1. Introduction
Tetanus is an acute, often fatal disease of humans caused by the tetanus neurotoxin (TeNT) produced by the bacterium
Clostridium tetani. Moreover, it is characterized by generalized rigidity and convulsive spasms of the skeletal muscles. The muscle stiffness usually involves lockjaw, the neck, and then becomes generalized. Hippocrates first described tetanus over 24 centuries ago [
1]. However, as one of the most potent toxins known, even today it causes paralytic death to hundreds of thousands of humans annually [
2]. As of December 2007, there were 47 countries remaining that had not eliminated neonatal tetanus (MNT) [
3].
The general principles for the treatment of tetanus are supportive, such as providing an artificial respirator, as well as passively giving human Tetanus Immunoglobulin (TIG) or antitoxins containing polyclonal antibodies (PAb) derived from an animal (e.g., horse) which had been immunized with an adjuvant tetanus toxoid [
4]. Due to the cost of TIG, only the horse products are available in most developing countries (e.g., Egypt) [
5]. Human anti-TeNT serum from immunized volunteers has limitations related to their small-scale production and the risk of infectious disease transmission. However, the use of antitoxin from no homologous species can generate side effects, including the risk of zoonosis and anaphylactic shock [
6].
A fully human specific antibody to TeNT, which can be produced in vitro without the immunization process, would be a better alternative to human anti-TeNT serum or antitoxins with large quantities and few side effects to humans [
7,
8]. Phage display technology is a tractable, economical, and rapid method to obtain human antibodies, in which a single-chain variable fragment (scFv) or antigen-binding fragment (Fab) antibody is expressed as a fusion with a coating protein on the surface of the phage [
9].
To date, several large human phage libraries have been reported, however, seldom regarding immune antibody libraries have been published, including that for neutralizing antibodies of Tetanus neurotoxin [
10,
11,
12].
Compared with the yield generated using hybridoma technology, additional antibodies can be derived from a recombinant immune library made with the material of a single immunized donor, and in vitro selection can enrich for rare antibody specificities. Furthermore, human immune or disease-associated antibody libraries have identified antibodies with very interesting properties unlikely to be present in nonimmune or synthetic libraries [
13].
In our previous work, a human immune scFv antibody phage display library consisting of 1 × 10
8 individual clones with high diversity had been generated and used to screen scFvs against the HC chain of TeNT (TeNT-Hc) [
14]. To construct the library, V
H and V
L genes were amplified from peripheral blood lymphocytes of five volunteers who have high antibody titers against tetanus toxoid.
The correct choice of antigen is very important in screening specific neutralizing TeNT antibodies from a phage display antibody library. TeNT is produced as a ~150-kDa protein that is cleaved to a di-chain protein, comprising an
N-terminal light chain (TeNT-L, ~50 kDa) and a
C-terminal heavy chain domain (H, ~100 kDa) linked through a single disulfide bond [
15]. TeNT light chain is a zinc metalloprotease that cleaves the neuronal SNARE protein VAMP2 [
16]. The TeNT heavy chain contains two functional domains: a translocation domain (TeNT-HN, ~50 kDa) and a
C-terminal receptor binding domain (TeNT-Hc, ~50 kDa). TeNT-L specificity of polyclonal and monoclonal antibodies may have a crucial role in full protection against tetanus toxin. However, the mechanism by which zinc metalloprotease activity of TeNT light chain is neutralized by antibodies is not fully understood. The enzymatic groove of holo-TeNT is masked. The enzymatic active site becomes un-masked after the TeNT light chain exits from endosome into the neuronal cell cytoplasm. The antibodies with TeNT protease inhibitory activity should be developed further into a non-toxic cell penetrating antibody (transbody) [
6].
TeNT-Hc is involved in binding to cellular receptors with completely non-toxic effects, and can induce protective antibodies in animals, which is considered to be a potential candidate for tetanus toxoid subunit vaccines [
17,
18,
19,
20]. The TeNT-Hc has been proved as the protective antigen and immunized with TeNT-Hc alone can achieve similar immune effect with full length tetanus toxoid does. Tetanus toxoid (TT) could induce the production of antibodies not only against TeNT-Hc but also against TeNT-HN and TeNT-L. However, the antibodies against TeNT-Hc induced by TT are the main neutralizing antibodies for protection.
Previously, we found that a soluble form of TeNT-Hc was expressed in
Escherichia coli, and approximately 333 mg/L (6.66 μmol/L) was acquired. The NIH mice vaccinated with TeNT-Hc adsorbed an aluminum hydroxide gel adjuvant and demonstrated a potency of 168 IU/mL, which is two times higher than the national standard for tetanus vaccines [
21].
The purpose of this study was to screen for specific neutralizing antibodies against TeNT from the human immune scFv antibody phage display library using purified TeNT-Hc and TeNT as the antigen. Three specific antibody clones with neutralizing activities were obtained, and the mixture of the three antibodies exhibited potent toxin-neutralizing properties of at least 20 LD50s/20 µg antibody mixture. The results indicated that using TeNT-Hc and TeNT as antigens to screen human antibodies for TeNT intoxication therapy from a human immunized antibody library was convenient and effective.
3. Discussion
The toxicity of tetanus toxin is among the most potent and lethal known toxins, with an estimated human lethal dose of less than 2.5 ng/kg. Tetanus is a fatal disease, resulting in an extremely high mortality rate between 40% and 78%, mainly due to respiratory failure [
26,
27]. Although tetanus is preventable by vaccination and post-exposure prophylaxis, the disease remains life-threatening, causing thousands of annual deaths worldwide in places in which the population is not extensively immunized [
28].
The treatment for human tetanus has remained unchanged since the early 1900s by passively administering animal-derived anti-tetanus serum containing polyclonal immune immunoglobulins (PAb) together with supportive measures (e.g., an artificial respirator) to the patient. Several obstacles are faced both for the production and use of the therapeutic immune serum derived from animals. The major ones include adverse reactions due to the foreignness of the heterologous proteins to the recipient immune system, the limited supply of the immune serum, and the batch-to-batch variation of the antitoxin potency. In many developing countries, the current treatment consists of an intravenous infusion of equine antibody-based antitoxin, which can be associated with side effects, including hypersensitivity (e.g., serum sickness and anaphylaxis).
Next-generation antitoxins are likely to be derived from human monoclonal antibodies mapping different TeNT epitopes. Fully human neutralizing antibodies were deemed to be the most ideal and effective form of TeNT therapy. Phage display technology is a tractable, economical, and rapid method to obtain human antibodies via a high throughput. Theoretically, if the library is sufficiently large in diversity and size, most antibodies can be selected from it.
Numerous monoclonal antibodies have been developed against TeNT and tested for their neutralizing activity. Some monoclonal antibodies were generated against TeNT using mouse hybridoma technology [
29,
30,
31], and some were selected from human phage libraries [
32,
33]. However, there are few existing reports describing selection from human immune libraries.
Antibodies with a high affinity are ideal candidates for clinical treatment. The affinities of the antibodies are correlated with the size of the repertoires. The larger the antibody library is, the higher the affinity of the antibodies that can be selected from it [
34,
35]. In our study, a human immunized scFv antibody phage display library of 1 × 10
8 was used. 2-7G, 2-2D, and S-4-7H antibodies with a KD of 1.50 × 10
−10 M, 7.82 × 10
−9 M and 2.25 × 10
−9 M were obtained. The three antibody clones can easily be associated with the antigen and are difficult to dissociate, which are essential characteristics for therapeutic antibodies.
The affinities of the three IgGs (2-7G, 2-2D and S-4-7H) increased by 2.9 to 10.1 times, compared with the scFv form. The antigen binding affinity of the IgG was significantly higher (lower KD) than for the corresponding scFv were also showed by James Marks’ group [
36]. MAbs S25 and 3D12 that were selected from phage libraries against botulinum toxoid improved their affinities by 18.7 and 656 times from scFv to lgG form respectively [
36]. Another example was mAb C10 which was selected from phage library against BoNT/A-Hc by our group [
9]. The KD value with C10 changed from 7.81 × 10
−9 M to 6.46 × 10
−11 M, increased by 120 times from scFv to lgG form. After IgG construction the affinity of corresponding scFv may increase, decrease or remain unchanged. In the current study, 2-7G, 2-2D and S-4-7H scFvs were all maintain special binding to TeNT-Hc and neutralizing potency in vivo. The antigen binding affinity of the IgG was significantly higher than that of the corresponding scFv, because of an increase in the association rate constant (kon), and decrease in the dissociation rate constant (koff), like 2-7G and 2-2D. However, not the same as 2-7G and 2-2D, S-4-7H which significantly decreased in the koff increased the affinity by 10 times. This was also showed in scFv of 3D12 which significantly decreased in the koff by 4 times [
36]. The affinity increased in scFv changed to lgG was also due to an increase in the stability of the molecule and hence an increase in the functional antibody concentration [
36]. This was also found in scFv re-constructed to diabody or disulfide-stabilized scFv in our experiment. Furthermore, another reason may be that IgG was glycosylated and scFv from
E. coli not, and glycosylation may affect antibody binding characteristics.
TeNT has AB structure-function properties: the A domain is a zinc metalloprotease, and the B domain encodes a translocation domain and
C-terminal receptor-binding domain (TeNT-Hc). Earlier studies demonstrated that TeNT-Hc bound to gangliosides via two carbohydrate-binding sites termed the lactose-binding site (the “W” pocket) and the sialic acid-binding site (the “R” pocket) [
37]. In this article, purified protective TeNT-Hc antigen was used as a selection antigen to screen neutralizing antibodies from the antibody phage display library. After three rounds of selection, more than 100 specific clones were obtained, and 12 clones were chosen based on the high OD values. Eight different sequences were identified from 12 clone sequences and three unique antibodies were mapped to different epitopes, whereas all others were mapped to the same epitope with the 2-7G. This may be due to the existence of a preponderant epitope on TeNT-Hc, which can bind to the phage antibodies with high affinity.
NGF-differentiated PC12 cells represent a widely used neuronal-like model system. After differentiation, they develop extended neuritis and an increased number of synaptic-like vesicles [
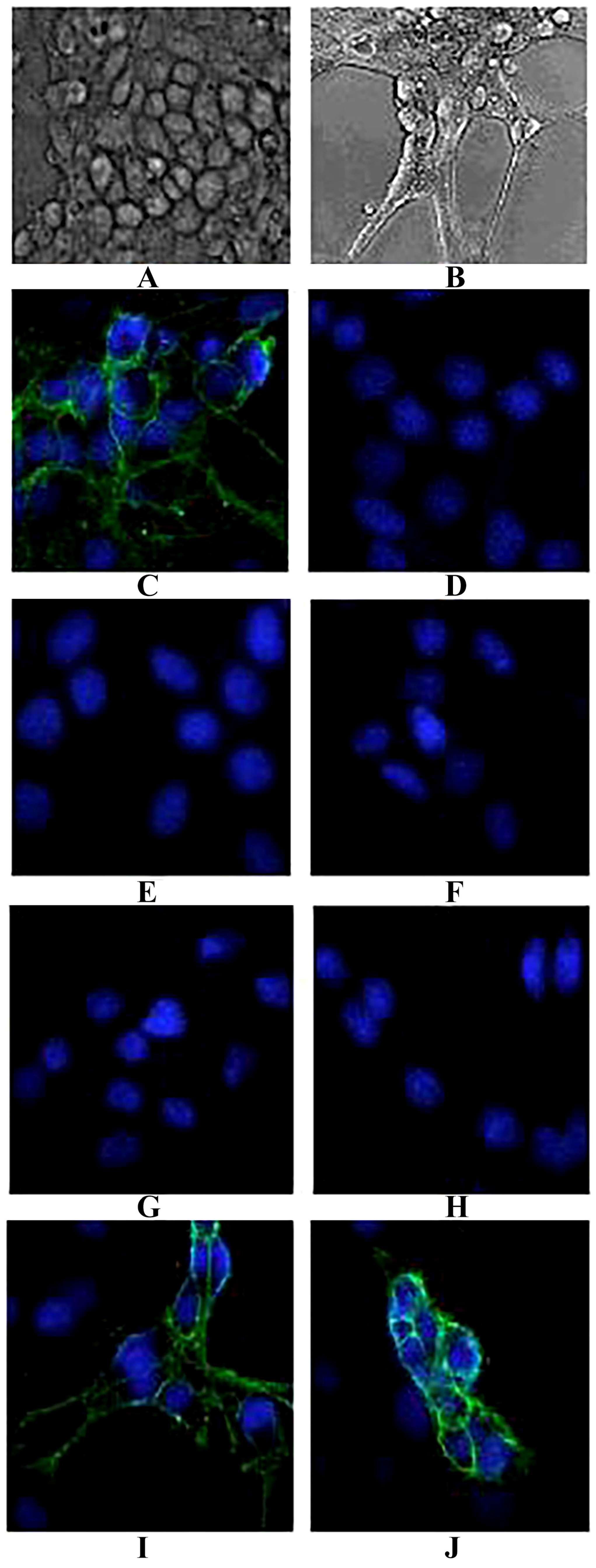
38]. In the present study, when the antibodies were cultured with TeNT-Hc prior to being added to the NGF-differentiated PC12 cells, 2-7G, 2-2D, and S-4-7H could effectively inhibit the binding of TeNT-Hc to PC-12. Each of these three clones has a greater potency of toxin neutralization in vitro.
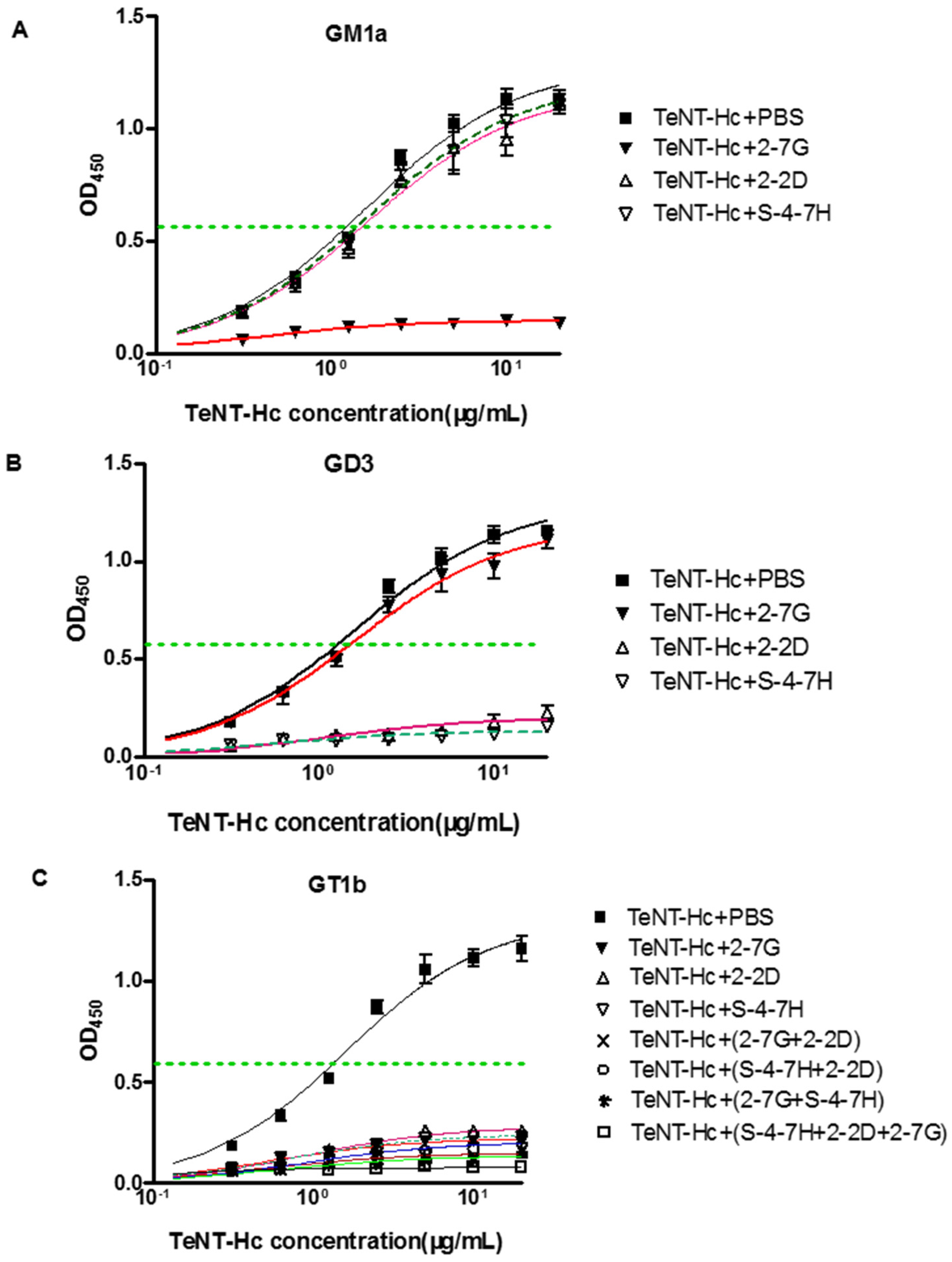
Previous studies have shown that GD3 bound the R pocket, and GM1a bound to the W pocket, whereas GT1b binds to both carbohydrate pockets of TeNT. Therefore, these gangliosides are useful tools for determining the roles of the W and R pockets in TeNT binding to neurons [
37]. Our results indicate that the 2-7G inhibits TeNT-Hc binding to the receptor via the carbohydrate-binding sites of the W pocket, and 2-2D as well as S-4-7H inhibits TeNT-Hc binding to the receptor via the carbohydrate-binding sites of the R pocket.
A dual-receptor model incorporating ganglioside receptors has been proposed to explain the distinct sites of action of TeNT [
39]. These models attempt to explain the experimental observations that are largely inconsistent with a sole ganglioside receptor (e.g., the difference in the binding affinity measured in vivo and in vitro) [
37]. Previous studies also demonstrated the simultaneous binding of two GT1b molecules to TeNT via the R and W binding sites, which was supported by the subsequent observation that GT1b can bind independently to the R or W pockets [
37]. This suggests that the observed binding to these gangliosides represents the simultaneous occupancy of both the W and R pockets of TeNT by two molecules of GT1b. The TeNT-Hc bound cells with high affinity only in the presence of gangliosides that together, can bind both the R and W carbohydrate-binding pockets [
37,
40,
41].
An interesting finding of this study was to test the synergistic neutralizing effect of the GT1b-binding inhibitory mAb. Cooperative binding is caused by conformational changes of the protein in which the binding of one antibody thermodynamically or entropically facilitates the binding of the next one. Such cooperative or synergistic effects have been reported for antibodies or scFvs binding to non-overlapping or independent epitopes of the tetanus toxin. Furthermore, another thing we concerned about was that when blocking specifically one carbohydrate-binding site of TeNT-Hc, the binding of GT1b to the other binding site was not visible in
Figure 5C. GM1a contained Gal4-GalNAc3- moieties, while GD3 contained Sia7-Sia6- moieties, and GT1b contained those two moieties so as to it could be bind to “R” and “W” pocket binding site at the same time [
37]. However, the structure of GT1b was not equal to the combination of GM1a and GD3. Occupation of two carbohydrate-binding pockets by gangliosides is required for high affinity binding of TeNT-Hc into PC12 Cells. Although TeNT-Hc did not bind to PPMP-treated PC12 (endogenous gangliosides of PC12 cells were depleted) cells preloaded with either GD3 or GM1a alone, TeNT-Hc bound PPMP-treated PC12 preloaded with a mixture of GD3/GM1a (1:1). TeNT-Hc also bound with high affinity PPMP-treated PC12 cells that were preloaded with GT1b. Interestingly, TeNT-Hc binding intensity in 2 mol of GT1b was higher level than that of GD3/GM1a (1 mol:1 mol). This showed that GT1b was more powerful than GD3/GM1a in promoting the combination of TeNT-Hc and PPMP-treated PC12 [
37]. Therefore we presumed the greater the contribution, the greater the inhibitory effect of the combination. That may be why antibody alone could inhibit the combination of TeNT-Hc and GT1b from 78.6% to 82.3%. In our future research, GD3/GM1a will be instead of GT1b to investigate the ability of antibody-mediated inhibition of TeNT-Hc binding to GM1a. Another reason may be the spatial structure which played an important role in the combination of TeNT-Hc and GT1b. The crystal structure of ganglioside-TeNT-Hc complexes shows cross-linking between the GT1b and TeNT-HC. A single ganglioside could bind simultaneously to more than one TeNT molecule [
42]. The inhibitory of one side of carbohydrate-binding pockets may affect the spatial structure of the cross-linking between GT1b and TeNT-HC, or even affect the combination of another GT1b to TeNT-HC.
The W pocket is a groove formed by Trp1289, His1271, and Tyr1290 [
42]. Trp1289 plays a dominant role in ganglioside interaction at the W pocket, thus the 2-7G inhibits TeNT-Hc binding to the receptor by recognizing an epitope that must encompass or lie near Trp1289. The R pocket consists of residues Asp1147, Arg1226, Asn1216, Asp1214, and Tyr1229, where the formation of a salt bridge between the carboxyl group of sialic acid and the guanidine group of Arg1226 is the most prominent feature [
42]. We performed co-injection Biacore experiments with pairs of scFvs, to determine whether antibodies could bind TeNT-Hc simultaneously. For 2-2D and S-4-7H only few changes in response were seen upon co-injection with theoretical Rmax values not reached, an indication that the scFvs were binding partly overlapping epitopes and hindering the binding of each other to TeNT-Hc. Its importance of Arg1226 is highlighted by the drastic drop in toxicity to 1.4% and the almost complete loss of binding in the synaptosome assay upon mutation of R1226 to leucine or phenylalanine. Placing a bulky phenyl ring at the opening of the binding pocket, as implemented by TeNT-Gly1215Phe, reduced the toxicity to 15% in the mouse phrenic nerve (MPN) assay, the binding to 18% in the synaptosome assay compared to the wild-type values. The mutation interferes with H-bonding between D1214 and N1216 and sialic acid, and partially shields the central R1226 residue, according to molecular modeling analyses [
40]. Accordingly, the 2-2D and S-4-7H epitopes map may encompass or lie near Arg1226 or Gly1215 respectively. In this study, 2-7G/2-2D and 2-7G/S-4-7H exhibits marked synergistic effects for the neutralization of TeNT-Hc binding to GT1b, while 2-2D/S-4-7H exhibits slightly synergistic effects with respect to neutralization.
The mixture of two or three antibodies was more effective than each single one for the neutralizing activities in vivo. Although the combination of 2-2D/S-4-7H and 2-7G could both protect 70% mice from death, the combination of 2-2D/S-4-7H exhibits slightly synergistic effects with respect to in vivo toxin neutralization than that of 2-7G. Any pair of mAbs that belonged to different carbohydrate-binding sites completely protected mice challenged with 20LD50s of toxin. The combination of 2-7G/2-2D and 2-7G/S-4-7H which belonged to different carbohydrate-binding sites exhibits marked more synergistic effects for the in vivo toxin neutralization than that of 2-2D/S-4-7H which belonged to the same carbohydrate-binding sites. All of the mice survived the challenge with 20 LD50s when given the mixture of the three antibodies.
As we know, with a similar structure to BoNT-Hc, TeNT-Hc consisted of a two-receptor model incorporating ganglioside and protein receptors. Similar findings of neutralizing antibodies by James Marks’ group also shown that botulinum neurotoxins/A, can be potently neutralized by an oligoclonal Ab consisting of only three mAbs (MAbs S25, 3D12 and C25) [
43,
44]. MAbs S25 and C25 were selected from phage libraries constructed using mice immunized with BoNT/A-HC, while mAb 3D12 was selected from humans immunized with pentavalent botulinum toxoid. The affinities of those three antibodies were from 7.3 × 10
−8 M to 1.1 × 10
−9 M. Like 2-2D and S-4-7H in this study, the S25 and 3D12 epitopes map within the
C-terminal subdomain, containing the putative sialo-ganglioside binding site. However, unlike 2-7G, C25 maps to a complex epitope that includes the majority of the Hc sequence, suggesting an epitope of adjacent
N- and
C-terminal subdomains. 50 micrograms of each single mAb prolonged the time to death but failed to protect mice challenged with 20 LD
50s. Any pair of mAbs completely protected mice challenged with 100 LD
50s of toxin. All mice receiving a mixture of all three mAbs (oligoclonal Ab) survived challenge with 500 LD
50s of toxin [
36].
The potential mechanism for potent tetanus toxin neutralization by oligoclonal Ab is the need to block multiple epitopes on the toxin binding domain surface that bind to cellular receptors. Broad interaction of the
C-terminal subdomain with cellular receptors is consistent with the mechanism of Botulinum Neurotoxin [
31]. Epitope mapping provides insight into why a single mAb cannot potently neutralize a toxin. Two spatially separated ganglioside binding sites have been observed in the co-crystal structure of the homologous tetanus toxin [
42], which that gangliosides are functional dual receptors for TeNT. Potent toxin neutralization would require blockade of this broad surface, which could not be covered by a single Ab. Administration of all three mAbs may more potently neutralize toxin by blocking a larger proportion of the binding surface. Furthermore, antibodies that recognize different carbohydrate-binding pockets could exhibit higher synergistic toxin neutralization activities than those that recognize the same carbohydrate-binding pockets.
Each antibody appeared to inhibit the binding of TeNT-Hc to PC-12 cells and ganglioside in vitro, whereas a greater potency of neutralizing activities in vivo was observed only when two or three antibodies were combined. In our opinion, three reasons may explain this phenomenon: (1) it has been suggested that different factors (e.g., ganglioside concentration, GPI-anchored proteins, and pH) of the microenvironment play a major role in TeNT binding and intoxication of neurons; (2) it may be due to a difference between the PC-12 cells treated with NGF and the targeted nerve cells in vivo. The PC-12 cell line is established from a rat pheochromocytoma and has many properties in common with primary sympathetic neurons. Although PC-12 cells can differentiate both morphologically and biologically into neuronal cells, it is not the same with the neurons in vivo, including the expression of the TeNT receptors; and (3) it may be due to the sensitivity of the in vitro assay, which is much less than an in vivo neutralizing test. There may be still a few proteins that bind TeNT-Hc to the PC-12 cells, which cannot be observed by the naked eye. Therefore, the result of the in vivo study is more credible than the PC-12 model performed in vitro.
Additional antibodies that map different epitopes especially against the light chain should be further screened to exploit more potent neutralizations. We have noted that more recent studies on human-like recombinant antibodies confirmed the neutralizing role antibodies against the light chain of related toxin (Botulimum toxin) [
45] and confirmed strong synergistic effect on combining the antibodies targeting different domains [
46]. The antitoxins are no longer effective once the toxin is taken up into the neurons which happen faster than the identification of a botulism case since patients develop symptoms more than three days after intoxication [
47]. Likewise, antibodies against the light chain of Tetanus Toxin may have a crucial role in treatment of tetanus toxin.
The acquisition of neutralizing antibodies against TeNT exploited a novel and method to screen for human therapeutic antibodies via the human immunized scFv antibody phage display library.
4. Materials and Methods
4.1. Animals
Female, specific pathogen-free (SPF) BALB/c mice, weighing 16 g to 22 g each, were purchased from Beijing Vital River Laboratory Animal Technology Co. Ltd., Beijing, China. All animals were raised under humanitarian conditions. These mice with free access to food and water were obtained from the Laboratory Animal Center, Academy of Military Medical and Sciences. All efforts were made to minimize suffering. After injection, mice were followed for the five days to check for any signs of paralysis or death. Loss of righting reflex was used as the humane end point of the experiment. Mice were monitored three times a day for their condition and for the occurrence of end point. Mice that presented loss of righting reflex were humanely euthanized and sacrificed. All animal experiments used in this study were performed according to the protocols approved by the Institutional Animal Care and Use Committee of Beijing Institution of Biotechnology (Identification code for mice: IACUC of AMMS-08-2015-011; Date of approval: 9 December 2015). The final death total was determined five days after injection.
4.2. Human Antibody Phage Display Library
The human immune scFv antibody phage display library used in this study was constructed in our laboratory from the immunoglobulin genes of five volunteers who exhibited high antibodies titers against tetanus toxoid [
14]. The details of the construction, the antibody diversity repertoire, and the other attributes of this phage library have been described elsewhere [
14]. Briefly, genes encoding the V
H1~V
H7, Vκ
1~Vκ
6, and V
λ1~V
λ10 families of human immunoglobulin were amplified using 44 primer pairs. The primer set used for PCR amplification of antibody genes appends an SfiI restriction site at the 5′ end of the scFv cassette and a NotI site at its 3′ end.
The VH and VL amplicons were molecularly linked at random via a (Gly4Ser) 3 polynucleotide linker to generate a repertoire of VH-linker-VL or scFv. The human scFv (huscFv) sequences were cloned into pCANTAB5E phagemid vectors, and the recombinant phagemids were introduced into competent suppressor TG1 E. coli cells. Finally, the library was comprised of 1 × 108 individual clones, and more than 95% of the phages in the library contained human scFv sequences in their genome.
4.3. Antigen Preparation
TeNT-Hc was expressed in
E. coli BL21 (DE3) and purified, while the immunogenicity of TeNT-Hc was tested via the subcutaneous immunization of individual NIH mice as previously described [
22]. A Bradford method kit (Tiangen Biotech, Beijing, China) was used to determine the concentration of the purified protein according to the manufacturer’s protocol.
4.4. Phage Preparation
The bacteria libraries from the human immunized scFv antibody library were diluted with blocking buffer (1× PBS containing 10% nonfat dry milk) and incubated at room temperature for 20 min. The diluted libraries were added to the 2× YT medium and the culture was incubated at 37 °C, 220 rpm to an OD600 of 0.5. M13KO7 helper phages (GE Healthcare, Waukesha, WI, USA) were added to the bacteria at an M.O.I. of 20:1 and left at 37 °C with no shaking for 30 min followed by gentle shaking at 150 rpm for another 30 min at 37 °C. The culture was then centrifuged, and the cell pellets were resuspended in 2TY-AG (100 µg/mL ampicillin, 1% glucose). After the addition of kanamycin to a final concentration of 50 µg/mL and IPTG (isopropyl thio-β-d-galactoride) to 0.1 mM, the bacteria were then cultured overnight at 30 °C, 200 rpm. The culture supernatant was purified and concentrated by PEG (20% polyethylene glycol 8000, 2.5 M NaCl) precipitations, and resuspended in PBS (pH 7.4).
4.5. Phage Display Biopanning
The immunotube (Nunc, Roskilde, Denmark) was coated with 200 nM TeNT-Hc or 200 nM pure active toxin (National Institutes for food and drug control, Beijing, China) in 0.05 M Na2CO3 buffer (pH 9.6) at 4 °C overnight. After rinsing twice with PBS, the immunotube was blocked with blocking buffer (1× PBS containing 3% nonfat dry milk) at room temperature for 2 h.
The pre-blocked antibody libraries were added to the immunotube and incubated at 37 °C for 90 min. The immunotube was then washed 10 times with PBST (PBS plus 0.1% Tween-20) followed by five rinses with PBS. The bound phages were eluted with 1.5 mL of 100 mM glycine-HCl (pH 2.2) for 15 min at room temperature and then neutralized with 1 M Tris-HCl to pH 7.4. The eluted phages were recovered by infecting the exponentially growing E. coli XL1-Blue. After culturing on Luria-Bertani (LB) medium-ATG (100 µg/mL ampicillin, 10 µg/mL tetracycline, 1% glucose) plates at 37 °C overnight, the XL1-Blue cells were scraped, and the phages were rescued by the helper phage M13KO7. The second and third panning rounds of the phage display scFv library were similar to that of the first round, except for the titers of the phage particles used and the concentrations of the coated antigen were lower than the first round. The titer of the unamplified third-round phage particles was determined on LB medium-CTG plates.
4.6. Screening for Specific scFvs
After three rounds of panning, the phage clones were screened by an enzyme-linked immunosorbent assay (ELISA). The 96-well ELISA plate (Costar, Corning, NY, USA) was coated with 40 nM TeNT-Hc, and also coated with 130 nM Yersinia pestis capsular F1, 54 nM Yersinia pestis capsular V protein (F1 and V, purified in our lab) and 91 nM interferon-omega (IFN-ω, purified in our lab) individually as negative antigens.50 μL of phage particles at a concentration of approximately 8 μM in 3% Marvel™ PBS (pH 7.4) were added, and the binding of the phages to the immobilized antigen was performed at 37 °C for 2 h. The unbound phages were removed by washing extensively with PBST. The plate wells were detected by horseradish peroxidase (HRP)-conjugated anti-M13 antibody (GE Healthcare), followed by the addition of peroxidase substrate, o-phenylenediamine dihydrochloride (OPD). 2 M H2SO4 was used to stop the reaction, and the results were monitored at OD492/630 with a microplate reader (Multiskin MK3, Shanghai Labsystems, Shanghai, China). The particular clones that were bound to TeNT-Hc were selected, and the corresponding phage plasmids were sequenced.
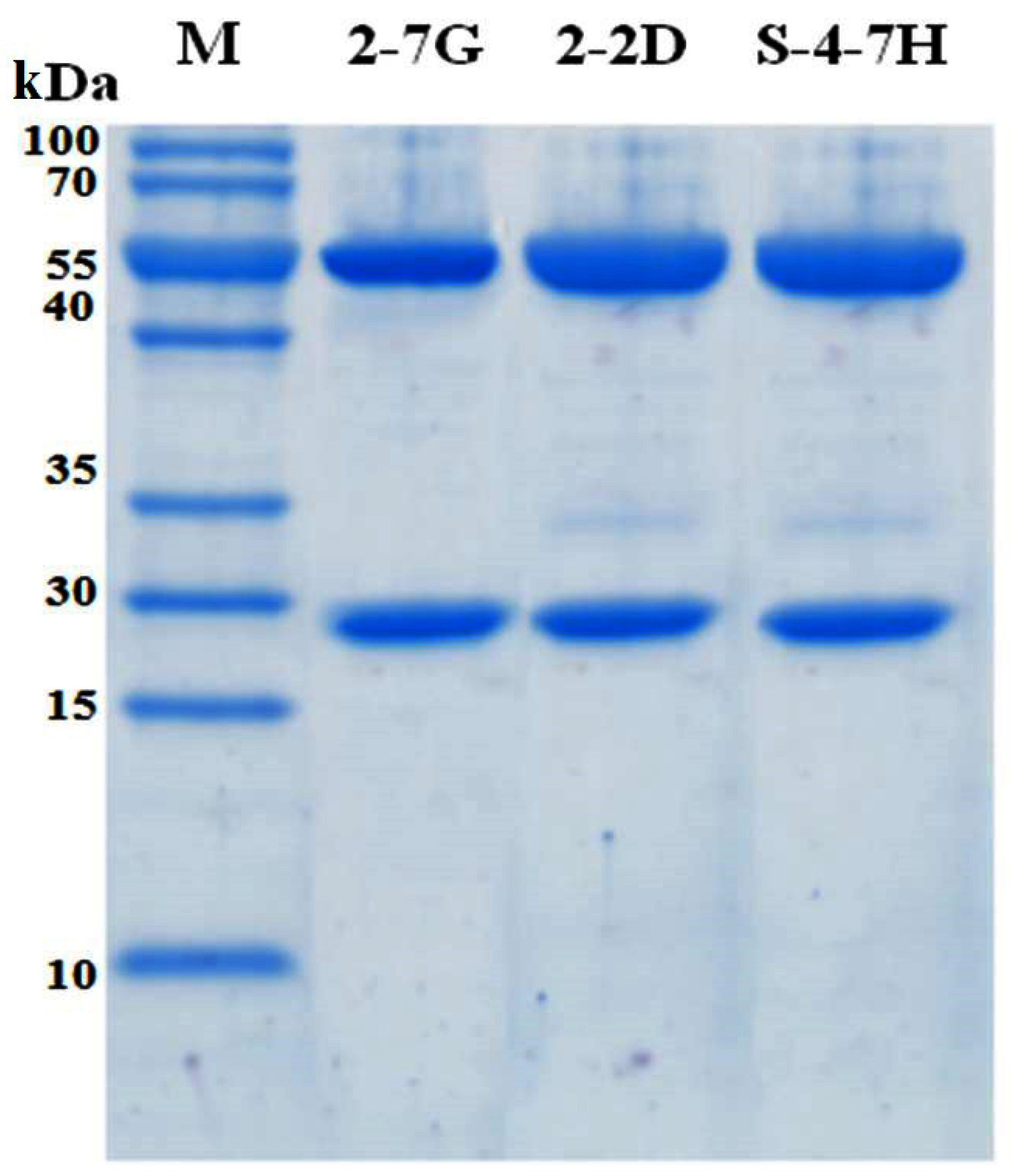
4.7. Subcloning and Expression of scFvs
The genes of immunopositive scFvs were cloned into the expression vector kil-SN through the restriction enzyme sites, SfiI and NotI. The kil-SN vector was reconstructed on the basis of pET22b (+) (Novagen, Madison, WI, USA), which contained the colicin release gene kil sequence, signal peptide pelB, E-tag, and 6× His. The recombinant plasmids were then transformed into the E. coli strains KS1000 (DE3) and cultured at 37 °C at 220 rpm to an OD600 of 0.5. After adding IPTG to a concentration of 0.005 mM, the culture was incubated at 30 °C and 220 rpm overnight for the detection of antibody expression. In the presence of kil proteins, the scFv fragment can be easily released from the periplasm into the growth medium. The supernatants containing scFv were harvested, and antibodies were purified with a HisTrap FF crude 1-mL column (GE Healthcare).
4.8. Epitope Mapping
A competitive binding ELISA was used to search for specific antibodies mapped to different epitopes of TeNT-Hc. After blocking, 50 µL of scFv at a concentration of approximately 8 μM was added to the plate coated with TeNT-Hc and incubated for 1 h [
48]. After washing, the supernatants containing phage-scFvs were then added and incubated for 1 h. An HRP-conjugated anti-M13 antibody was used to detect antigen recognition, and the results were detected as above. The wells, which the
E. coli-expressed scFvs were not added to, were used as the negative controls. If two detected antibodies mapped to the same or similar epitope, inhibition would be detected. The inhibition efficiency (IE) was counted using the following formula: IE (%) = (1 − OD492/630 of experimental well/OD492/630 of negative control well) 100%. IE ≥ 50% was considered an inhibition (+), 20% < IE < 50% was considered partial inhibition, and IE ≤ 20% was considered a non-inhibition.
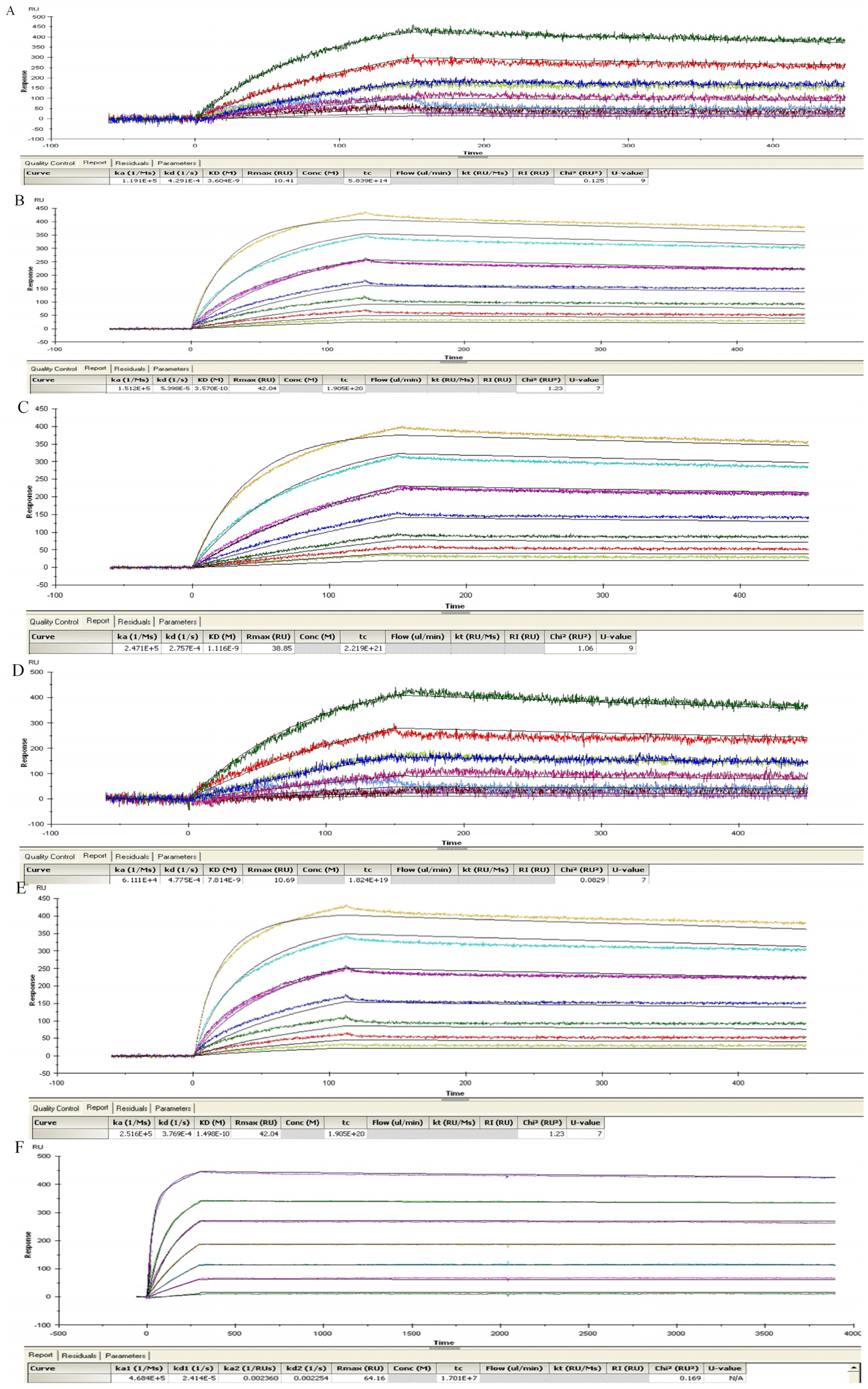
4.9. ScFv SPR Kinetic Measurements
Antibody affinity was determined by surface plasmon resonance spectroscopy using a BIAcore T200 instrument (GE Healthcare-Biacore, Uppsala, Sweden). ScFv or lgG was coated at a maximum of 3500 RU (7000 RU for lgG) on a CM5 chip (BR100012, GE Healthcare-Biacore) via amine coupling according to the manufacturer’s instructions. A flow rate of 20 µL/min was maintained during each measurement and volumes of 100 µL of activated TeNT (125 nM to 0.1 nM) dissolved in HBS-EP buffer (BR100188, GE Healthcare-Biacore) were tested. After each dilution, the chip was regenerated with 1.5 µM glycine buffer (BR100354, GE Healthcare-Biacore) injected for 30 s at 10 µL/min. Affinities were calculated using the BIAevaluation software (GE Healthcare-Biacore) according to the Langmuir 1:1 adsorption model (except for 2-7G-lgG using bivalent model) and were verified by internal consistency test [
49]. The number of technical replicates was equal to 3 and mean +/− standard deviations of Kon, Koff and KD were given.
4.10. Competition Studies Using SPR Analysis
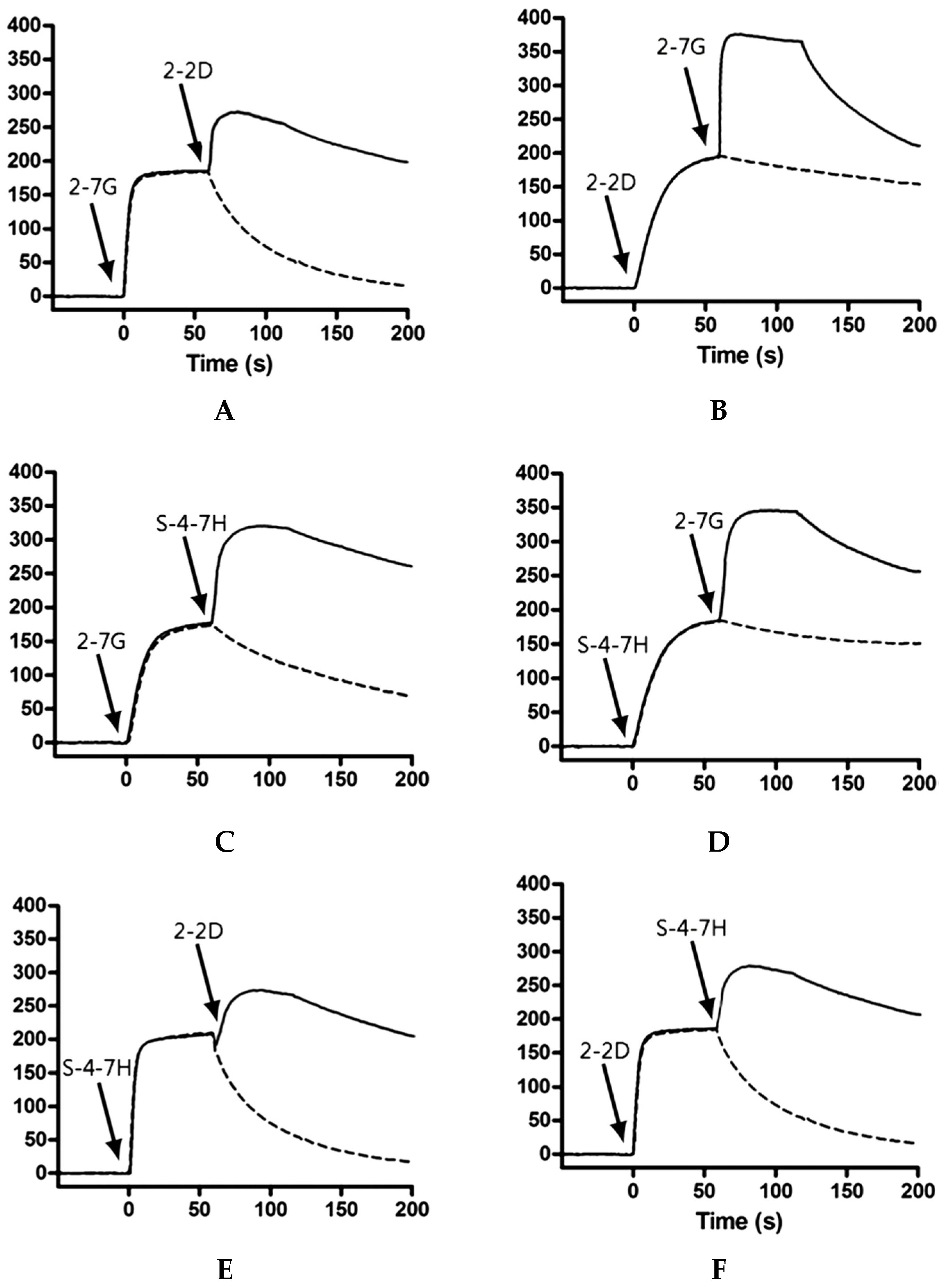
We used Biacore co-injection experiments to determine whether our mAbs could bind unique, nonoverlapping epitopes on TeNT-Hc. Briefly, 80 μL of the first mAb diluted in HBS-EP buffer to a concentration of 20× its KD value was injected over 10,098 RUs of immobilized TeNT-Hc at 40 μL/min. Following injection of the first mAb, buffer or a second mAb (80 μL total volume, at 20× KD) was injected at 40 μL/min over the TeNT-Hc surface already saturated with the first mAb. Data were collected on all possible paired combinations of 2-7G, 2-2D, and S-4-7H, in both orientations (i.e., each mAb acted as the first and second mAb). For every mAb the percentage of binding to a specified amount of TeNT-Hc was determined. The Maximal RU mAb is determined by the highest response obtained while the first Ab binding to the immobilized TeNT-Hc. The percentage of binding of a certain mAb to TeNT-Hc was expressed relative to the maximal binding as obtained with any of the presenting mAb according to the following equation:
A percentage of binding of the second mAb, in relation to the TeNT-Hc presenting first mAb, of <10% of the maximal binding was defined as inhibition in binding. Binding of the second mAb of <50% and >10% of the maximal binding was arbitrarily defined as partial inhibition in binding. A binding of >50% of the maximal binding was defined as concurrent binding [
50].
4.11. IgG4 Construction
The heavy and light chain expression vectors were used to cotransfect FreeStyle
TM 293-F cells (Invitrogen, Carlsbad, CA, USA) for instantaneous expression as a previously described procedure [
9]. Supernatants containing IgG4 were collected and purified with the use of a HiTrap protein An FF 1-mL column (GE Healthcare).
VH genes of scFvs were amplified using PCR from the E. coli expression vector kil-SN-scFv with the primer pairs CAGTGTGAAGTTCAACTG GTTCAAAGTGGTG and GTTCAGCTAGCGCTCGACACGGTCACCAGAGTGC. DNA digested with NheI was ligated into plasmid H293 (constructed by our lab, containing the CH domain of IgG4), which was digested with BstZ17I and NheI. The clones containing the correct VH were identified by DNA sequencing. The Vλ genes of scFvs were amplified from the E. coli expression vectors kil-SN-scFv with the primer pairs CCTGGGCCAGC TACGAACTGA CCCAGCCG, and CATTAAGCTTGGTGCCACCGCCAAACAC. DNA, digested with HindIII, was ligated into plasmid L293-Cλ (constructed by our lab, containing the Cλ domain), which was digested with EcoRV and HindIII, and the clones containing the correct Vλ were identified via DNA sequencing.
4.12. In Vitro Inhibition of PC-12 Cell Binding
The rat pheochromocytoma cell line (PC12) was cultured as described previously [
51]. For crosslinking experiments, cells were seeded in 12-well plates (Costar-Corning, Cambridge, MA, USA) at a density of 25,000 cells/well. After 24 h, the medium was supplemented with 75 ng/mL nerve growth factor (NGF, produced by our lab). NGF-treated rat PC12 were prepared and fixed with 4% (
w/
v) paraformaldehyde in 100 mM sodium phosphate buffer (pH 7.4) for 30 min at room temperature. The antigenicity of certain surface receptors was not affected by this fixation procedure. Cells were incubated with 40 nM TeNT-Hc and mixed with or without the purified IgG4 antibodies (the final concentration was 40 nM) for 1 h. Mouse polyclonal anti-TeNT-Hc antibodies (produced in our lab) were used as the positive controls. Bovine serum albumin (BSA) was used as negative controls. The inhibition efficiency of the binding between TeNT-Hc and PC-12 cells was measured with the mouse polyclonal anti- TeNT antibodies (1:5000, Zhongshan Goldenbridge, Beijing, China) followed by FITC-labeled anti-mouse IgG antibodies (1:5000, Zhongshan Goldenbridge, China). Evan’s blue dye was used to stain the nuclei. The fluorescence was observed by an inverted Epi-fluorescence microscope (Eclipse TS300, Nikon, Amsterdam, Netherlands).
4.13. Assessment of mAb Inhibitory Activity on the TeNT Binding to Ganglioside
The ability of the mAbs to inhibit the binding of TeNT to ganglioside GT1b/GM1a/GD3 was assessed by a modification of a previously described procedure [
31,
52]. Briefly, a serial concentrations of TeNT-Hc (expressed by our laboratory, from 6 nM to 400 nM) was chosen for use in the assay, and incubated with an equal volume of anti-TeNT mAb (66.7 nM) for 2 h at room temperature. Microtiter ELISA plates were coated with ganglioside GT1b/GM1a/GD3 (Sigma, Saint Louis, MO, USA) (10 μg/mL in methanol, 100 μL/well) and the plates were left at room temperature overnight to allow for the evaporation of methanol. The plates were then blocked with 1% BSA/PBS for 2 h at room temperature. After washing four times with PBS-Tween 20, 100 μL of each TeNT-Hc/antibody mixture (preincubated for 2–3 h) was added to the wells and incubated for 2 h. The plates were washed four times and then incubated 2 h with HRP-conjugated human anti TeNT polyclonal antibodies. Following the addition of the TMB substrate solution and then adding the stop solution, the ODs were measured at 450 nm by a multiscan ELISA reader. The half-maximal binding of TeNT-Hc to gangliosides GT1b/GM1a/GD3 were calculated.
4.14. In Vivo Toxin Neutralization
In vivo toxin neutralization was studied with the help of a mouse assay in which the toxin and 20 µg of the antibodies were premixed and injected intraperitoneally (i.p.). TeNT neurotoxin was purchased from National Institutes for food and drug control, China. The protein content was 5 mg/mL (33.3 μM) with a specific toxicity in mice of 2 × 107 LD50/mg protein. Every ten mice were classified in one group. The time-to-death and the numbers of surviving mice were determined. Using the horse polyclonal anti-TeNT-Hc antibodies showed 2000 IU/mL (Zhongshan Goldenbridge, Beijing, China) as a positive control. 20 µg of the appropriate IgG4 was added to 20 mouse LD50s of TeNT neurotoxin to a total volume of 0.5 mL of PBS and incubated at 37 °C for 30 min. For the Ab pairs, 10 µg of each Ab was added. For a combination of the three Abs, 6.6 µg of each Ab was added. The mixture was then i.p. injected into female Balb/c mice (16–22 g). Mice were studied in nine groups and were observed three times a day. The final death total was determined five days after injection.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}