
The Pathogenetic Effect of Natural and Bacterial Toxins on Atopic Dermatitis


Abstract
:1. Introduction
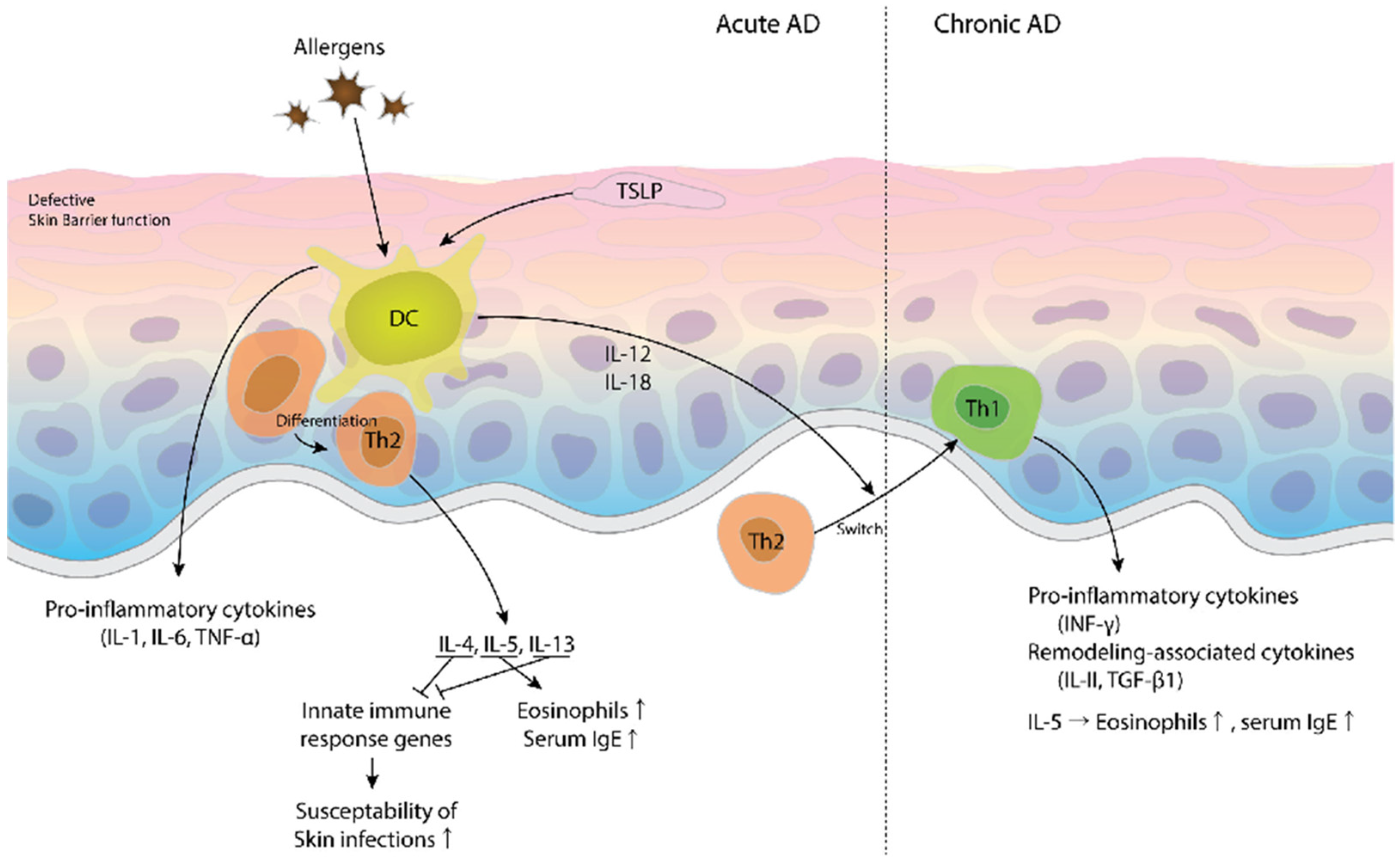
2. Pathogenesis of AD from an Immunologic Point of View
3. Pathogenesis of AD from a Non-Immunologic Point of View
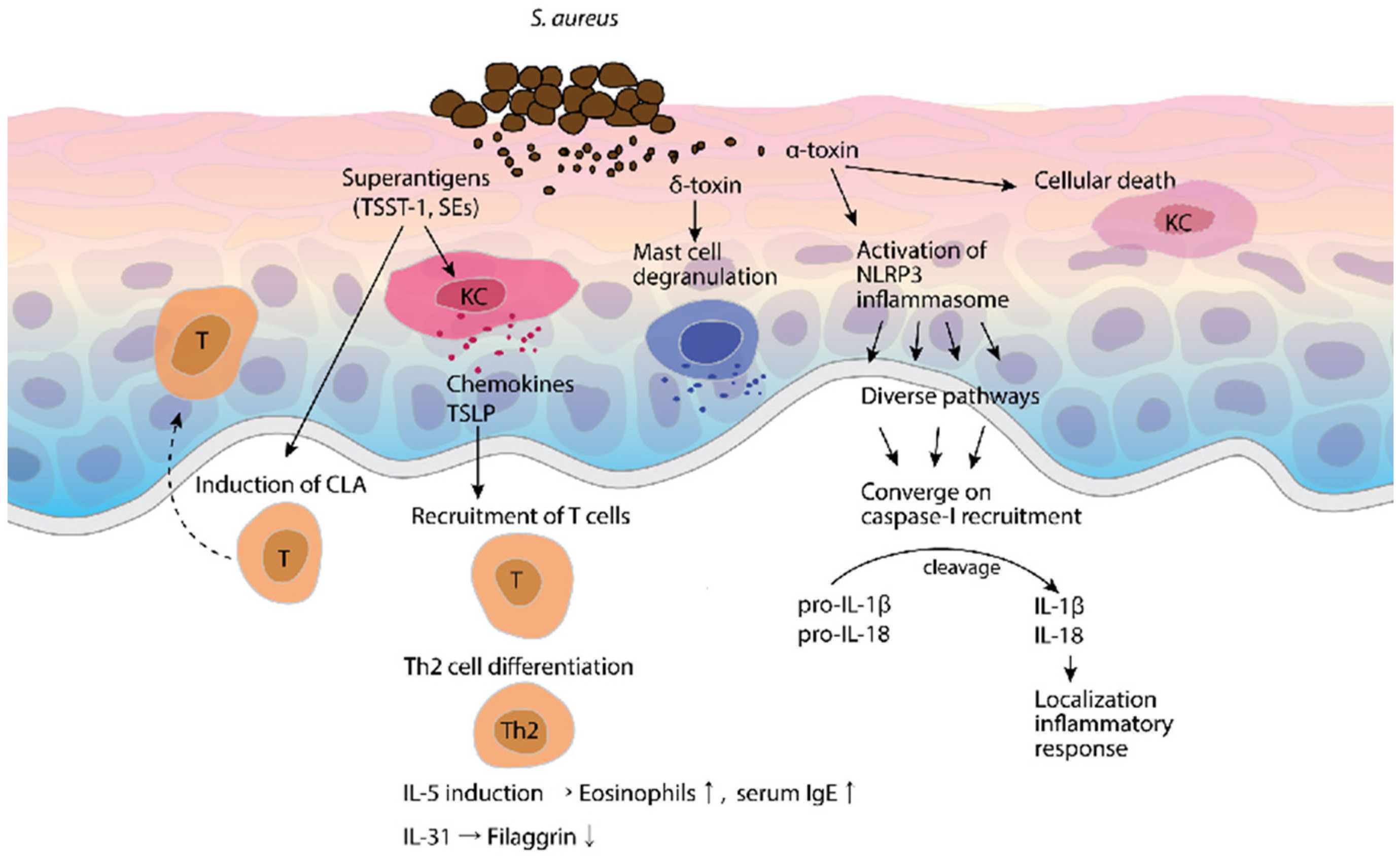
4. The Effect of S. aureus and Its Toxins on AD
5. Toxins That Inhibit AD and Their Inhibitory Mechanisms
6. Inflammasome Expression and Function in AD
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bieber, T. Atopic dermatitis. N. Engl. J. Med. 2008, 358, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.; Bieber, T. Atopic dermatitis. Lancet 2003, 361, 151–160. [Google Scholar] [CrossRef]
- Hagermark, O.; Hokfelt, T.; Pernow, B. Flare and itch induced by substance P in human skin. J. Investig. Dermatol. 1978, 71, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Rukwied, R.; Lischetzki, G.; McGlone, F.; Heyer, G.; Schmelz, M. Mast cell mediators other than histamine induce pruritus in atopic dermatitis patients: A dermal microdialysis study. Br. J. Dermatol. 2000, 142, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Harvima, I.T.; Nilsson, G. Mast cells as regulators of skin inflammation and immunity. Acta Derm.-Venereol. 2011, 91, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Flohr, C.; Mann, J. New insights into the epidemiology of childhood atopic dermatitis. Allergy 2014, 69, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I.; Hanifin, J.; Simpson, E.L. Climatic factors are associated with childhood eczema prevalence in the united states. J. Investig. Dermatol. 2013, 133, 1752–1759. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.; Flohr, C. How epidemiology has challenged 3 prevailing concepts about atopic dermatitis. J. Allergy Clin. Immunol. 2006, 118, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Novak, N. Pathogenesis of atopic dermatitis. Clin. Exp. Allergy 2015, 45, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.B. Toxic interaction between Th2 cytokines and Staphylococcus aureus in atopic dermatitis. J. Investig. Dermatol. 2014, 134, 2069–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuer, K.; S, H.A.; Kapp, A.; Werfel, T. Staphylococcus aureus: Colonizing features and influence of an antibacterial treatment in adults with atopic dermatitis. Br. J. Dermatol. 2002, 147, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Kim, C.R.; Huh, I.S.; Jung, M.Y.; Seo, E.Y.; Park, J.H.; Lee, D.Y.; Yang, J.M. Staphylococcus aureus colonization in acute and chronic skin lesions of patients with atopic dermatitis. Ann. Dermatol. 2013, 25, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Totte, J.E.; van der Feltz, W.T.; Hennekam, M.; van Belkum, A.; van Zuuren, E.J.; Pasmans, S.G. Prevalence and odds of Staphylococcus aureus carriage in atopic dermatitis: A systematic review and meta-analysis. Br. J. Dermatol. 2016, 175, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Chinen, J.; Shearer, W.T. Advances in basic and clinical immunology in 2007. J. Allergy Clin. Immunol. 2008, 122, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, W.R.; An, H.J.; Kim, J.Y.; Chung, H.; Han, S.M.; Lee, M.L.; Lee, K.G.; Pak, S.C.; Park, K.K. Bee venom ameliorates compound 48/80-induced atopic dermatitis-related symptoms. Int. J. Clin. Exp. Pathol. 2013, 6, 2896–2903. [Google Scholar] [PubMed]
- Homey, B.; Steinhoff, M.; Ruzicka, T.; Leung, D.Y. Cytokines and chemokines orchestrate atopic skin inflammation. J. Allergy Clin. Immunol. 2006, 118, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Schuepbach-Mallepell, S.; Philippe, V.; Bruggen, M.C.; Watanabe, H.; Roques, S.; Baldeschi, C.; Gaide, O. Antagonistic effect of the inflammasome on thymic stromal lymphopoietin expression in the skin. J. Allergy Clin. Immunol. 2013, 132, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Grigoryev, D.N.; Howell, M.D.; Watkins, T.N.; Chen, Y.C.; Cheadle, C.; Boguniewicz, M.; Barnes, K.C.; Leung, D.Y. Vaccinia virus-specific molecular signature in atopic dermatitis skin. J. Allergy Clin. Immunol. 2010, 125, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Girardin, S.E.; Jehanno, M.; Mengin-Lecreulx, D.; Sansonetti, P.J.; Alzari, P.M.; Philpott, D.J. Identification of the critical residues involved in peptidoglycan detection by Nod1. J. Biol. Chem. 2005, 280, 38648–38656. [Google Scholar] [CrossRef] [PubMed]
- Traidl-Hoffmann, C.; Mariani, V.; Hochrein, H.; Karg, K.; Wagner, H.; Ring, J.; Mueller, M.J.; Jakob, T.; Behrendt, H. Pollen-associated phytoprostanes inhibit dendritic cell interleukin-12 production and augment T helper type 2 cell polarization. J. Exp. Med. 2005, 201, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Shreffler, W.G.; Castro, R.R.; Kucuk, Z.Y.; Charlop-Powers, Z.; Grishina, G.; Yoo, S.; Burks, A.W.; Sampson, H.A. The major glycoprotein allergen from Arachis hypogaea, Ara h 1, is a ligand of dendritic cell-specific ICAM-grabbing nonintegrin and acts as a Th2 adjuvant in vitro. J. Immunol. 2006, 177, 3677–3685. [Google Scholar] [CrossRef] [PubMed]
- Spergel, J.M.; Mizoguchi, E.; Brewer, J.P.; Martin, T.R.; Bhan, A.K.; Geha, R.S. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J. Clin. Investig. 1998, 101, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Novak, N. An update on the role of human dendritic cells in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2012, 129, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Boguniewicz, M.; Leung, D.Y. Atopic dermatitis: A disease of altered skin barrier and immune dysregulation. Immunol. Rev. 2011, 242, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Nomura, I.; Gao, B.; Boguniewicz, M.; Darst, M.A.; Travers, J.B.; Leung, D.Y. Distinct patterns of gene expression in the skin lesions of atopic dermatitis and psoriasis: A gene microarray analysis. J. Allergy Clin. Immunol. 2003, 112, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, J.S.; Cho, D.H.; Park, H.J. Molecular mechanisms of cutaneous inflammatory disorder: Atopic dermatitis. Int. J. Mol. Sci. 2016, 17, 1234. [Google Scholar] [CrossRef] [PubMed]
- Ong, P.Y.; Ohtake, T.; Brandt, C.; Strickland, I.; Boguniewicz, M.; Ganz, T.; Gallo, R.L.; Leung, D.Y. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med. 2002, 347, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Schittek, B. The antimicrobial skin barrier in patients with atopic dermatitis. Curr. Probl. Dermatol. 2011, 41, 54–67. [Google Scholar] [PubMed]
- Hasannejad, H.; Takahashi, R.; Kimishima, M.; Hayakawa, K.; Shiohara, T. Selective impairment of toll-like receptor 2-mediated proinflammatory cytokine production by monocytes from patients with atopic dermatitis. J. Allergy Clin. Immunol. 2007, 120, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Deng, X.; Chen, W.; Xu, J.; Chen, S.; Zhong, H.; Hao, F. Toll-like receptor 2 agonist Pam3CSK4 up-regulates FcepsilonRI receptor expression on monocytes from patients with severe extrinsic atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.F.; Artis, D. Sensing the outside world: TSLP regulates barrier immunity. Nat. Immunol. 2010, 11, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.; Boguniewicz, M.; Howell, M.D.; Nomura, I.; Hamid, Q.A. New insights into atopic dermatitis. J. Clin. Investig. 2004, 113, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Leung, D.Y.; Molet, S.; Boguniewicz, M.; Taha, R.; Christodoulopoulos, P.; Fukuda, T.; Elias, J.A.; Hamid, Q.A. Polarized in vivo expression of IL-11 and IL-17 between acute and chronic skin lesions. J. Allergy Clin. Immunol. 2003, 111, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Taha, R.A.; Leung, D.Y.; Ghaffar, O.; Boguniewicz, M.; Hamid, Q. In vivo expression of cytokine receptor mrna in atopic dermatitis. J. Allergy Clin. Immunol. 1998, 102, 245–250. [Google Scholar] [CrossRef]
- Teraki, Y.; Sakurai, A.; Izaki, S. IL-13/IL-22-coproducing T cells, a novel subset, are increased in atopic dermatitis. J. Allergy Clin. Immunol. 2013, 132, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Nomura, I.; Goleva, E.; Howell, M.D.; Hamid, Q.A.; Ong, P.Y.; Hall, C.F.; Darst, M.A.; Gao, B.; Boguniewicz, M.; Travers, J.B.; et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J. Immunol. 2003, 171, 3262–3269. [Google Scholar] [CrossRef] [PubMed]
- Albanesi, C.; Fairchild, H.R.; Madonna, S.; Scarponi, C.; De Pita, O.; Leung, D.Y.; Howell, M.D. IL-4 and IL-13 negatively regulate TNF-alpha- and IFN-gamma-induced beta-defensin expression through STAT-6, suppressor of cytokine signaling (SOCS)-1, and SOCS-3. J. Immunol. 2007, 179, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Wollenberg, A.; Gallo, R.L.; Flaig, M.; Streib, J.E.; Wong, C.; Pavicic, T.; Boguniewicz, M.; Leung, D.Y. Cathelicidin deficiency predisposes to eczema herpeticum. J. Allergy Clin. Immunol. 2006, 117, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Brauweiler, A.M.; Goleva, E.; Leung, D.Y. Th2 cytokines increase Staphylococcus aureus alpha toxin-induced keratinocyte death through the signal transducer and activator of transcription 6 (STAT6). J. Investig. Dermatol. 2014, 134, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Parker, M.L.; Mustelin, T.; Ranade, K. Past, present, and future for biologic intervention in atopic dermatitis. Allergy 2015, 70, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Braathen, L.R.; Simon, H.U. Eosinophils and atopic dermatitis. Allergy 2004, 59, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Molfino, N.A.; Gossage, D.; Kolbeck, R.; Parker, J.M.; Geba, G.P. Molecular and clinical rationale for therapeutic targeting of interleukin-5 and its receptor. Clin. Exp. Allergy 2012, 42, 712–737. [Google Scholar] [CrossRef] [PubMed]
- Jeong, C.W.; Ahn, K.S.; Rho, N.K.; Park, Y.D.; Lee, D.Y.; Lee, J.H.; Lee, E.S.; Yang, J.M. Differential in vivo cytokine mRNA expression in lesional skin of intrinsic vs. Extrinsic atopic dermatitis patients using semiquantitative RT-PCR. Clin. Exp. Allergy 2003, 33, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Spergel, J.M.; Mizoguchi, E.; Oettgen, H.; Bhan, A.K.; Geha, R.S. Roles of Th1 and Th2 cytokines in a murine model of allergic dermatitis. J. Clin. Investig. 1999, 103, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Omori, M.; Gyarmati, D.; Zhou, B.; Aye, T.; Brewer, A.; Comeau, M.R.; Campbell, D.J.; Ziegler, S.F. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J. Exp. Med. 2005, 202, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Oyoshi, M.K.; Larson, R.P.; Ziegler, S.F.; Geha, R.S. Mechanical injury polarizes skin dendritic cells to elicit a T(H)2 response by inducing cutaneous thymic stromal lymphopoietin expression. J. Allergy Clin. Immunol. 2010, 126, 976–984. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Oyoshi, M.K.; Garibyan, L.; Kumar, L.; Ziegler, S.F.; Geha, R.S. TSLP acts on infiltrating effector T cells to drive allergic skin inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 11875–11880. [Google Scholar] [CrossRef] [PubMed]
- Jessup, H.K.; Brewer, A.W.; Omori, M.; Rickel, E.A.; Budelsky, A.L.; Yoon, B.R.; Ziegler, S.F.; Comeau, M.R. Intradermal administration of thymic stromal lymphopoietin induces a T cell- and eosinophil-dependent systemic Th2 inflammatory response. J. Immunol. 2008, 181, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; The, L.; Batia, L.M.; Beattie, K.; Katibah, G.E.; McClain, S.P.; Pellegrino, M.; Estandian, D.M.; Bautista, D.M. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 2013, 155, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, U.; Hvid, M.; Johansen, C.; Buchner, M.; Folster-Holst, R.; Deleuran, M.; Vestergaard, C. TSLP, IL-31, Il-33 and sST2 are new biomarkers in endophenotypic profiling of adult and childhood atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Muller, A.; Lauerma, A.I.; Pivarcsi, A.; Soto, H.; Kemeny, L.; Alenius, H.; Dieu-Nosjean, M.C.; Meller, S.; Rieker, J.; et al. IL-31: A new link between t cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol. 2006, 117, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, M.H.; Hasan, Z.E.; Shaheen, K.Y. Serum measurement of interleukin-31 (IL-31) in paediatric atopic dermatitis: Elevated levels correlate with severity scoring. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, O.; Furue, M.; Nakagawa, H.; Shiramoto, M.; Hanada, R.; Matsuki, S.; Imayama, S.; Kato, M.; Hasebe, I.; Taira, K.; et al. The first trial of CIM331, a humanized antihuman interleukin-31 receptor A antibody, in healthy volunteers and patients with atopic dermatitis to evaluate safety, tolerability and pharmacokinetics of a single dose in a randomized, double-blind, placebo-controlled study. Br. J. Dermatol. 2016, 174, 296–304. [Google Scholar] [PubMed]
- Lee, J.H.; Cho, D.H.; Park, H.J. IL-18 and cutaneous inflammatory diseases. Int. J. Mol. Sci. 2015, 16, 29357–29369. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Aihara, M.; Kirino, M.; Harada, I.; Komori-Yamaguchi, J.; Yamaguchi, Y.; Nagashima, Y.; Ikezawa, Z. Interleukin-18 is elevated in the horny layer in patients with atopic dermatitis and is associated with Staphylococcus aureus colonization. Br. J. Dermatol. 2011, 164, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Terada, M.; Tsutsui, H.; Imai, Y.; Yasuda, K.; Mizutani, H.; Yamanishi, K.; Kubo, M.; Matsui, K.; Sano, H.; Nakanishi, K. Contribution of IL-18 to atopic-dermatitis-like skin inflammation induced by Staphylococcus aureus product in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 8816–8821. [Google Scholar] [CrossRef] [PubMed]
- Orfali, R.L.; Sato, M.N.; Takaoka, R.; Azor, M.H.; Rivitti, E.A.; Hanifin, J.M.; Aoki, V. Atopic dermatitis in adults: Evaluation of peripheral blood mononuclear cells proliferation response to Staphylococcus aureus enterotoxins A and B and analysis of interleukin-18 secretion. Exp. Dermatol. 2009, 18, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Choi, J.K.; Jung, H.J.; Park, K.H.; Jang, Y.H.; Lee, W.J.; Lee, S.J.; Kim, S.H.; Kang, H.Y.; Kim, J.M.; et al. Effects of topical application of a recombinant staphylococcal enterotoxin A on DNCB and dust mite extract-induced atopic dermatitis-like lesions in a murine model. Eur. J. Dermatol. 2014, 24, 186–193. [Google Scholar] [PubMed]
- Zedan, K.; Rasheed, Z.; Farouk, Y.; Alzolibani, A.A.; Bin Saif, G.; Ismail, H.A.; Al Robaee, A.A. Immunoglobulin E, interleukin-18 and interleukin-12 in patients with atopic dermatitis: Correlation with disease activity. J. Clin. Diagn. Res. 2015, 9, WC01–WC05. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, A.; Di Meglio, P.; Nestle, F.O. A role for Th17 cells in the immunopathogenesis of atopic dermatitis? J. Investig. Dermatol. 2008, 128, 2569–2571. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Mainardy, J.; Heratizadeh, A.; Satzger, I.; Werfel, T. Staphylococcal exotoxins induce interleukin 22 in human Th22 cells. Int. Arch. Allergy Immunol. 2014, 165, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; Krueger, J.G.; Guttman-Yassky, E. Skin barrier and immune dysregulation in atopic dermatitis: An evolving story with important clinical implications. J. Allergy Clin. Immunol. Pract. 2014, 2, 371–379; quiz 380–381. [Google Scholar] [CrossRef] [PubMed]
- Koga, C.; Kabashima, K.; Shiraishi, N.; Kobayashi, M.; Tokura, Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J. Investig. Dermatol. 2008, 128, 2625–2630. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; Esaki, H.; Gonzalez, J.; Malajian, D.; Shemer, A.; Noda, S.; Talasila, S.; Berry, A.; Gray, J.; Becker, L.; et al. Early pediatric atopic dermatitis shows only a cutaneous lymphocyte antigen (CLA)(+) TH2/TH1 cell imbalance, whereas adults acquire CLA(+) TH22/TC22 cell subsets. J. Allergy Clin. Immunol. 2015, 136, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Trifari, S.; Kaplan, C.D.; Tran, E.H.; Crellin, N.K.; Spits, H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat. Immunol. 2009, 10, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Geiger, R.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat. Immunol. 2009, 10, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Auriemma, M.; Vianale, G.; Amerio, P.; Reale, M. Cytokines and T cells in atopic dermatitis. Eur. Cytokine Netw. 2013, 24, 37–44. [Google Scholar] [PubMed]
- Czarnowicki, T.; Gonzalez, J.; Shemer, A.; Malajian, D.; Xu, H.; Zheng, X.; Khattri, S.; Gilleaudeau, P.; Sullivan-Whalen, M.; Suarez-Farinas, M.; et al. Severe atopic dermatitis is characterized by selective expansion of circulating TH2/TC2 and TH22/TC22, but not TH17/TC17, cells within the skin-homing T-cell population. J. Allergy Clin. Immunol. 2015, 136, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Krueger, J.G.; Guttman-Yassky, E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J. Allergy Clin. Immunol. 2015, 135, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Proksch, E.; Folster-Holst, R.; Brautigam, M.; Sepehrmanesh, M.; Pfeiffer, S.; Jensen, J.M. Role of the epidermal barrier in atopic dermatitis. J. Dtsch. Dermatol. Ges. 2009, 7, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Cork, M.J.; Danby, S.G.; Vasilopoulos, Y.; Hadgraft, J.; Lane, M.E.; Moustafa, M.; Guy, R.H.; Macgowan, A.L.; Tazi-Ahnini, R.; Ward, S.J. Epidermal barrier dysfunction in atopic dermatitis. J. Investig. Dermatol. 2009, 129, 1892–1908. [Google Scholar] [CrossRef] [PubMed]
- O'Regan, G.M.; Sandilands, A.; McLean, W.H.; Irvine, A.D. Filaggrin in atopic dermatitis. J. Allergy Clin. Immunol. 2008, 122, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Roelandt, T.; Heughebaert, C.; Hachem, J.P. Proteolytically active allergens cause barrier breakdown. J. Investig. Dermatol. 2008, 128, 1878–1880. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.K.; Kim, H.J.; Youm, J.K.; Ahn, S.K.; Choi, E.H.; Sohn, M.H.; Kim, K.E.; Hong, J.H.; Shin, D.M.; Lee, S.H. Mite and cockroach allergens activate protease-activated receptor 2 and delay epidermal permeability barrier recovery. J. Investig. Dermatol. 2008, 128, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Arlian, L.G.; Neal, J.S.; Morgan, M.S.; Vyszenski-Moher, D.L.; Rapp, C.M.; Alexander, A.K. Reducing relative humidity is a practical way to control dust mites and their allergens in homes in temperate climates. J. Allergy Clin. Immunol. 2001, 107, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Proksch, E.; Elias, P.M. Epidermal barrier in atopic dermatitis. In Atopic Dermatitis; Marcel Dekker: New York, NY, USA, 2002; pp. 123–143. [Google Scholar]
- Thyssen, J.P. Atopic dermatitis, filaggrin mutations and irritant contact dermatitis. Br. J. Dermatol. 2013, 168, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Thyssen, J.P.; Kezic, S. Causes of epidermal filaggrin reduction and their role in the pathogenesis of atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.N.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Illig, T.; Baurecht, H.; Irvine, A.D.; Rodriguez, E.; Diaz-Lacava, A.; Klopp, N.; Wagenpfeil, S.; Zhao, Y.; Liao, H.; et al. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J. Allergy Clin. Immunol. 2006, 118, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Marenholz, I.; Nickel, R.; Ruschendorf, F.; Schulz, F.; Esparza-Gordillo, J.; Kerscher, T.; Gruber, C.; Lau, S.; Worm, M.; Keil, T.; et al. Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. J. Allergy Clin. Immunol. 2006, 118, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Irvine, A.D.; McLean, W.H.; Leung, D.Y. Filaggrin mutations associated with skin and allergic diseases. N. Engl. J. Med. 2011, 365, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.P.; Moran, T.; Floudas, A.; Wurlod, F.; Kaszlikowska, A.; Salimi, M.; Quinn, E.M.; Oliphant, C.J.; Nunez, G.; McManus, R.; et al. Spontaneous atopic dermatitis is mediated by innate immunity, with the secondary lung inflammation of the atopic march requiring adaptive immunity. J. Allergy Clin. Immunol. 2016, 137, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.J.; Asai, Y.; Cordell, H.J.; Campbell, L.E.; Zhao, Y.; Liao, H.; Northstone, K.; Henderson, J.; Alizadehfar, R.; Ben-Shoshan, M.; et al. Loss-of-function variants in the filaggrin gene are a significant risk factor for peanut allergy. J. Allergy Clin. Immunol. 2011, 127, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilopoulos, Y.; Cork, M.J.; Murphy, R.; Williams, H.C.; Robinson, D.A.; Duff, G.W.; Ward, S.J.; Tazi-Ahnini, R. Genetic association between an AACC insertion in the 3’UTR of the stratum corneum chymotryptic enzyme gene and atopic dermatitis. J. Investig. Dermatol. 2004, 123, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Folster-Holst, R.; Stoll, M.; Koch, W.A.; Hampe, J.; Christophers, E.; Schreiber, S. Lack of association of SPINK5 polymorphisms with nonsyndromic atopic dermatitis in the population of northern Germany. Br. J. Dermatol. 2005, 152, 1365–1367. [Google Scholar] [CrossRef] [PubMed]
- Walley, A.J.; Chavanas, S.; Moffatt, M.F.; Esnouf, R.M.; Ubhi, B.; Lawrence, R.; Wong, K.; Abecasis, G.R.; Jones, E.Y.; Harper, J.I.; et al. Gene polymorphism in Netherton and common atopic disease. Nat. Genet. 2001, 29, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Hubiche, T.; Ged, C.; Benard, A.; Leaute-Labreze, C.; McElreavey, K.; de Verneuil, H.; Taieb, A.; Boralevi, F. Analysis of SPINK5, KLK7 and FLG genotypes in a french atopic dermatitis cohort. Acta Derm.-Venereol. 2007, 87, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Baurecht, H.; Wagenpfeil, S.; Henderson, J.; Novak, N.; Sandilands, A.; Chen, H.; Rodriguez, E.; O’Regan, G.M.; Watson, R.; et al. Analysis of the individual and aggregate genetic contributions of previously identified serine peptidase inhibitor kazal type 5 (SPINK5), kallikrein-related peptidase 7 (KLK7), and filaggrin (FLG) polymorphisms to eczema risk. J. Allergy Clin. Immunol. 2008, 122, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.J.; Apter, A.J.; Gupta, J.; Hoffstad, O.; Papadopoulos, M.; Campbell, L.E.; Sandilands, A.; McLean, W.H.; Rebbeck, T.R.; Mitra, N. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J. Allergy Clin. Immunol. 2012, 130, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Gao, P.; Kim, B.E.; Lesley, L.J.; Streib, J.E.; Taylor, P.A.; Zaccaro, D.J.; Boguniewicz, M.; Beck, L.A.; Hanifin, J.M.; et al. The signal transducer and activator of transcription 6 gene (STAT6) increases the propensity of patients with atopic dermatitis toward disseminated viral skin infections. J. Allergy Clin. Immunol. 2011, 128, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Gittler, J.K.; Shemer, A.; Suarez-Farinas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.F. Thymic stromal lymphopoietin and allergic disease. J. Allergy Clin. Immunol. 2012, 130, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Igyarto, B.Z.; Honda, T.; Egawa, G.; Otsuka, A.; Hara-Chikuma, M.; Watanabe, N.; Ziegler, S.F.; Tomura, M.; Inaba, K.; et al. Langerhans cells are critical in epicutaneous sensitization with protein antigen via thymic stromal lymphopoietin receptor signaling. J. Allergy Clin. Immunol. 2012, 129, 1048–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; Debenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol. 2007, 120, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Leung, D.Y.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol. 2008, 126, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Fairchild, H.R.; Kim, B.E.; Bin, L.; Boguniewicz, M.; Redzic, J.S.; Hansen, K.C.; Leung, D.Y. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J. Investig. Dermatol. 2008, 128, 2248–2258. [Google Scholar] [CrossRef] [PubMed]
- Zaniboni, M.C.; Samorano, L.P.; Orfali, R.L.; Aoki, V. Skin barrier in atopic dermatitis: Beyond filaggrin. Anais Bras. Dermatol. 2016, 91, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M. Claudin-based barrier in simple and stratified cellular sheets. Curr. Opin. Cell Biol. 2002, 14, 531–536. [Google Scholar] [CrossRef]
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight junction defects in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Batista, D.I.; Perez, L.; Orfali, R.L.; Zaniboni, M.C.; Samorano, L.P.; Pereira, N.V.; Sotto, M.N.; Ishizaki, A.S.; Oliveira, L.M.; Sato, M.N.; et al. Profile of skin barrier proteins (filaggrin, claudins 1 and 4) and Th1/Th2/Th17 cytokines in adults with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Yokouchi, M.; Kubo, A.; Kawasaki, H.; Yoshida, K.; Ishii, K.; Furuse, M.; Amagai, M. Epidermal tight junction barrier function is altered by skin inflammation, but not by filaggrin-deficient stratum corneum. J. Dermatol. Sci. 2015, 77, 28–36. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, A.; Slifka, M.K.; Rafaels, N.M.; Kuo, I.H.; Georas, S.N.; Boguniewicz, M.; Hata, T.; Schneider, L.C.; Hanifin, J.M.; Gallo, R.L.; et al. Reductions in claudin-1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J. Allergy Clin. Immunol. 2011, 128, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Cogen, A.L.; Nizet, V.; Gallo, R.L. Skin microbiota: A source of disease or defence? Br. J. Dermatol. 2008, 158, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; Green, E.D.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214.[Green Version]
- Bjorksten, B.; Sepp, E.; Julge, K.; Voor, T.; Mikelsaar, M. Allergy development and the intestinal microflora during the first year of life. J. Allergy Clin. Immunol. 2001, 108, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Seite, S.; Flores, G.E.; Henley, J.B.; Martin, R.; Zelenkova, H.; Aguilar, L.; Fierer, N. Microbiome of affected and unaffected skin of patients with atopic dermatitis before and after emollient treatment. J. Drugs Dermatol. 2014, 13, 1365–1372. [Google Scholar] [PubMed]
- Kennedy, E.A.; Connolly, J.; J, O.B.H.; Fallon, P.G.; McLean, W.I.; Murray, D.; Jo, J.H.; Segre, J.A.; Kong, H.H.; Irvine, A.D. Skin microbiome prior to development of atopic dermatitis: Early colonization with commensal Staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J. Allergy Clin. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wickens, K.; Black, P.; Stanley, T.V.; Mitchell, E.; Barthow, C.; Fitzharris, P.; Purdie, G.; Crane, J. A protective effect of Lactobacillus rhamnosus HN001 against eczema in the first 2 years of life persists to age 4 years. Clin. Exp. Allergy 2012, 42, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Min, T.K.; Lee, H.W.; Pyun, B.Y. Efficacy of probiotic therapy on atopic dermatitis in children: A randomized, double-blind, placebo-controlled trial. Allergy Asthma Immunol. Res. 2014, 6, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Kim, B.; Ban, J.; Lee, J.; Kim, B.J.; Choi, B.S.; Hwang, S.; Ahn, K.; Kim, J. A randomized trial of Lactobacillus plantarum CJLP133 for the treatment of atopic dermatitis. Pediatr. Allergy Immunol. 2012, 23, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Gore, C.; Custovic, A.; Tannock, G.W.; Munro, K.; Kerry, G.; Johnson, K.; Peterson, C.; Morris, J.; Chaloner, C.; Murray, C.S.; et al. Treatment and secondary prevention effects of the probiotics Lactobacillus paracasei or Bifidobacterium lactis on early infant eczema: Randomized controlled trial with follow-up until age 3 years. Clin. Exp. Allergy 2012, 42, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.G.; Li, T.H.; Peng, H.J. Lactobacillus salivarius plus fructo-oligosaccharide is superior to fructo-oligosaccharide alone for treating children with moderate to severe atopic dermatitis: A double-blind, randomized, clinical trial of efficacy and safety. Br. J. Dermatol. 2012, 166, 129–136. [Google Scholar] [CrossRef] [PubMed]
- van der Aa, L.B.; Lutter, R.; Heymans, H.S.; Smids, B.S.; Dekker, T.; van Aalderen, W.M.; Sillevis Smitt, J.H.; Knippels, L.M.; Garssen, J.; Nauta, A.J.; et al. No detectable beneficial systemic immunomodulatory effects of a specific synbiotic mixture in infants with atopic dermatitis. Clin. Exp. Allergy 2012, 42, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Orfali, R.L.; Sato, M.N.; Santos, V.G.; Titz, T.O.; Brito, C.A.; Duarte, A.J.; Takaoka, R.; Aoki, V. Staphylococcal enterotoxin B induces specific IgG4 and IgE antibody serum levels in atopic dermatitis. Int. J. Dermatol. 2015, 54, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Jinnestal, C.L.; Belfrage, E.; Back, O.; Schmidtchen, A.; Sonesson, A. Skin barrier impairment correlates with cutaneous Staphylococcus aureus colonization and sensitization to skin-associated microbial antigens in adult patients with atopic dermatitis. Int. J. Dermatol. 2014, 53, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Darabi, K.; Hostetler, S.G.; Bechtel, M.A.; Zirwas, M. The role of Malassezia in atopic dermatitis affecting the head and neck of adults. J. Am. Acad. Dermatol. 2009, 60, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Sonesson, A.; Bartosik, J.; Christiansen, J.; Roscher, I.; Nilsson, F.; Schmidtchen, A.; Back, O. Sensitization to skin-associated microorganisms in adult patients with atopic dermatitis is of importance for disease severity. Acta Derm.-Venereol. 2013, 93, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Buentke, E.; Heffler, L.C.; Wallin, R.P.; Lofman, C.; Ljunggren, H.G.; Scheynius, A. The allergenic yeast Malassezia furfur induces maturation of human dendritic cells. Clin. Exp. Allergy 2001, 31, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Higaki, S.; Morohashi, M.; Yamagishi, T.; Hasegawa, Y. Comparative study of Staphylococci from the skin of atopic dermatitis patients and from healthy subjects. Int. J. Dermatol. 1999, 38, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.H.; Oh, J.; Deming, C.; Conlan, S.; Grice, E.A.; Beatson, M.A.; Nomicos, E.; Polley, E.C.; Komarow, H.D.; Murray, P.R.; et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012, 22, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Schlievert, P.M.; Strandberg, K.L.; Lin, Y.C.; Peterson, M.L.; Leung, D.Y. Secreted virulence factor comparison between methicillin-resistant and methicillin-sensitive Staphylococcus aureus, and its relevance to atopic dermatitis. J. Allergy Clin. Immunol. 2010, 125, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Oscherwitz, J.; Cease, K.; Chan, S.; Munoz-Planillo, R.; Hasegawa, M.; Villaruz, A.; Cheung, G.; McGavin, M.; Otto, M. Staphylococcus δ-toxin promotes allergic skin disease by inducing mast cell degranulation. Nature 2013, 503, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Leppla, S.H.; Moayeri, M. Bacterial exotoxins and the inflammasome. Front. Immunol. 2015, 6, 570. [Google Scholar] [CrossRef] [PubMed]
- Bohach, G.A.; Fast, D.J.; Nelson, R.D.; Schlievert, P.M. Staphylococcal and streptococcal pyrogenic toxins involved in toxic shock syndrome and related illnesses. Crit. Rev. Microbiol. 1990, 17, 251–272. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.; Peterson, M.; Schlievert, P.; Fischetti, V.; Novick, R.; Ferretti, J.; Portnoy, D.; Rood, J. Toxins and superantigens of group A Streptococci. Gram-Posit. Pathog. 2006, 47–58. [Google Scholar]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Lina, G.; Bohach, G.A.; Nair, S.P.; Hiramatsu, K.; Jouvin-Marche, E.; Mariuzza, R. Standard nomenclature for the superantigens expressed by Staphylococcus. J. Infect. Dis. 2004, 189, 2334–2336. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, C.; Marquardt, Y.; Czaja, K.; Wenzel, J.; Frank, J.; Luscher-Firzlaff, J.; Luscher, B.; Baron, J.M. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J. Allergy Clin. Immunol. 2012, 129, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Macias, E.S.; Pereira, F.A.; Rietkerk, W.; Safai, B. Superantigens in dermatology. J. Am. Acad. Dermatol. 2011, 64, 455–472; quiz 473–474. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y. Atopic dermatitis: New insights and opportunities for therapeutic intervention. J. Allergy Clin. Immunol. 2000, 105, 860–876. [Google Scholar] [CrossRef] [PubMed]
- Ezepchuk, Y.V.; Leung, D.Y.; Middleton, M.H.; Bina, P.; Reiser, R.; Norris, D.A. Staphylococcal toxins and protein a differentially induce cytotoxicity and release of tumor necrosis factor-alpha from human keratinocytes. J. Investig. Dermatol. 1996, 107, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.B.; Leung, D.Y.; Johnson, C.; Schlievert, P.; Marques, M.; Cosgrove, J.; Clay, K.L. Augmentation of staphylococcal alpha-toxin signaling by the epidermal platelet-activating factor receptor. J. Investig. Dermatol. 2003, 120, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Choi, E.B.; Min, T.K.; Kim, J.H.; Kim, M.H.; Jeon, S.G.; Lee, B.J.; Gho, Y.S.; Jee, Y.K.; Pyun, B.Y.; et al. An important role of alpha-hemolysin in extracellular vesicles on the development of atopic dermatitis induced by Staphylococcus aureus. PLoS ONE 2014, 9, e100499. [Google Scholar]
- Kawasaki, H.; Nagao, K.; Kubo, A.; Hata, T.; Shimizu, A.; Mizuno, H.; Yamada, T.; Amagai, M. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J. Allergy Clin. Immunol. 2012, 129, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Miajlovic, H.; Fallon, P.G.; Irvine, A.D.; Foster, T.J. Effect of filaggrin breakdown products on growth of and protein expression by Staphylococcus aureus. J. Allergy Clin. Immunol. 2010, 126, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Bin, L.; Kim, B.E.; Brauweiler, A.; Goleva, E.; Streib, J.; Ji, Y.; Schlievert, P.M.; Leung, D.Y. Staphylococcus aureus alpha-toxin modulates skin host response to viral infection. J. Allergy Clin. Immunol. 2012, 130, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal enterotoxins. Toxins 2010, 2, 2177–2197. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; He, R.; Oyoshi, M.; Geha, R.S. Animal models of atopic dermatitis. J. Investig. Dermatol. 2009, 129, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.Y.; Jung, C.J.; Huang, C.N.; Huang, Y.C.; Lien, H.T.; Wang, W.B.; Wang, L.F.; Chia, J.S. A legume product fermented by Saccharomyces cerevisiae modulates cutaneous atopic dermatitis-like inflammation in mice. BMC Complement. Altern. Med. 2014, 14, 194. [Google Scholar] [CrossRef] [PubMed]
- Han, N.R.; Kang, S.W.; Moon, P.D.; Jang, J.B.; Kim, H.M.; Jeong, H.J. Genuine traditional Korean medicine, Naju Jjok (Chung-Dae, Polygonum tinctorium) improves 2,4-dinitrofluorobenzene-induced atopic dermatitis-like lesional skin. Phytomedicine 2014, 21, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.R.; Choi, J.; Kim, J.; Kim, H.; Kang, H.; Kim, E.H.; Chang, J.H.; Kim, Y.E.; Choi, Y.J.; Lee, K.W.; et al. 20-O-beta-d-glucopyranosyl-20(S)-protopanaxadiol-fortified ginseng extract attenuates the development of atopic dermatitis-like symptoms in NC/Nga mice. J. Ethnopharmacol. 2014, 151, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Kim, K.; Choi, I.; Ko, S.G. Topical herbal application in the management of atopic dermatitis: A review of animal studies. Mediat. Inflamm. 2014, 752103. [Google Scholar] [CrossRef] [PubMed]
- Bandara, M.; Arun, S.J.; Allanson, M.; Widyarini, S.; Chai, Z.; Reeve, V.E. Topical isoflavonoids reduce experimental cutaneous inflammation in mice. Immunol. Cell Biol. 2010, 88, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Kogiso, M.; Mitsuya, K.; Komatsu, T.; Yamamoto, S. Genistein suppresses development of spontaneous atopic-like dermatitis in NC/Nga mice. J. Nutr. Sci. Vitaminol. 2006, 52, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Cho, S.H. Effects of Korean red ginseng extract on the prevention of atopic dermatitis and its mechanism on early lesions in a murine model. J. Ethnopharmacol. 2013, 145, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, S.H.; Son, D.J.; Oh, K.W.; Kim, K.H.; Song, H.S.; Kim, G.J.; Oh, G.T.; Yoon, D.Y.; Hong, J.T. Antiarthritic effect of bee venom: Inhibition of inflammation mediator generation by suppression of NF-kappaB through interaction with the p50 subunit. Arthritis Rheum. 2004, 50, 3504–3515. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.G.; Cho, H.J.; Bae, Y.S.; Park, K.K.; Choe, J.Y.; Chung, I.K.; Kim, M.; Yeo, J.H.; Park, K.H.; Lee, Y.S.; et al. Bee venom suppresses LPS-mediated NO/iNOS induction through inhibition of PKC-alpha expression. J. Ethnopharmacol. 2009, 123, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Park, J.H.; Kim, K.H.; Lee, W.R.; Chang, Y.C.; Park, K.K.; Lee, K.G.; Han, S.M.; Yeo, J.H.; Pak, S.C. Bee venom inhibits hepatic fibrosis through suppression of pro-fibrogenic cytokine expression. Am. J. Chin. Med. 2010, 38, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y.; Kwon, Y.B.; Kim, H.W.; Roh, D.H.; Seo, H.S.; Han, H.J.; Lee, H.J.; Beitz, A.J.; Hwang, S.W.; Lee, J.H. Peripheral bee venom’s anti-inflammatory effect involves activation of the coeruleospinal pathway and sympathetic preganglionic neurons. Neurosci. Res. 2007, 59, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lee, K.; Yeo, J.; Kim, W.; Park, K. Biological effects of treatment of an animal skin wound with honeybee (Apis mellifera. L) venom. J. Plast. Reconstr. Aesthet. Surg. 2011, 64, e67–e72. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.R.; Kim, K.H.; An, H.J.; Kim, J.Y.; Chang, Y.C.; Chung, H.; Park, Y.Y.; Lee, M.L.; Park, K.K. The protective effects of melittin on Propionibacterium acnes-induced inflammatory responses in vitro and in vivo. J. Investig. Dermatol. 2014, 134, 1922–1930. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Hong, D.R.; Lee, E.H. Inhibition of mast cell-dependent anaphylactic reactions by the pigment of Polygonum tinctorium (Chung-Dae) in rats. Gen. Pharmacol. 1998, 31, 361–365. [Google Scholar] [CrossRef]
- Micallef, M.J.; Iwaki, K.; Ishihara, T.; Ushio, S.; Aga, M.; Kunikata, T.; Koya-Miyata, S.; Kimoto, T.; Ikeda, M.; Kurimoto, M. The natural plant product tryptanthrin ameliorates dextran sodium sulfate-induced colitis in mice. Int. Immunopharmacol. 2002, 2, 565–578. [Google Scholar] [CrossRef]
- Han, N.R.; Moon, P.D.; Kim, H.M.; Jeong, H.J. Tryptanthrin ameliorates atopic dermatitis through down-regulation of TSLP. Arch. Biochem. Biophys. 2014, 542, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Jin, S.W.; Park, B.H.; Kim, H.G.; Khanal, T.; Han, H.J.; Hwang, Y.P.; Choi, J.M.; Chung, Y.C.; Hwang, S.K.; et al. Cultivated ginseng inhibits 2,4-dinitrochlorobenzene-induced atopic dermatitis-like skin lesions in NC/Nga mice and TNF-alpha/IFN-gamma-induced TARC activation in HaCaT cells. Food Chem. Toxicol. 2013, 56, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Yang, A.W.; Xue, C.C.; Li, C.G.; Pang, C.; Zhang, W.; Williams, H.C. Chinese herbal medicine for atopic eczema. Cochrane Database Syst. Rev. 2013, CD008642. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O'Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Kankkunen, P.; Rintahaka, J.; Aalto, A.; Leino, M.; Majuri, M.L.; Alenius, H.; Wolff, H.; Matikainen, S. Trichothecene mycotoxins activate inflammatory response in human macrophages. J. Immunol. 2009, 182, 6418–6425. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yanagi, Y.; Ichinohe, T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 2012, 8, e1002857. [Google Scholar] [CrossRef] [PubMed]
- Palm, N.W.; Medzhitov, R. Role of the inflammasome in defense against venoms. Proc. Natl. Acad. Sci. USA 2013, 110, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Contassot, E.; Beer, H.D.; French, L.E. Interleukin-1, inflammasomes, autoinflammation and the skin. Swiss Med. Wkly 2012, 142, w13590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dokmeci, E.; Herrick, C.A. The immune system and atopic dermatitis. Semin. Cutan. Med. Surg. 2008, 27, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Craven, R.R.; Gao, X.; Allen, I.C.; Gris, D.; Bubeck Wardenburg, J.; McElvania-Tekippe, E.; Ting, J.P.; Duncan, J.A. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 2009, 4, e7446. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Franchi, L.; Miller, L.S.; Nunez, G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the NLRP3 inflammasome. J. Immunol. 2009, 183, 3942–3948. [Google Scholar] [CrossRef] [PubMed]
- Kistowska, M.; Fenini, G.; Jankovic, D.; Feldmeyer, L.; Kerl, K.; Bosshard, P.; Contassot, E.; French, L.E. Malassezia yeasts activate the NLRP3 inflammasome in antigen-presenting cells via Syk-kinase signalling. Exp. Dermatol. 2014, 23, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Sayama, K.; Tohyama, M.; Shirakata, Y.; Hanakawa, Y.; Tokumaru, S.; Yang, L.; Hirakawa, S.; Hashimoto, K. Mite allergen is a danger signal for the skin via activation of inflammasome in keratinocytes. J. Allergy Clin. Immunol. 2011, 127, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.; Champagne, C.; Morizot, A.; Lapointe, J.M.; Saleh, M. The inflammatory caspases-1 and -11 mediate the pathogenesis of dermatitis in sharpin-deficient mice. J. Immunol. 2015, 195, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Kabesch, M.; Peters, W.; Carr, D.; Leupold, W.; Weiland, S.K.; von Mutius, E. Association between polymorphisms in caspase recruitment domain containing protein 15 and allergy in two German populations. J. Allergy Clin. Immunol. 2003, 111, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Klopp, N.; Rummler, L.; Wagenpfeil, S.; Baurecht, H.J.; Gauger, A.; Darsow, U.; Jakob, T.; Novak, N.; Schafer, T.; et al. Association of CARD15 polymorphisms with atopy-related traits in a population-based cohort of Caucasian adults. Clin. Exp. Allergy 2005, 35, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Hysi, P.; Kabesch, M.; Moffatt, M.F.; Schedel, M.; Carr, D.; Zhang, Y.; Boardman, B.; von Mutius, E.; Weiland, S.K.; Leupold, W.; et al. NOD1 variation, immunoglobulin E and asthma. Hum. Mol. Genet. 2005, 14, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Baumert, K.; Heratizadeh, A.; Satzger, I.; Werfel, T. Impaired NLRP3 inflammasome expression and function in atopic dermatitis due to Th2 milieu. Allergy 2014, 69, 1058–1067. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Toxin/Natural Material | Results and Mechanism | References |
|---|---|---|
| Saccharomyces cerevisiae legume fermented product | Skin: thickness ↓, eosinophil ↓, IL-5 ↓, IL-13 ↓, CXCL11 ↓ Lymph node: IL-4 ↓, IL-17A ↓ | [143] |
| Bee venom | Skin: scratching ↓, mast cell degranulation ↓ TNF-α ↓, IL-1β ↓ | [15] |
| Polygonum tinctorium (Naju Jjok) | Skin: thickness ↓, inflammatory cells ↓, TSLP ↓ Serum: IL-4 ↓, IgE ↓ | [144] |
| Ginseng extract | Skin: IL-4 ↓, IL-5 ↓, IL-13 ↓, IFN-γ ↓, TNF-α ↓, CCL17 ↓ Serum: IgE ↓, CCL17↓ | [145] |
| Korean red ginseng extract | Skin: thickness ↓, water loss ↓, inflammatory cells ↓ TNF- α ↓, TSLP ↓, Serum: IgE ↓ | [149] |
| Herbs | See reference Suppression of Th2 response | [146] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, K.-D.; Pak, S.C.; Park, K.-K. The Pathogenetic Effect of Natural and Bacterial Toxins on Atopic Dermatitis. Toxins 2017, 9, 3. https://doi.org/10.3390/toxins9010003
Park K-D, Pak SC, Park K-K. The Pathogenetic Effect of Natural and Bacterial Toxins on Atopic Dermatitis. Toxins. 2017; 9(1):3. https://doi.org/10.3390/toxins9010003
Chicago/Turabian StylePark, Kyung-Duck, Sok Cheon Pak, and Kwan-Kyu Park. 2017. "The Pathogenetic Effect of Natural and Bacterial Toxins on Atopic Dermatitis" Toxins 9, no. 1: 3. https://doi.org/10.3390/toxins9010003
APA StylePark, K. -D., Pak, S. C., & Park, K. -K. (2017). The Pathogenetic Effect of Natural and Bacterial Toxins on Atopic Dermatitis. Toxins, 9(1), 3. https://doi.org/10.3390/toxins9010003