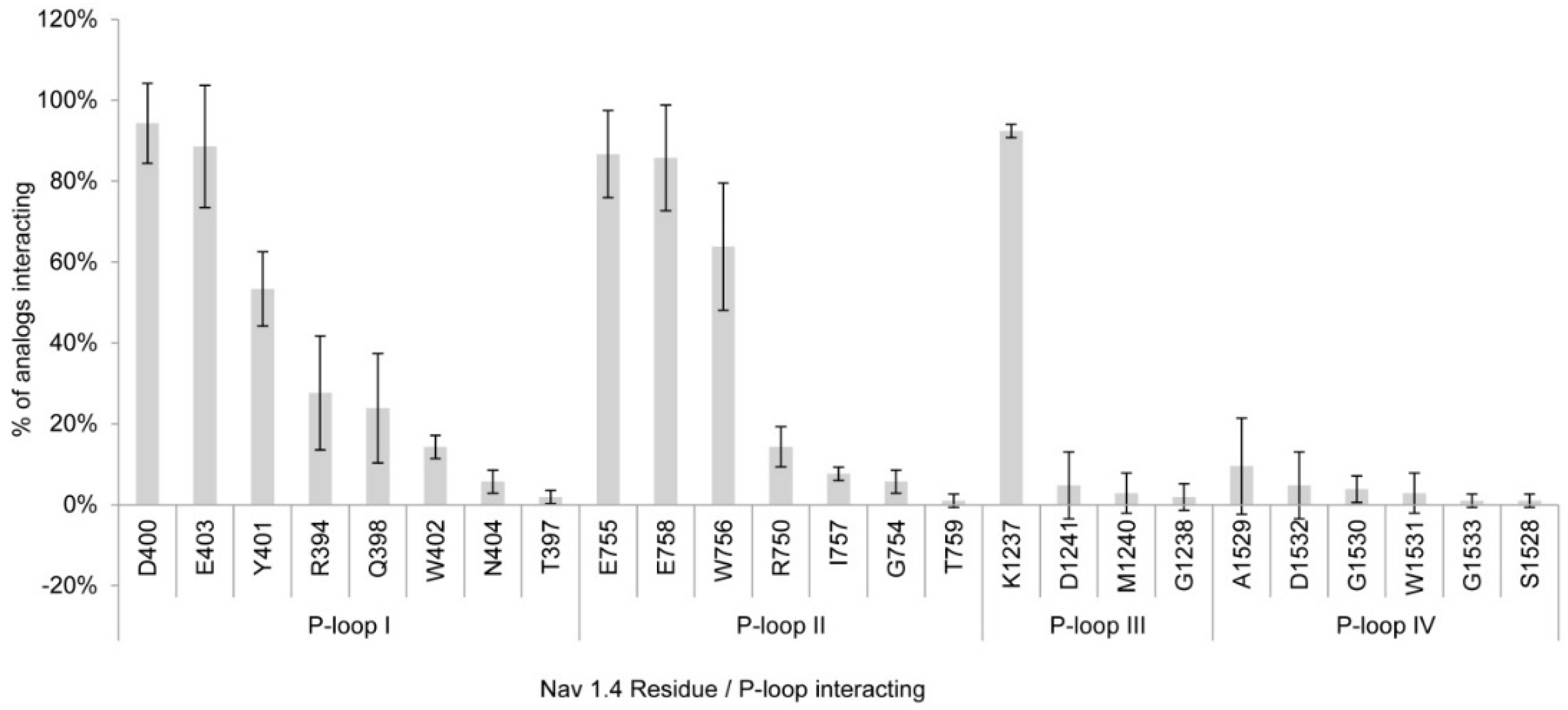
2.1.1. Nav1.4 Model 1
2.1.2. Nav1.4 Model 2
2.1.3. Comparison between Model 1 and Model 2
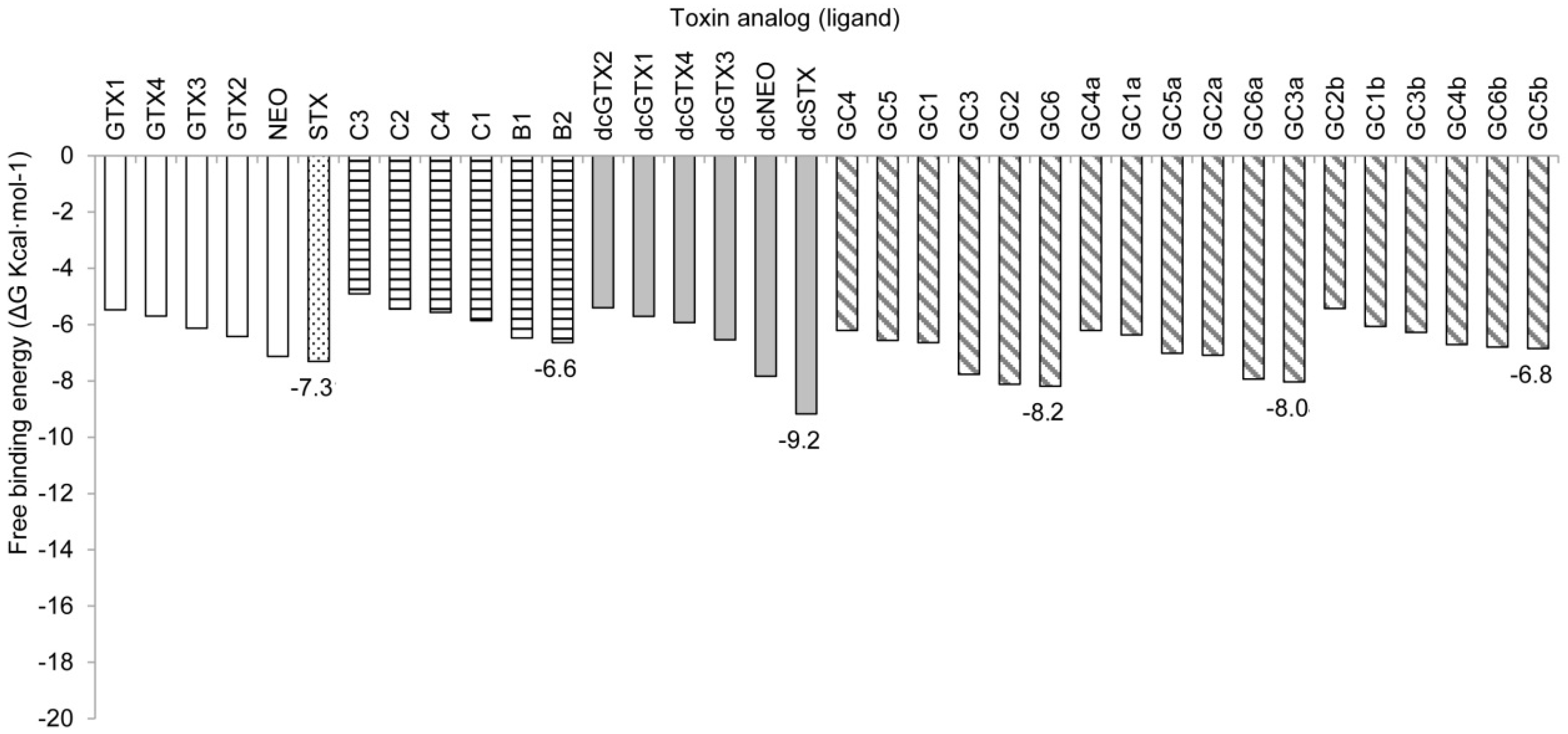
2.2. Binding Free Energy
2.2.1. Nav1.4 Model 1
2.2.2. Nav1.4 Model 2
3. Discussion
3.1. Interactions with Nav Residues
3.2. Free-Binding Energy and Specific Toxicity
4. Conclusions
5. Materials and Methods
5.1. Voltage-Gated Sodium Channel Models
5.2. Molecular Docking Simulations
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Nav | voltage-gated sodium channel |
| PSTs | paralytic shellfish toxins |
| PSP | paralytic shellfish poisoning |
| TTX | tetrodotoxin |
References
- Llewellyn, L.E. Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat. Prod. Rep. 2006, 23, 200–222. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.; Walker, S. Active groups of saxitoxin and tetrodotoxin as deduced from actions of saxitoxin analogues on frog muscle and squid axon. J. Physiol. 1982, 323, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M. Toxic red tides and harmful algal blooms: A practical challenge in coastal oceanography. Rev. Geophys. 1995, 33, 1189–1200. [Google Scholar] [CrossRef]
- Cembella, A. Ecophysiology and metabolism of paralytic shellfish toxins in marine microalgae. In Physiological Ecology of Harmful Algal Blooms; Anderson, D.M., Cembella, A.D., Hallegraeff, G.M., Eds.; NATO-Advanced Study Institute Series; Springer-Verlag: Berlin, Germany, 1998; pp. 381–404. [Google Scholar]
- Oshima, Y.; Hasegawa, M.; Yasumoto, T.; Hallegraeff, G.; Blackburn, S. Dinoflagellate Gymnodinium catenatum as the source of paralytic shellfish toxins in Tasmanian shellfish. Toxicon 1987, 25, 1105–1111. [Google Scholar] [CrossRef]
- Yotsu-Yamashita, M.; Kim, Y.H.; Dudley, S.C., Jr.; Choudhary, G.; Pfahnl, A.; Oshima, Y.; Daly, J.W. The structure of zetekitoxin AB, a saxitoxin analog from the panamanian golden frog Atelopus zeteki: A potent sodium-channel blocker. Proc. Natl. Acad. Sci. USA 2004, 101, 4346–4351. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Marine biotoxins in shellfish—Saxitoxin group scientific opinion of the panel on contaminants in the food chain. EFSA J. 2009, 1019, 1–3. [Google Scholar]
- Negri, A.; Stirling, D.; Quilliam, M.; Blackburn, S.; Bolch, C.; Burton, I.; Eaglesham, G.; Thomas, K.; Walter, J.; Willis, R. Three novel hydroxybenzoate saxitoxin analogues isolated from the dinoflagellate Gymnodinium catenatum. Chem. Res. Toxicol. 2003, 16, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.P.; Bolch, C.J.S.; Geier, S.; Green, D.H.; Park, T.-G.; Blackburn, S.I. Widespread presence of hydrophobic paralytic shellfish toxins in Gymnodinium catenatum. Harmful Algae 2007, 6, 774–780. [Google Scholar] [CrossRef]
- Vale, P. Complex profiles of hydrophobic paralytic shellfish poisoning compounds in Gymnodinium catenatum identified by liquid chromatography with fluorescence detection and mass spectrometry. J. Chromatogr. A 2008, 1195, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Bustillos-Guzmán, J.; Vale, P.; Band-Schmidt, C.J. Presence of benzoate type toxins in Gymnodinium catenatum Graham isolated from the Mexican Pacific. Toxicon 2011, 57, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Durán-Riveroll, L.M.; Peralta-Cruz, J.; Bustillos-Guzmán, J.J.; Band-Schmidt, C.J. Presencia de toxinas tipo benzoato en una cepa de Gymnodinium catenatum (Dinophyceae) aislada de Manzanillo, Colima, México. Hidrobiológica 2013, 23, 169–175. [Google Scholar]
- Vale, P. Hydrolysis of hydroxybenzoate saxitoxin analogues originating from Gymnodinium catenatum. Food Chem. 2011, 125, 1160–1165. [Google Scholar] [CrossRef]
- Vale, P. New saxitoxin analogues in the marine environment: Developments in toxin chemistry, detection and biotransformation during the 2000s. Phytochem. Rev. 2010, 9, 525–535. [Google Scholar] [CrossRef]
- Choudhary, G.; Aliste, M.P.; Tieleman, D.P.; French, R.J.; Dudley, J.; Samuel, C. Docking of μ-conotoxin GIIIA in the sodium channel outer vestibule. Channels 2007, 1, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Guy, H.R.; Conti, F. Pursuing the structure and function of voltage-gated channels. Trends Neurosci. 1990, 13, 201–206. [Google Scholar] [PubMed]
- Llewellyn, L.; Negri, A.; Quilliam, M. High affinity for the rat brain sodium channel of newly discovered hydroxybenzoate saxitoxin analogues from the dinoflagellate Gymnodinium catenatum. Toxicon 2004, 43, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.; Kuyucak, S. Molecular dynamics study of binding of µ-conotoxin GIIIA to the voltage-gated sodium channel Nav1.4. PLoS ONE 2014, 9, e105300. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Suzuki, H.; Numa, S.; Stühmer, W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 1989, 259, 213–216. [Google Scholar] [CrossRef]
- Terlau, H.; Heinemann, S.H.; Stühmer, W.; Pusch, M.; Conti, F.; Imoto, K.; Numa, S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991, 293, 93–96. [Google Scholar] [CrossRef]
- Tubaro, A.; Sosa, S.; Hungerford, J. Toxicology and Diversity of Marine Toxins; Elsevier: New York, NY, USA, 2012; pp. 896–936. [Google Scholar]
- Bello, M.; Martínez-Archundia, M.; Correa-Basurto, J. Automated docking for novel drug discovery. Expert Opin. Drug Discov. 2013, 8, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Arulalapperumal, V.; Sakkiah, S.; Thangapandian, S. Ligand based pharmacophore identification and molecular docking studies for Grb2 inhibitors. Korean Chem. Soc. 2012, 33, 1707. [Google Scholar] [CrossRef]
- Munday, R. Toxicology of seafood toxins: A critical review. In Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection; CRC PRESS: Boca Raton, FL, USA, 2014; p. 197. [Google Scholar]
- Brooijmans, N. Docking methods, ligand design, and validating data sets in the structural genomic era. In Structural Bioinformatics; John Wiley and Sons Inc.: New York, NY, USA, 2009; pp. 635–663. [Google Scholar]
- Gordon, D.; Chen, R.; Chung, S.-H. Computational methods of studying the binding of toxins from venomous animals to biological ion channels: Theory and applications. Physiol. Rev. 2013, 93, 767–802. [Google Scholar] [CrossRef] [PubMed]
- Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins 2002, 47, 409–443. [Google Scholar] [CrossRef] [PubMed]
- Brooijmans, N.; Kuntz, I.D. Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 335–373. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Ulucan Acan, Ö. Molecular Modeling of Biomolecular Associations and Quantifying Allosteric Effects. Ph.D. Thesis, Universität des Saarlandes, Saarbrücken, Germany, 2015. [Google Scholar]
- Choudhary, G.; Shang, L.; Li, X.; Dudley, S.C. Energetic localization of saxitoxin in its channel binding site. Biophys. J. 2002, 83, 912–919. [Google Scholar] [CrossRef]
- Lee, H.; Lee, G.; Jeon, J.; Cho, M. Vibrational spectroscopic determination of local solvent electric field, solute-solvent electrostatic interaction energy, and their fluctuation amplitudes. J. Phys. Chem. A 2011, 116, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Uppalapati, S.R.; Kingston, J.J.; Qureshi, I.A.; Murali, H.S.; Batra, H.V. In Silico, in vitro and in vivo analysis of binding affinity between N and C-domains of Clostridium perfringens alpha toxin. PLoS ONE 2013, 8, e82024. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.; Wekell, M.; Kentalla, L. Application of HPLC for the determination of PSP toxins in shellfish. J. Food Sci. 1985, 50, 26–29. [Google Scholar] [CrossRef]
- Oshima, Y. Post-column derivatization HPLC methods for paralytic shellfish poisons. In Manual on Harmful Marine Microalgae; Hallegraeff, G.M., Anderson, D.M., Cembella, A.D., Eds.; United Nations Educational: Paris, France, 1995; pp. 81–94. [Google Scholar]
- Chen, R.; Chung, S.H. Binding modes of μ-conotoxin to the bacterial sodium channel (NavAb). Biophys. J. 2012, 102, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Fayer, M.D. Dynamics of water interacting with interfaces, molecules, and ions. Acc. Chem. Res. 2011, 45, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, L.E. Predictive toxinology: An initial foray using calculated molecular descriptors to describe toxicity using saxitoxins as a model. Toxicon 2007, 50, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Alfonso, A.; Vieytes, M.R.; Botana, L.M. Evaluation of toxicity equivalent factors of paralytic shellfish poisoning toxins in seven human sodium channels types by an automated high throughput electrophysiology system. Arch. Toxicol. 2016, 90, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Lipkind, G.M.; Fozzard, H.A. Kcsa crystal structure as framework for a molecular model of the Na+ channel pore. Biochemistry 2000, 39, 8161–8170. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. Gaussview, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Montgomery, J., Jr.; Vreven, T.; Kudin, K.; Burant, J. Gaussian 03, revision C. 02; Gaussian: Wallingford, CT, USA, 2004. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Catterall, W.A.; Cestele, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]












| Toxin Analog | Relative Toxicity |
|---|---|
| STX | 1.0 |
| NEO | 1.0 |
| GTX1 | 1.0 |
| GTX2 | 0.4 |
| GTX3 | 0.6 |
| GTX4 | 0.7 |
| dcSTX | 1.0 |
| dcGTX1 | 0.5 * |
| dcGTX2 | 0.2 |
| dcGTX3 | 0.4 |
| dcGTX4 | 0.5 * |
| dcNEO | 0.4 |
| B1 | 0.1 |
| B2 | 0.1 |
| C1 | 0.01 ** |
| C2 | 0.1 |
| C3 | 0.01 ** |
| C4 | 0.1 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durán-Riveroll, L.M.; Cembella, A.D.; Band-Schmidt, C.J.; Bustillos-Guzmán, J.J.; Correa-Basurto, J. Docking Simulation of the Binding Interactions of Saxitoxin Analogs Produced by the Marine Dinoflagellate Gymnodinium catenatum to the Voltage-Gated Sodium Channel Nav1.4. Toxins 2016, 8, 129. https://doi.org/10.3390/toxins8050129
Durán-Riveroll LM, Cembella AD, Band-Schmidt CJ, Bustillos-Guzmán JJ, Correa-Basurto J. Docking Simulation of the Binding Interactions of Saxitoxin Analogs Produced by the Marine Dinoflagellate Gymnodinium catenatum to the Voltage-Gated Sodium Channel Nav1.4. Toxins. 2016; 8(5):129. https://doi.org/10.3390/toxins8050129
Chicago/Turabian StyleDurán-Riveroll, Lorena M., Allan D. Cembella, Christine J. Band-Schmidt, José J. Bustillos-Guzmán, and José Correa-Basurto. 2016. "Docking Simulation of the Binding Interactions of Saxitoxin Analogs Produced by the Marine Dinoflagellate Gymnodinium catenatum to the Voltage-Gated Sodium Channel Nav1.4" Toxins 8, no. 5: 129. https://doi.org/10.3390/toxins8050129
APA StyleDurán-Riveroll, L. M., Cembella, A. D., Band-Schmidt, C. J., Bustillos-Guzmán, J. J., & Correa-Basurto, J. (2016). Docking Simulation of the Binding Interactions of Saxitoxin Analogs Produced by the Marine Dinoflagellate Gymnodinium catenatum to the Voltage-Gated Sodium Channel Nav1.4. Toxins, 8(5), 129. https://doi.org/10.3390/toxins8050129