
Structure–Function Relationships Underlying the Capacity of Bordetella Adenylate Cyclase Toxin to Disarm Host Phagocytes

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
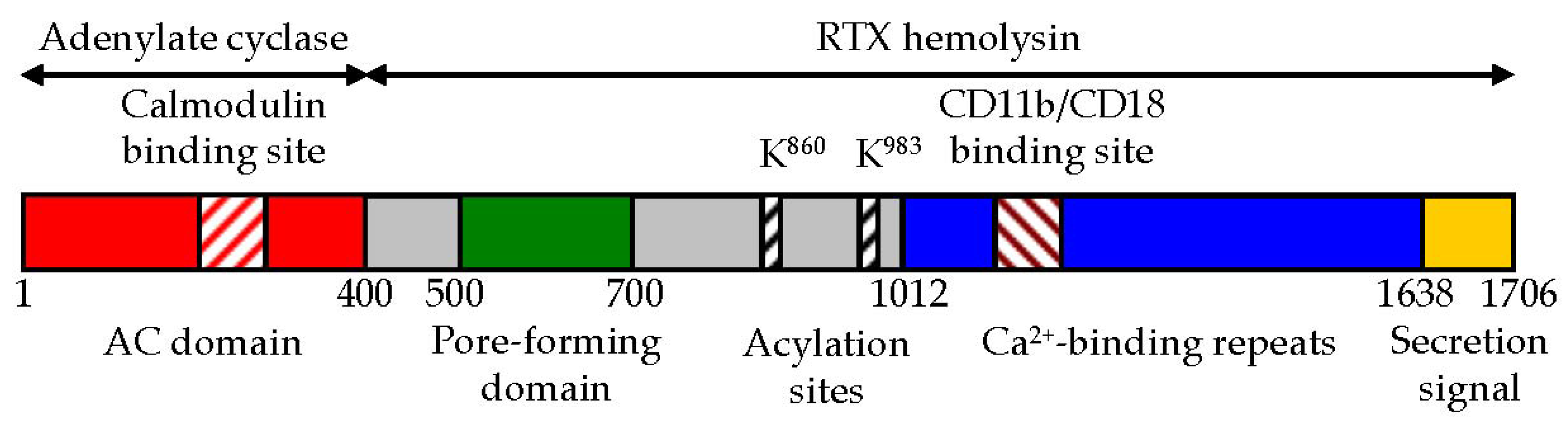
2. CyaA Structure
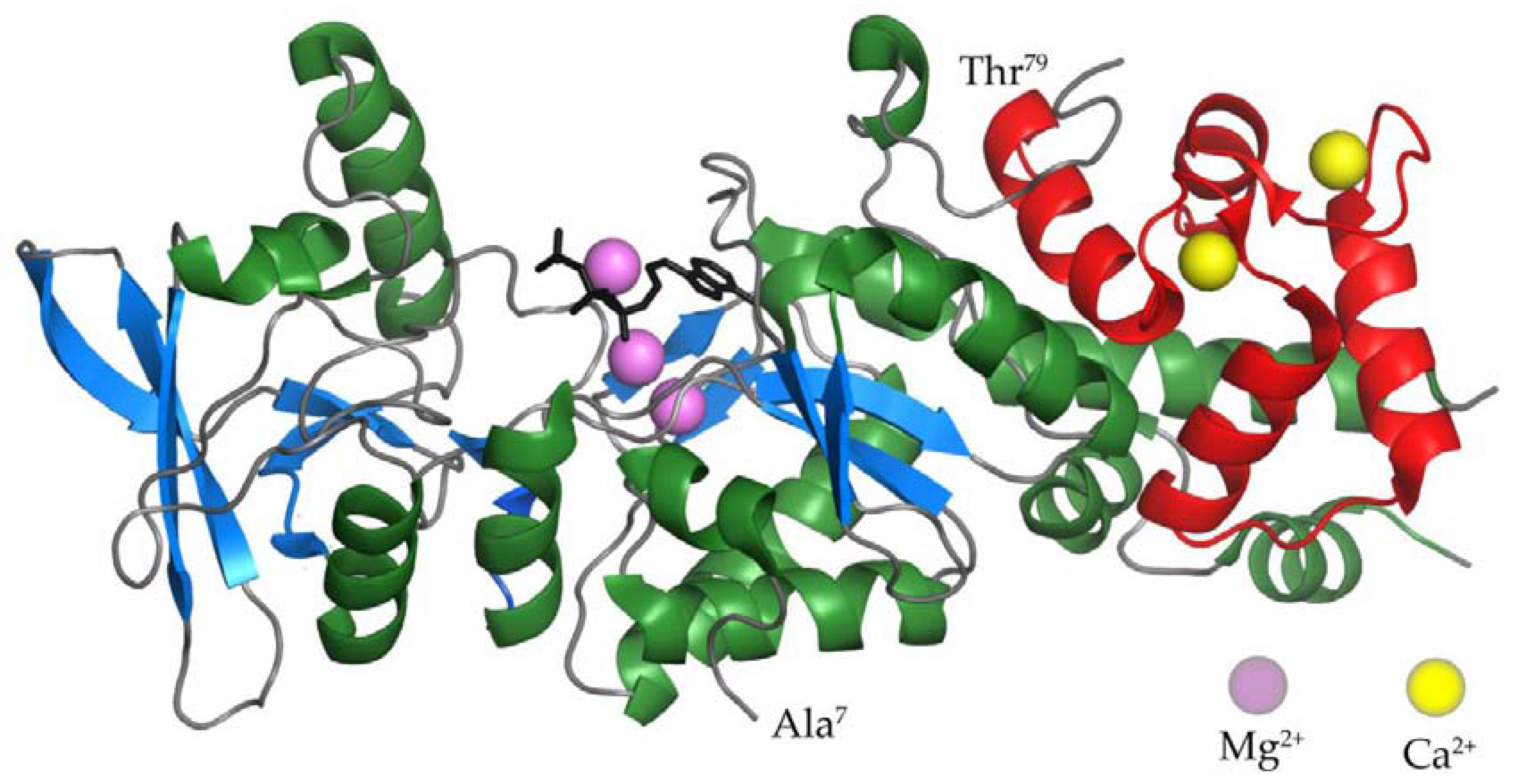
2.1. The AC Domain of CyaA
2.2. The AC to Hly-Linking Segment
2.3. The Pore-Forming Domain of CyaA
2.4. The Activation Domain of CyaA
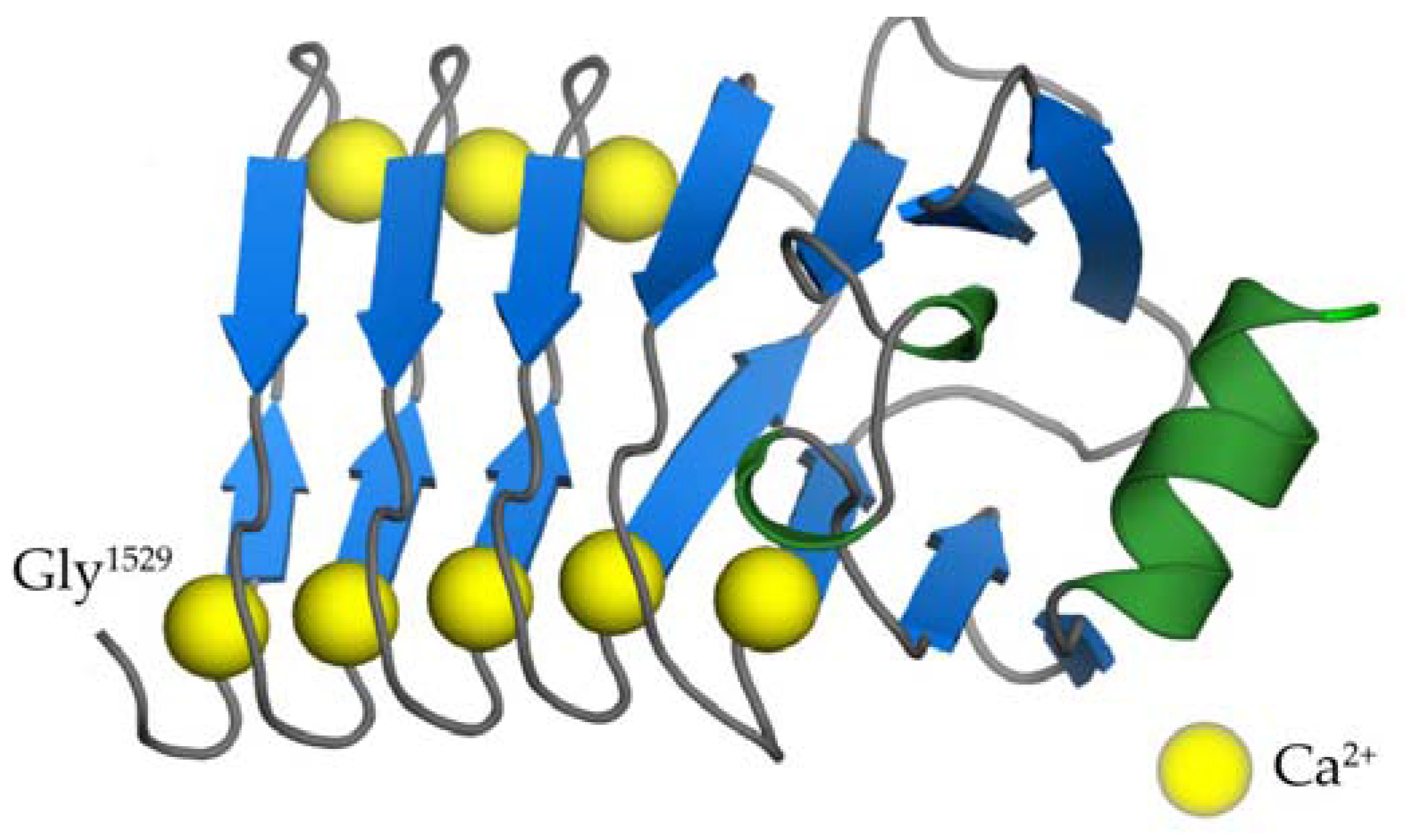
2.5. The RTX Domain of CyaA
2.6. The C-Terminal Secretion Signal of CyaA
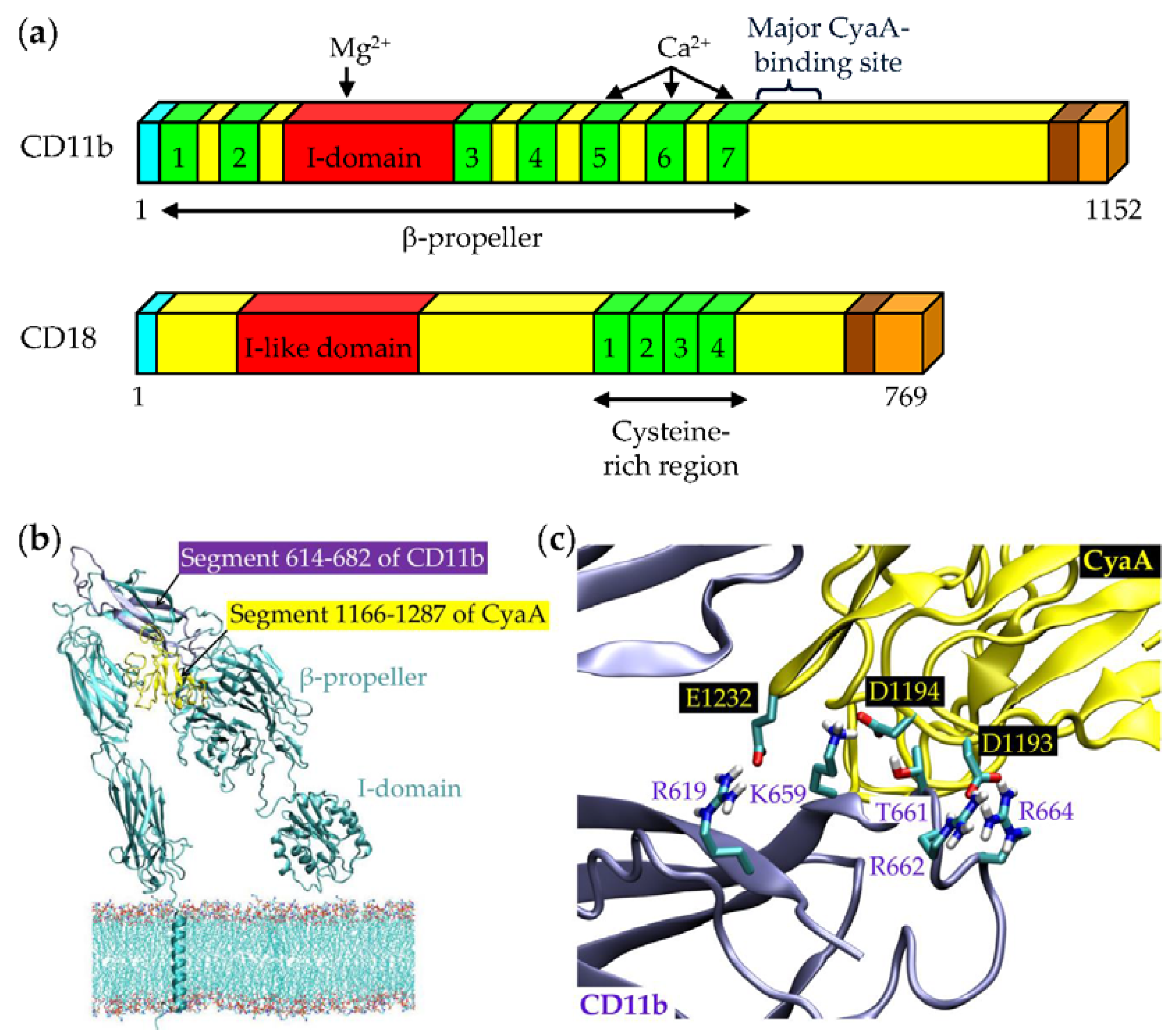
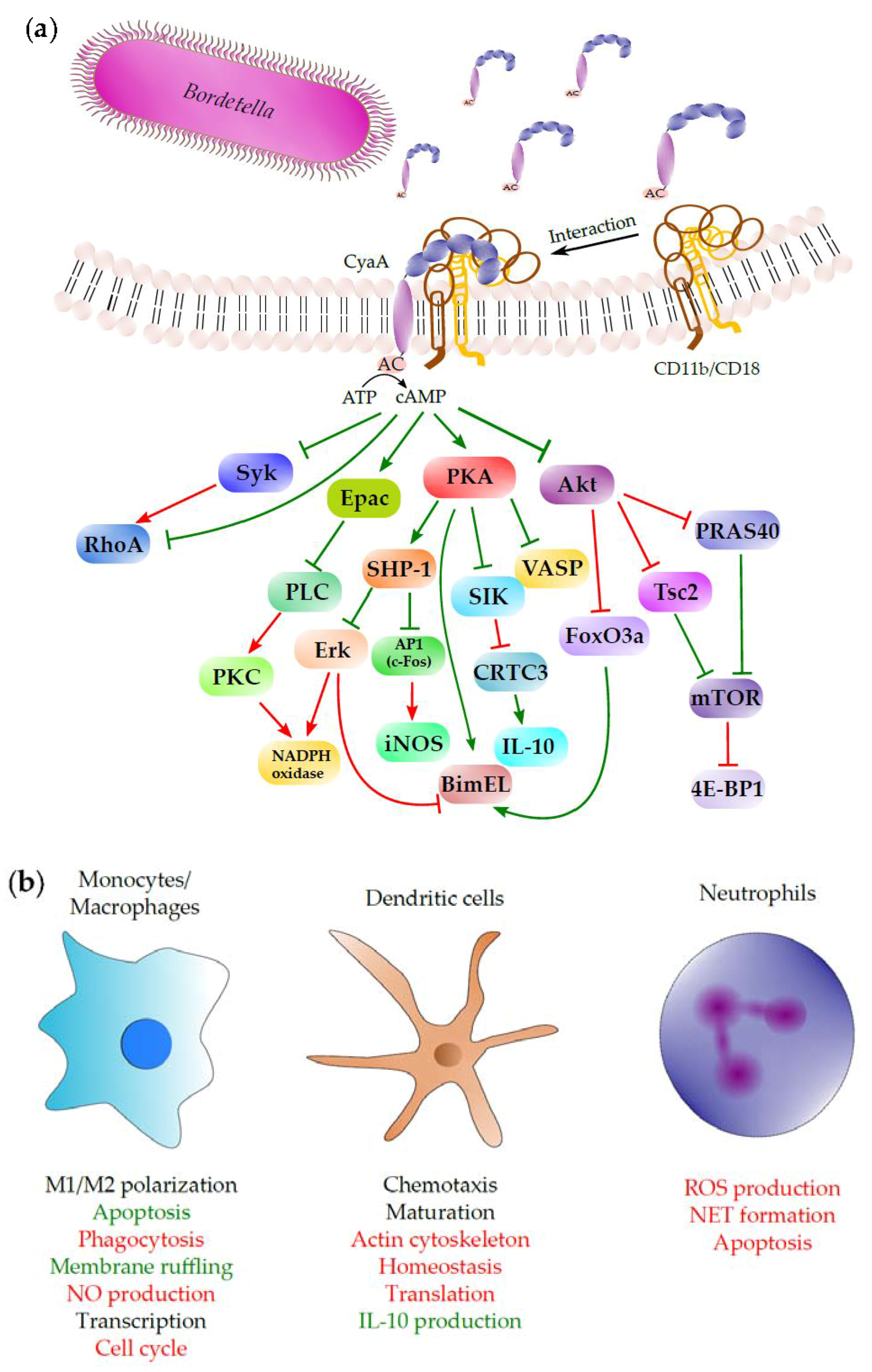
3. Binding Interaction of CyaA with the Complement Receptor 3 on the Surface of Phagocytic Cells
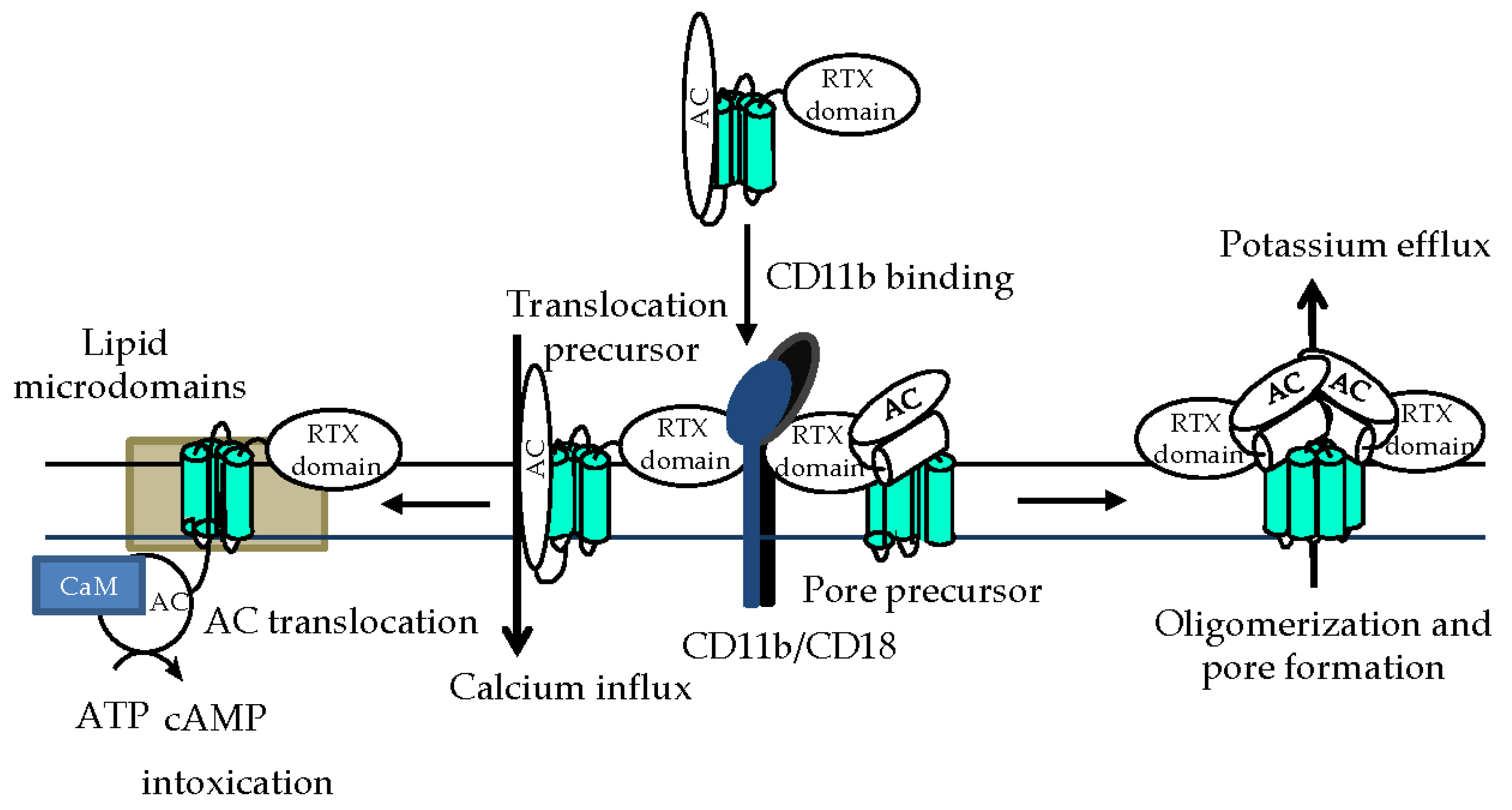
4. Phagocyte Membrane Penetration and Permeabilization by CyaA
5. Subversion of Spleen Tyrosine Kinase (Syk)-Mediated Signaling by CyaA-Produced cAMP
6. Subversion of Airway Sentinel Cell Functions by CyaA
6.1. Alveolar Macrophages
6.2. Modulation of Macrophage Functions by CyaA/cAMP Signaling
6.3. CyaA/cAMP-Triggered Reprogramming of Gene Expression in Macrophages
6.4. Induction of Macrophage Apoptosis by CyaA
6.5. Intraepithelial Dendritic Cells
6.6. CyaA Action on Neutrophils
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Melvin, J.A.; Scheller, E.V.; Miller, J.F.; Cotter, P.A. Bordetella pertussis pathogenesis: Current and future challenges. Nat. Rev. Microbiol. 2014, 12, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.H.T.; Duclos, P.; Nelson, E.A.S.; Hutubessy, R.C.W. An update of the global burden of pertussis in children younger than 5 years: A modelling study. Lancet Infect. Dis. 2017, 17, 974–980. [Google Scholar] [CrossRef]
- Cherry, J.D. Pertussis in young infants throughout the world. Clin. Infect. Dis. 2016, 63, S119–S122. [Google Scholar] [CrossRef] [PubMed]
- Vojtova, J.; Kamanova, J.; Sebo, P. Bordetella adenylate cyclase toxin: A swift saboteur of host defense. Curr. Opin. Microbiol. 2006, 9, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Sebo, P.; Osicka, R.; Masin, J. Adenylate cyclase toxin-hemolysin relevance for pertussis vaccines. Expert Rev. Vaccines 2014, 13, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Linhartova, I.; Bumba, L.; Masin, J.; Basler, M.; Osicka, R.; Kamanova, J.; Prochazkova, K.; Adkins, I.; Hejnova-Holubova, J.; Sadilkova, L.; et al. RTX proteins: A highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.; Sebo, P.; Bellalou, J.; Ladant, D. Interaction of calcium with Bordetella pertussis adenylate cyclase toxin. Characterization of multiple calcium-binding sites and calcium-induced conformational changes. J. Biol. Chem. 1995, 270, 26370–26376. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Osicka, R.; Bumba, L.; Sebo, P. Bordetella adenylate cyclase toxin: A unique combination of a pore-forming moiety with a cell-invading adenylate cyclase enzyme. Pathog. Dis. 2015, 73, ftv075. [Google Scholar] [CrossRef] [PubMed]
- Glaser, P.; Sakamoto, H.; Bellalou, J.; Ullmann, A.; Danchin, A. Secretion of cyclolysin, the calmodulin-sensitive adenylate cyclase-haemolysin bifunctional protein of Bordetella pertussis. EMBO J. 1988, 7, 3997–4004. [Google Scholar] [PubMed]
- Iwaki, M.; Ullmann, A.; Sebo, P. Identification by in vitro complementation of regions required for cell-invasive activity of Bordetella pertussis adenylate cyclase toxin. Mol. Microbiol. 1995, 17, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Benz, R.; Maier, E.; Ladant, D.; Ullmann, A.; Sebo, P. Adenylate cyclase toxin (CyaA) of Bordetella pertussis. Evidence for the formation of small ion-permeable channels and comparison with HlyA of Escherichia coli. J. Biol. Chem. 1994, 269, 27231–27239. [Google Scholar] [PubMed]
- Hackett, M.; Guo, L.; Shabanowitz, J.; Hunt, D.F.; Hewlett, E.L. Internal lysine palmitoylation in adenylate cyclase toxin from Bordetella pertussis. Science 1994, 266, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Hackett, M.; Walker, C.B.; Guo, L.; Gray, M.C.; Van Cuyk, S.; Ullmann, A.; Shabanowitz, J.; Hunt, D.F.; Hewlett, E.L.; Sebo, P. Hemolytic, but not cell-invasive activity, of adenylate cyclase toxin is selectively affected by differential fatty-acylation in Escherichia coli. J. Biol. Chem. 1995, 270, 20250–20253. [Google Scholar] [CrossRef] [PubMed]
- Osicka, R.; Osickova, A.; Basar, T.; Guermonprez, P.; Rojas, M.; Leclerc, C.; Sebo, P. Delivery of CD8(+) T-cell epitopes into major histocompatibility complex class I antigen presentation pathway by Bordetella pertussis adenylate cyclase: Delineation of cell invasive structures and permissive insertion sites. Infect. Immun. 2000, 68, 247–256. [Google Scholar] [PubMed]
- Bumba, L.; Masin, J.; Macek, P.; Wald, T.; Motlova, L.; Bibova, I.; Klimova, N.; Bednarova, L.; Veverka, V.; Kachala, M.; et al. Calcium-driven folding of RTX domain beta-rolls ratchets translocation of RTX proteins through type I secretion ducts. Mol. Cell 2016, 62, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Sebo, P.; Ladant, D. Repeat sequences in the Bordetella pertussis adenylate cyclase toxin can be recognized as alternative carboxy-proximal secretion signals by the Escherichia coli alpha-haemolysin translocator. Mol. Microbiol. 1993, 9, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Khelef, N.; Blouin, E.; Rieu, P.; Ricciardi-Castagnoli, P.; Guiso, N.; Ladant, D.; Leclerc, C. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18). J. Exp. Med. 2001, 193, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Osicka, R.; Osickova, A.; Hasan, S.; Bumba, L.; Cerny, J.; Sebo, P. Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. eLife 2015, 4, e10766. [Google Scholar] [CrossRef] [PubMed]
- Bellalou, J.; Sakamoto, H.; Ladant, D.; Geoffroy, C.; Ullmann, A. Deletions affecting hemolytic and toxin activities of Bordetella pertussis adenylate cyclase. Infect. Immun. 1990, 58, 3242–3247. [Google Scholar] [PubMed]
- Gordon, V.M.; Young, W.W., Jr.; Lechler, S.M.; Gray, M.C.; Leppla, S.H.; Hewlett, E.L. Adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Different processes for interaction with and entry into target cells. J. Biol. Chem. 1989, 264, 14792–14796. [Google Scholar] [PubMed]
- Holubova, J.; Kamanova, J.; Jelinek, J.; Tomala, J.; Masin, J.; Kosova, M.; Stanek, O.; Bumba, L.; Michalek, J.; Kovar, M.; et al. Delivery of large heterologous polypeptides across the cytoplasmic membrane of antigen-presenting cells by the Bordetella RTX hemolysin moiety lacking the adenylyl cyclase domain. Infect. Immun. 2012, 80, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.; Cook, G.H.; Goldhammer, A.R.; Berkowitz, S.A. Calmodulin activates prokaryotic adenylate cyclase. Proc. Natl. Acad. Sci. USA 1980, 77, 3841–3844. [Google Scholar] [CrossRef] [PubMed]
- Confer, D.L.; Eaton, J.W. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 1982, 217, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Ladant, D.; Ullmann, A. Bordetella pertussis adenylate cyclase: A toxin with multiple talents. Trends Microbiol. 1999, 7, 172–176. [Google Scholar] [CrossRef]
- Szabo, G.; Gray, M.C.; Hewlett, E.L. Adenylate cyclase toxin from Bordetella pertussis produces ion conductance across artificial lipid bilayers in a calcium- and polarity-dependent manner. J. Biol. Chem. 1994, 269, 22496–22499. [Google Scholar] [PubMed]
- Ehrmann, I.E.; Gray, M.C.; Gordon, V.M.; Gray, L.S.; Hewlett, E.L. Hemolytic activity of adenylate cyclase toxin from Bordetella pertussis. FEBS Lett. 1991, 278, 79–83. [Google Scholar] [PubMed]
- Basler, M.; Masin, J.; Osicka, R.; Sebo, P. Pore-forming and enzymatic activities of Bordetella pertussis adenylate cyclase toxin synergize in promoting lysis of monocytes. Infect. Immun. 2006, 74, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Fiser, R.; Masin, J.; Basler, M.; Krusek, J.; Spulakova, V.; Konopasek, I.; Sebo, P. Third activity of Bordetella adenylate cyclase (AC) toxin-hemolysin. Membrane translocation of AC domain polypeptide promotes calcium influx into CD11b+ monocytes independently of the catalytic and hemolytic activities. J. Biol. Chem. 2007, 282, 2808–2820. [Google Scholar] [CrossRef] [PubMed]
- Cerny, O.; Kamanova, J.; Masin, J.; Bibova, I.; Skopova, K.; Sebo, P. Bordetella pertussis adenylate cyclase toxin blocks induction of bactericidal nitric oxide in macrophages through cAMP-dependent activation of the SHP-1 phosphatase. J. Immunol. 2015, 194, 4901–4913. [Google Scholar] [CrossRef] [PubMed]
- Cerny, O.; Anderson, K.E.; Stephens, L.R.; Hawkins, P.T.; Sebo, P. cAMP signaling of adenylate cyclase toxin blocks the oxidative burst of neutrophils through Epac-mediated inhibition of phospholipase C activity. J. Immunol. 2017, 198, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Kamanova, J.; Kofronova, O.; Masin, J.; Genth, H.; Vojtova, J.; Linhartova, I.; Benada, O.; Just, I.; Sebo, P. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J. Immunol. 2008, 181, 5587–5597. [Google Scholar] [CrossRef] [PubMed]
- Dunne, A.; Ross, P.J.; Pospisilova, E.; Masin, J.; Meaney, A.; Sutton, C.E.; Iwakura, Y.; Tschopp, J.; Sebo, P.; Mills, K.H. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J. Immunol. 2010, 185, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Fiser, R.; Masin, J.; Bumba, L.; Pospisilova, E.; Fayolle, C.; Basler, M.; Sadilkova, L.; Adkins, I.; Kamanova, J.; Cerny, J.; et al. Calcium influx rescues adenylate cyclase-hemolysin from rapid cell membrane removal and enables phagocyte permeabilization by toxin pores. PLoS Pathog. 2012, 8, e1002580. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.; Szabo, G.; Otero, A.S.; Gray, L.; Hewlett, E. Distinct mechanisms for K+ efflux, intoxication, and hemolysis by Bordetella pertussis AC toxin. J. Biol. Chem. 1998, 273, 18260–18267. [Google Scholar] [CrossRef] [PubMed]
- Svedova, M.; Masin, J.; Fiser, R.; Cerny, O.; Tomala, J.; Freudenberg, M.; Tuckova, L.; Kovar, M.; Dadaglio, G.; Adkins, I.; et al. Pore-formation by adenylate cyclase toxoid activates dendritic cells to prime CD8+ and CD4+ T cells. Immunol. Cell Biol. 2016, 94, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Wald, T.; Petry-Podgorska, I.; Fiser, R.; Matousek, T.; Dedina, J.; Osicka, R.; Sebo, P.; Masin, J. Quantification of potassium levels in cells treated with Bordetella adenylate cyclase toxin. Anal. Biochem. 2014, 450, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Bachelet, M.; Richard, M.J.; Francois, D.; Polla, B.S. Mitochondrial alterations precede Bordetella pertussis-induced apoptosis. FEMS Immunol. Med. Microbiol. 2002, 32, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, E.L.; Donato, G.M.; Gray, M.C. Macrophage cytotoxicity produced by adenylate cyclase toxin from Bordetella pertussis: More than just making cyclic amp! Mol. Microbiol. 2006, 59, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Khelef, N.; Guiso, N. Induction of macrophage apoptosis by Bordetella pertussis adenylate cyclase-hemolysin. FEMS Microbiol. Lett. 1995, 134, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Barzu, O.; Danchin, A. Adenylyl cyclases: A heterogeneous class of ATP-utilizing enzymes. Prog. Nucleic Acid Res. Mol. Biol. 1994, 49, 241–283. [Google Scholar] [PubMed]
- Shen, Y.; Zhukovskaya, N.L.; Guo, Q.; Florian, J.; Tang, W.J. Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J. 2005, 24, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.J.; Guo, Q. The adenylyl cyclase activity of anthrax edema factor. Mol. Asp. Med. 2009, 30, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Morrow, K.A.; Frank, D.W.; Balczon, R.; Stevens, T. The Pseudomonas aeruginosa exoenzyme Y: A promiscuous nucleotidyl cyclase edema factor and virulence determinant. Handb. Exp. Pharmacol. 2017, 238, 67–85. [Google Scholar] [PubMed]
- Yahr, T.L.; Vallis, A.J.; Hancock, M.K.; Barbieri, J.T.; Frank, D.W. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc. Natl. Acad. Sci. USA 1998, 95, 13899–13904. [Google Scholar] [CrossRef] [PubMed]
- Belyy, A.; Raoux-Barbot, D.; Saveanu, C.; Namane, A.; Ogryzko, V.; Worpenberg, L.; David, V.; Henriot, V.; Fellous, S.; Merrifield, C.; et al. Actin activates Pseudomonas aeruginosa ExoY nucleotidyl cyclase toxin and ExoY-like effector domains from MARTX toxins. Nat. Commun. 2016, 7, 13582. [Google Scholar] [CrossRef] [PubMed]
- Ladant, D. Interaction of Bordetella pertussis adenylate cyclase with calmodulin. Identification of two separated calmodulin-binding domains. J. Biol. Chem. 1988, 263, 2612–2618. [Google Scholar] [PubMed]
- Rogel, A.; Schultz, J.E.; Brownlie, R.M.; Coote, J.G.; Parton, R.; Hanski, E. Bordetella pertussis adenylate cyclase: Purification and characterization of the toxic form of the enzyme. EMBO J. 1989, 8, 2755–2760. [Google Scholar] [PubMed]
- Gottle, M.; Dove, S.; Kees, F.; Schlossmann, J.; Geduhn, J.; Konig, B.; Shen, Y.; Tang, W.J.; Kaever, V.; Seifert, R. Cytidylyl and uridylyl cyclase activity of Bacillus anthracis edema factor and Bordetella pertussis CyaA. Biochemistry 2010, 49, 5494–5503. [Google Scholar] [CrossRef] [PubMed]
- Glaser, P.; Elmaoglou-Lazaridou, A.; Krin, E.; Ladant, D.; Barzu, O.; Danchin, A. Identification of residues essential for catalysis and binding of calmodulin in Bordetella pertussis adenylate cyclase by site-directed mutagenesis. EMBO J. 1989, 8, 967–972. [Google Scholar] [PubMed]
- Ladant, D.; Michelson, S.; Sarfati, R.; Gilles, A.M.; Predeleanu, R.; Barzu, O. Characterization of the calmodulin-binding and of the catalytic domains of Bordetella pertussis adenylate cyclase. J. Biol. Chem. 1989, 264, 4015–4020. [Google Scholar] [PubMed]
- Munier, H.; Bouhss, A.; Krin, E.; Danchin, A.; Gilles, A.M.; Glaser, P.; Barzu, O. The role of histidine 63 in the catalytic mechanism of Bordetella pertussis adenylate cyclase. J. Biol. Chem. 1992, 267, 9816–9820. [Google Scholar] [PubMed]
- Glaser, P.; Munier, H.; Gilles, A.M.; Krin, E.; Porumb, T.; Barzu, O.; Sarfati, R.; Pellecuer, C.; Danchin, A. Functional consequences of single amino acid substitutions in calmodulin-activated adenylate cyclase of Bordetella pertussis. EMBO J. 1991, 10, 1683–1688. [Google Scholar] [PubMed]
- Guo, Q.; Shen, Y.; Lee, Y.S.; Gibbs, C.S.; Mrksich, M.; Tang, W.J. Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 2005, 24, 3190–3201. [Google Scholar] [CrossRef] [PubMed]
- Bouhss, A.; Krin, E.; Munier, H.; Gilles, A.M.; Danchin, A.; Glaser, P.; Barzu, O. Cooperative phenomena in binding and activation of Bordetella pertussis adenylate cyclase by calmodulin. J. Biol. Chem. 1993, 268, 1690–1694. [Google Scholar] [PubMed]
- Guo, Q.; Jureller, J.E.; Warren, J.T.; Solomaha, E.; Florian, J.; Tang, W.J. Protein-protein docking and analysis reveal that two homologous bacterial adenylyl cyclase toxins interact with calmodulin differently. J. Biol. Chem. 2008, 283, 23836–23845. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.I.; Goebel, E.; Hariraju, D.; Finley, N.L. Mutation in the beta-hairpin of the Bordetella pertussis adenylate cyclase toxin modulates N-lobe conformation in calmodulin. Biochem. Biophys. Res. Commun. 2014, 453, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.I.; Emerson, C.C.; Johns, C.W.; Finley, N.L. Interaction with adenylate cyclase toxin from Bordetella pertussis affects the metal binding properties of calmodulin. FEBS Open Bio 2017, 7, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Bumba, L.; Masin, J.; Fiser, R.; Sebo, P. Bordetella adenylate cyclase toxin mobilizes its beta2 integrin receptor into lipid rafts to accomplish translocation across target cell membrane in two steps. PLoS Pathog. 2010, 6, e1000901. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.S.; Yi, X.B.; Gray, M.C.; Szabo, G.; Hewlett, E.L. Membrane depolarization prevents cell invasion by Bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 1995, 270, 9695–9697. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lee, Y.S.; Soelaiman, S.; Bergson, P.; Lu, D.; Chen, A.; Beckingham, K.; Grabarek, Z.; Mrksich, M.; Tang, W.J. Physiological calcium concentrations regulate calmodulin binding and catalysis of adenylyl cyclase exotoxins. EMBO J. 2002, 21, 6721–6732. [Google Scholar] [CrossRef] [PubMed]
- Subrini, O.; Sotomayor Perez, A.C.; Hessel, A.; Spiaczka-Karst, J.; Selwa, E.; Sapay, N.; Veneziano, R.; Pansieri, J.; Chopineau, J.; Ladant, D.; et al. Characterization of a membrane-active peptide from the Bordetella pertussis CyaA toxin. J. Biol. Chem. 2013, 288, 32585–32598. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Osickova, A.; Sukova, A.; Fiser, R.; Halada, P.; Bumba, L.; Linhartova, I.; Osicka, R.; Sebo, P. Negatively charged residues of the segment linking the enzyme and cytolysin moieties restrict the membrane-permeabilizing capacity of adenylate cyclase toxin. Sci. Rep. 2016, 6, 29137. [Google Scholar] [CrossRef] [PubMed]
- Karst, J.C.; Barker, R.; Devi, U.; Swann, M.J.; Davi, M.; Roser, S.J.; Ladant, D.; Chenal, A. Identification of a region that assists membrane insertion and translocation of the catalytic domain of Bordetella pertussis CyaA toxin. J. Biol. Chem. 2012, 287, 9200–9212. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.C.; Lee, S.J.; Gray, L.S.; Zaretzky, F.R.; Otero, A.S.; Szabo, G.; Hewlett, E.L. Translocation-specific conformation of adenylate cyclase toxin from Bordetella pertussis inhibits toxin-mediated hemolysis. J. Bacteriol. 2001, 183, 5904–5910. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Fiser, R.; Linhartova, I.; Osicka, R.; Bumba, L.; Hewlett, E.L.; Benz, R.; Sebo, P. Differences in purinergic amplification of osmotic cell lysis by the pore-forming RTX toxins Bordetella pertussis CyaA and Actinobacillus pleuropneumoniae ApxIA: The role of pore size. Infect. Immun. 2013, 81, 4571–4582. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Knapp, O.; Masin, J.; Fiser, R.; Maier, E.; Benz, R.; Sebo, P.; Osicka, R. Segments crucial for membrane translocation and pore-forming activity of Bordetella adenylate cyclase toxin. J. Biol. Chem. 2007, 282, 12419–12429. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Roderova, J.; Osickova, A.; Novak, P.; Bumba, L.; Fiser, R.; Sebo, P.; Osicka, R. The conserved tyrosine residue 940 plays a key structural role in membrane interaction of Bordetella adenylate cyclase toxin. Sci. Rep. 2017, 7, 9330. [Google Scholar] [CrossRef] [PubMed]
- Osickova, A.; Osicka, R.; Maier, E.; Benz, R.; Sebo, P. An amphipathic alpha-helix including glutamates 509 and 516 is crucial for membrane translocation of adenylate cyclase toxin and modulates formation and cation selectivity of its membrane channels. J. Biol. Chem. 1999, 274, 37644–37650. [Google Scholar] [PubMed]
- Osickova, A.; Masin, J.; Fayolle, C.; Krusek, J.; Basler, M.; Pospisilova, E.; Leclerc, C.; Osicka, R.; Sebo, P. Adenylate cyclase toxin translocates across target cell membrane without forming a pore. Mol. Microbiol. 2010, 75, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.A.; Hewlett, E.L.; Myers, G.A.; Falkow, S. Pertussis toxin and extracytoplasmic adenylate cyclase as virulence factors of Bordetella pertussis. J. Infect. Dis. 1984, 150, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Rogel, A.; Hanski, E. Distinct steps in the penetration of adenylate cyclase toxin of Bordetella pertussis into sheep erythrocytes. Translocation of the toxin across the membrane. J. Biol. Chem. 1992, 267, 22599–22605. [Google Scholar] [PubMed]
- Rogel, A.; Meller, R.; Hanski, E. Adenylate cyclase toxin from Bordetella pertussis. The relationship between induction of cAMP and hemolysis. J. Biol. Chem. 1991, 266, 3154–3161. [Google Scholar] [PubMed]
- Masin, J.; Konopasek, I.; Svobodova, J.; Sebo, P. Different structural requirements for adenylate cyclase toxin interactions with erythrocyte and liposome membranes. Biochim. Biophys. Acta 2004, 1660, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Bauche, C.; Chenal, A.; Knapp, O.; Bodenreider, C.; Benz, R.; Chaffotte, A.; Ladant, D. Structural and functional characterization of an essential RTX subdomain of Bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 2006, 281, 16914–16926. [Google Scholar] [CrossRef] [PubMed]
- Chenal, A.; Guijarro, J.I.; Raynal, B.; Delepierre, M.; Ladant, D. RTX calcium binding motifs are intrinsically disordered in the absence of calcium: Implication for protein secretion. J. Biol. Chem. 2009, 284, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; Maier, E.; Polleichtner, G.; Masin, J.; Sebo, P.; Benz, R. Channel formation in model membranes by the adenylate cyclase toxin of Bordetella pertussis: Effect of calcium. Biochemistry 2003, 42, 8077–8084. [Google Scholar] [CrossRef] [PubMed]
- Fiser, R.; Konopasek, I. Different modes of membrane permeabilization by two RTX toxins: HlyA from Escherichia coli and CyaA from Bordetella pertussis. Biochim. Biophys. Acta 2009, 1788, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Requero, M.A.; Masin, J.; Konopasek, I.; Goni, F.M.; Sebo, P.; Ostolaza, H. Membrane restructuring by Bordetella pertussis adenylate cyclase toxin, a member of the RTX toxin family. J. Bacteriol. 2004, 186, 3760–3765. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; Maier, E.; Masin, J.; Sebo, P.; Benz, R. Pore formation by the Bordetella adenylate cyclase toxin in lipid bilayer membranes: Role of voltage and pH. Biochim. Biophys. Acta 2008, 1778, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.M.; Weiss, A.A.; Ehrmann, I.E.; Gray, M.C.; Hewlett, E.L.; Goodwin, M.S. Bordetella pertussis adenylate cyclase toxin and hemolytic activities require a second gene, cyaC, for activation. J. Bacteriol. 1991, 173, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Sebo, P.; Glaser, P.; Sakamoto, H.; Ullmann, A. High-level synthesis of active adenylate cyclase toxin of Bordetella pertussis in a reconstructed Escherichia coli system. Gene 1991, 104, 19–24. [Google Scholar] [CrossRef]
- Havlicek, V.; Higgins, L.; Chen, W.; Halada, P.; Sebo, P.; Sakamoto, H.; Hackett, M. Mass spectrometric analysis of recombinant adenylate cyclase toxin from Bordetella pertussis strain 18323/pHSP9. J. Mass Spectrom. JMS 2001, 36, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Basar, T.; Havlicek, V.; Bezouskova, S.; Halada, P.; Hackett, M.; Sebo, P. The conserved lysine 860 in the additional fatty-acylation site of Bordetella pertussis adenylate cyclase is crucial for toxin function independently of its acylation status. J. Biol. Chem. 1999, 274, 10777–10783. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Basler, M.; Knapp, O.; El-Azami-El-Idrissi, M.; Maier, E.; Konopasek, I.; Benz, R.; Leclerc, C.; Sebo, P. Acylation of lysine 860 allows tight binding and cytotoxicity of Bordetella adenylate cyclase on CD11b-expressing cells. Biochemistry 2005, 44, 12759–12766. [Google Scholar] [CrossRef] [PubMed]
- Basar, T.; Havlicek, V.; Bezouskova, S.; Hackett, M.; Sebo, P. Acylation of lysine 983 is sufficient for toxin activity of Bordetella pertussis adenylate cyclase. Substitutions of alanine 140 modulate acylation site selectivity of the toxin acyltransferase CyaC. J. Biol. Chem. 2001, 276, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Baumann, U. Crystal structure of the 50 kda metallo protease from Serratia marcescens. J. Mol. Biol. 1994, 242, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Baumann, U.; Wu, S.; Flaherty, K.M.; McKay, D.B. Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: A two-domain protein with a calcium binding parallel beta roll motif. EMBO J. 1993, 12, 3357–3364. [Google Scholar] [PubMed]
- Meier, R.; Drepper, T.; Svensson, V.; Jaeger, K.E.; Baumann, U. A calcium-gated lid and a large beta-roll sandwich are revealed by the crystal structure of extracellular lipase from Serratia marcescens. J. Biol. Chem. 2007, 282, 31477–31483. [Google Scholar] [CrossRef] [PubMed]
- Cannella, S.E.; Ntsogo Enguene, V.Y.; Davi, M.; Malosse, C.; Sotomayor Perez, A.C.; Chamot-Rooke, J.; Vachette, P.; Durand, D.; Ladant, D.; Chenal, A. Stability, structural and functional properties of a monomeric, calcium-loaded adenylate cyclase toxin, CyaA, from Bordetella pertussis. Sci. Rep. 2017, 7, 42065. [Google Scholar] [CrossRef] [PubMed]
- El-Azami-El-Idrissi, M.; Bauche, C.; Loucka, J.; Osicka, R.; Sebo, P.; Ladant, D.; Leclerc, C. Interaction of Bordetella pertussis adenylate cyclase with CD11b/CD18: Role of toxin acylation and identification of the main integrin interaction domain. J. Biol. Chem. 2003, 278, 38514–38521. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Stapleton, J.A.; Klesmith, J.R.; Hewlett, E.L.; Whitehead, T.A.; Maynard, J.A. Fine epitope mapping of two antibodies neutralizing the Bordetella adenylate cyclase toxin. Biochemistry 2017, 56, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Masure, H.R.; Au, D.C.; Gross, M.K.; Donovan, M.G.; Storm, D.R. Secretion of the Bordetella pertussis adenylate cyclase from Escherichia coli containing the hemolysin operon. Biochemistry 1990, 29, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Holland, I.B.; Peherstorfer, S.; Kanonenberg, K.; Lenders, M.; Reimann, S.; Schmitt, L. Type I protein secretion-deceptively simple yet with a wide range of mechanistic variability across the family. EcoSal Plus 2016, 7. [Google Scholar] [CrossRef]
- Thomas, S.; Holland, I.B.; Schmitt, L. The type 1 secretion pathway—The hemolysin system and beyond. Biochim. Biophys. Acta 2014, 1843, 1629–1641. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, D.P.; Hernandez, B.; Durand, D.; Hourdel, V.; Sotomayor-Perez, A.C.; Vachette, P.; Ghomi, M.; Chamot-Rooke, J.; Ladant, D.; Brier, S.; et al. Structural models of intrinsically disordered and calcium-bound folded states of a protein adapted for secretion. Sci. Rep. 2015, 5, 14223. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor-Perez, A.C.; Ladant, D.; Chenal, A. Disorder-to-order transition in the CyaA toxin RTX domain: Implications for toxin secretion. Toxins 2014, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Szilvay, G.R.; Blenner, M.A.; Shur, O.; Cropek, D.M.; Banta, S. A fret-based method for probing the conformational behavior of an intrinsically disordered repeat domain from Bordetella pertussis adenylate cyclase. Biochemistry 2009, 48, 11273–11282. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.H.; Weidtkamp-Peters, S.; Kleinschrodt, D.; Jaeger, K.E.; Smits, S.H.; Schmitt, L. Directionality of substrate translocation of the hemolysin A type I secretion system. Sci. Rep. 2015, 5, 12470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.C.; Ross, W.; Kim, K.; Hewlett, E.L. Characterization of binding of adenylate cyclase toxin to target cells by flow cytometry. Infect. Immun. 1999, 67, 4393–4399. [Google Scholar] [PubMed]
- Hanski, E. Invasive adenylate cyclase toxin of Bordetella pertussis. Trends Biochem. Sci. 1989, 14, 459–463. [Google Scholar] [CrossRef]
- Vojtova, J.; Kofronova, O.; Sebo, P.; Benada, O. Bordetella adenylate cyclase toxin induces a cascade of morphological changes of sheep erythrocytes and localizes into clusters in erythrocyte membranes. Microsc. Res. Tech. 2006, 69, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.D.; Symes, P.; Conboy, M.; Weiss, A.A.; Hewlett, E.L. Inhibition of monocyte oxidative responses by Bordetella pertussis adenylate cyclase toxin. J. Immunol. 1987, 139, 2749–2754. [Google Scholar] [PubMed]
- Gueirard, P.; Druilhe, A.; Pretolani, M.; Guiso, N. Role of adenylate cyclase-hemolysin in alveolar macrophage apoptosis during Bordetella pertussis infection in vivo. Infect. Immun. 1998, 66, 1718–1725. [Google Scholar] [PubMed]
- Harvill, E.T.; Cotter, P.A.; Yuk, M.H.; Miller, J.F. Probing the function of Bordetella bronchiseptica adenylate cyclase toxin by manipulating host immunity. Infect. Immun. 1999, 67, 1493–1500. [Google Scholar] [PubMed]
- Ambagala, T.C.; Ambagala, A.P.; Srikumaran, S. The leukotoxin of Pasteurella haemolytica binds to beta(2) integrins on bovine leukocytes. FEMS Microbiol. Lett. 1999, 179, 161–167. [Google Scholar] [PubMed]
- Jeyaseelan, S.; Hsuan, S.L.; Kannan, M.S.; Walcheck, B.; Wang, J.F.; Kehrli, M.E.; Lally, E.T.; Sieck, G.C.; Maheswaran, S.K. Lymphocyte function-associated antigen 1 is a receptor for Pasteurella haemolytica leukotoxin in bovine leukocytes. Infect. Immun. 2000, 68, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Lally, E.T.; Kieba, I.R.; Sato, A.; Green, C.L.; Rosenbloom, J.; Korostoff, J.; Wang, J.F.; Shenker, B.J.; Ortlepp, S.; Robinson, M.K.; et al. RTX toxins recognize a beta2 integrin on the surface of human target cells. J. Biol. Chem. 1997, 272, 30463–30469. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Clinkenbeard, K.D.; Ritchey, J.W. Bovine CD18 identified as a species specific receptor for Pasteurella haemolytica leukotoxin. Vet. Microbiol. 1999, 67, 91–97. [Google Scholar] [CrossRef]
- Arnaout, M.A. Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood 1990, 75, 1037–1050. [Google Scholar] [PubMed]
- Mazzone, A.; Ricevuti, G. Leukocyte CD11/CD18 integrins: Biological and clinical relevance. Haematologica 1995, 80, 161–175. [Google Scholar] [PubMed]
- Eby, J.C.; Gray, M.C.; Warfel, J.M.; Paddock, C.D.; Jones, T.F.; Day, S.R.; Bowden, J.; Poulter, M.D.; Donato, G.M.; Merkel, T.J.; et al. Quantification of the adenylate cyclase toxin of Bordetella pertussis in vitro and during respiratory infection. Infect. Immun. 2013, 81, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.N.; Cerny, O.; Linhartova, I.; Masin, J.; Osicka, R.; Sebo, P. cAMP signalling of Bordetella adenylate cyclase toxin through the SHP-1 phosphatase activates the BimEL-Bax pro-apoptotic cascade in phagocytes. Cell. Microbiol. 2016, 18, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Corbi, A.L.; Kishimoto, T.K.; Miller, L.J.; Springer, T.A. The human leukocyte adhesion glycoprotein Mac-1 (complement receptor type 3, CD11b) alpha subunit. Cloning, primary structure, and relation to the integrins, von Willebrand factor and factor B. J. Biol. Chem. 1988, 263, 12403–12411. [Google Scholar] [PubMed]
- Hickstein, D.D.; Hickey, M.J.; Ozols, J.; Baker, D.M.; Back, A.L.; Roth, G.J. cDNA sequence for the alpha M subunit of the human neutrophil adherence receptor indicates homology to integrin alpha subunits. Proc. Natl. Acad. Sci. USA 1989, 86, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Garcia-Aguilar, J.; Bickford, J.K.; Corbi, A.L.; Springer, T.A. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J. Cell Biol. 1993, 120, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Oxvig, C.; Springer, T.A. Experimental support for a beta-propeller domain in integrin alpha-subunits and a calcium binding site on its lower surface. Proc. Natl. Acad. Sci. USA 1998, 95, 4870–4875. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.O.; Rieu, P.; Arnaout, M.A.; Liddington, R. Crystal structure of the A domain from the alpha subunit of integrin CR3 (CD11b/CD18). Cell 1995, 80, 631–638. [Google Scholar] [CrossRef]
- Michishita, M.; Videm, V.; Arnaout, M.A. A novel divalent cation-binding site in the a domain of the beta 2 integrin cr3 (cd11b/cd18) is essential for ligand binding. Cell 1993, 72, 857–867. [Google Scholar] [CrossRef]
- Gahmberg, C.G.; Tolvanen, M.; Kotovuori, P. Leukocyte adhesion-structure and function of human leukocyte beta2-integrins and their cellular ligands. Eur. J. Biochem. 1997, 245, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Osickova, A.; Bumba, L.; Novak, P.; Sebo, P.; Osicka, R. Interaction of Bordetella adenylate cyclase toxin with complement receptor 3 involves multivalent glycan binding. FEBS Lett. 2015, 589, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Asada, M.; Furukawa, K.; Kantor, C.; Gahmberg, C.G.; Kobata, A. Structural study of the sugar chains of human leukocyte cell adhesion molecules CD11/CD18. Biochemistry 1991, 30, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Morova, J.; Osicka, R.; Masin, J.; Sebo, P. RTX cytotoxins recognize beta2 integrin receptors through N-linked oligosaccharides. Proc. Natl. Acad. Sci. USA 2008, 105, 5355–5360. [Google Scholar] [CrossRef] [PubMed]
- Hirai, M.; Iwase, H.; Hayakawa, T.; Koizumi, M.; Takahashi, H. Determination of asymmetric structure of ganglioside-DPPC mixed vesicle using SANS, SAXS, and DLS. Biophys. J. 2003, 85, 1600–1610. [Google Scholar] [CrossRef]
- Wald, T.; Osickova, A.; Masin, J.; Liskova, P.M.; Petry-Podgorska, I.; Matousek, T.; Sebo, P.; Osicka, R. Transmembrane segments of complement receptor 3 do not participate in cytotoxic activities but determine receptor structure required for action of Bordetella adenylate cyclase toxin. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef] [PubMed]
- Veneziano, R.; Rossi, C.; Chenal, A.; Devoisselle, J.M.; Ladant, D.; Chopineau, J. Bordetella pertussis adenylate cyclase toxin translocation across a tethered lipid bilayer. Proc. Natl. Acad. Sci. USA 2013, 110, 20473–20478. [Google Scholar] [CrossRef] [PubMed]
- Powthongchin, B.; Angsuthanasombat, C. Effects on haemolytic activity of single proline substitutions in the Bordetella pertussis CyaA pore-forming fragment. Arch. Microbiol. 2009, 191, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Betsou, F.; Sebo, P.; Guiso, N. CyaC-mediated activation is important not only for toxic but also for protective activities of Bordetella pertussis adenylate cyclase-hemolysin. Infect. Immun. 1993, 61, 3583–3589. [Google Scholar] [PubMed]
- Vojtova-Vodolanova, J.; Basler, M.; Osicka, R.; Knapp, O.; Maier, E.; Cerny, J.; Benada, O.; Benz, R.; Sebo, P. Oligomerization is involved in pore formation by Bordetella adenylate cyclase toxin. FASEB J. 2009, 23, 2831–2843. [Google Scholar] [CrossRef] [PubMed]
- Bejerano, M.; Nisan, I.; Ludwig, A.; Goebel, W.; Hanski, E. Characterization of the C-terminal domain essential for toxic activity of adenylate cyclase toxin. Mol. Microbiol. 1999, 31, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Kurehong, C.; Powthongchin, B.; Thamwiriyasati, N.; Angsuthanasombat, C. Functional significance of the highly conserved Glu(570) in the putative pore-forming helix 3 of the Bordetella pertussis haemolysin toxin. Toxicon 2011, 57, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Anthis, N.J.; Campbell, I.D. The tail of integrin activation. Trends Biochem. Sci. 2011, 36, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M. The leucocyte beta2 (CD18) integrins: The structure, functional regulation and signalling properties. Biosci. Rep. 2012, 32, 241–269. [Google Scholar] [CrossRef] [PubMed]
- Kinashi, T. Adhere upright: A switchblade-like extension of beta2 integrins. Immunity 2006, 25, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.H.; Springer, T.A. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Yalamanchili, P.; Lu, C.; Oxvig, C.; Springer, T.A. Folding and function of I domain-deleted Mac-1 and lymphocyte function-associated antigen-1. J. Biol. Chem. 2000, 275, 21877–21882. [Google Scholar] [CrossRef] [PubMed]
- Jakus, Z.; Fodor, S.; Abram, C.L.; Lowell, C.A.; Mocsai, A. Immunoreceptor-like signaling by beta 2 and beta 3 integrins. Trends Cell Biol. 2007, 17, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Mocsai, A.; Zhou, M.; Meng, F.; Tybulewicz, V.L.; Lowell, C.A. Syk is required for integrin signaling in neutrophils. Immunity 2002, 16, 547–558. [Google Scholar] [CrossRef]
- Mocsai, A.; Abram, C.L.; Jakus, Z.; Hu, Y.; Lanier, L.L.; Lowell, C.A. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat. Immunol. 2006, 7, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Mocsai, A.; Ruland, J.; Tybulewicz, V.L. The Syk tyrosine kinase: A crucial player in diverse biological functions. Nat. Rev. Immunol. 2010, 10, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Schymeinsky, J.; Mocsai, A.; Walzog, B. Neutrophil activation via beta2 integrins (CD11/CD18): Molecular mechanisms and clinical implications. Thromb. Haemost. 2007, 98, 262–273. [Google Scholar] [PubMed]
- Crowley, M.T.; Costello, P.S.; Fitzer-Attas, C.J.; Turner, M.; Meng, F.; Lowell, C.; Tybulewicz, V.L.; DeFranco, A.L. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J. Exp. Med. 1997, 186, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, F.; Brumell, J.; Al-Alawi, N.; Latour, S.; Cheng, A.; Veillette, A.; Grinstein, S.; Pawson, T. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol. Cell Biol. 1998, 18, 4209–4220. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tohyama, Y.; Kadono, T.; He, J.; Miah, S.M.; Hazama, R.; Tanaka, C.; Tohyama, K.; Yamamura, H. Protein-tyrosine kinase Syk is required for pathogen engulfment in complement-mediated phagocytosis. Blood 2006, 107, 4554–4562. [Google Scholar] [CrossRef] [PubMed]
- Caron, E.; Hall, A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science 1998, 282, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Lenz, D.H.; Weingart, C.L.; Weiss, A.A. Phagocytosed Bordetella pertussis fails to survive in human neutrophils. Infect. Immun. 2000, 68, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Willeke, T.; Schymeinsky, J.; Prange, P.; Zahler, S.; Walzog, B. A role for Syk-kinase in the control of the binding cycle of the beta2 integrins (CD11/CD18) in human polymorphonuclear neutrophils. J. Leukoc. Biol. 2003, 74, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevrey, J.C.; Isaac, B.M.; Cox, D. Syk is required for monocyte/macrophage chemotaxis to CX3CL1 (fractalkine). J. Immunol. 2005, 175, 3737–3745. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, M.; Lofgren, R.; Zheng, L.; Stendahl, O. Tumour necrosis factor-alpha potentiates CR3-induced respiratory burst by activating p38 MAP kinase in human neutrophils. Immunology 2001, 103, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Van Ziffle, J.A.; Lowell, C.A. Neutrophil-specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood 2009, 114, 4871–4882. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.L.; Fiederlein, R.L.; Glasser, L.; Galgiani, J.N. Bordetella pertussis adenylate cyclase: Effects of affinity-purified adenylate cyclase on human polymorphonuclear leukocyte functions. Infect. Immun. 1987, 55, 135–140. [Google Scholar] [PubMed]
- Eby, J.C.; Gray, M.C.; Hewlett, E.L. Cyclic AMP-mediated suppression of neutrophil extracellular trap formation and apoptosis by the Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 2014, 82, 5256–5269. [Google Scholar] [CrossRef] [PubMed]
- Guth, A.M.; Janssen, W.J.; Bosio, C.M.; Crouch, E.C.; Henson, P.M.; Dow, S.W. Lung environment determines unique phenotype of alveolar macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L936–L946. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Pinilla-Vera, M.; Xiong, Z.; Zhao, Y.; Zhao, J.; Donahoe, M.P.; Barge, S.; Horne, W.T.; Kolls, J.K.; McVerry, B.J.; Birukova, A.; et al. Full spectrum of LPS activation in alveolar macrophages of healthy volunteers by whole transcriptomic profiling. PLoS ONE 2016, 11, e0159329. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.; Mai, J.; Cai, S.; Jeyaseelan, S. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect. Immun. 2009, 77, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Skopova, K.; Tomalova, B.; Kanchev, I.; Rossmann, P.; Svedova, M.; Adkins, I.; Bibova, I.; Tomala, J.; Masin, J.; Guiso, N.; et al. Cyclic AMP-elevating capacity of adenylate cyclase toxin-hemolysin is sufficient for lung infection but not for full virulence of Bordetella pertussis. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.L.; Nordensson, K.; Wilson, L.; Akporiaye, E.T.; Yocum, D.E. Uptake and intracellular survival of Bordetella pertussis in human macrophages. Infect. Immun. 1992, 60, 4578–4585. [Google Scholar] [PubMed]
- Valdez, H.A.; Oviedo, J.M.; Gorgojo, J.P.; Lamberti, Y.; Rodriguez, M.E. Bordetella pertussis modulates human macrophage defense gene expression. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.Y.; Dickinson, P.; Sing, G.; Craigon, M.; Ghazal, P.; Parton, R.; Coote, J.G. Transcriptional responses of murine macrophages to the adenylate cyclase toxin of Bordetella pertussis. Microb. Pathog. 2008, 44, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.C.; Hewlett, E.L. Cell cycle arrest induced by the bacterial adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Cell. Microbiol. 2011, 13, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Park, H.J.; Jeon, S.H.; Lee, C.; Seong, R.H.; Park, S.H.; Hong, S. Ubiquitous over-expression of chromatin remodeling factor SRG3 ameliorates the T cell-mediated exacerbation of EAE by modulating the phenotypes of both dendritic cells and macrophages. PLoS ONE 2015, 10, e0132329. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.; Fabrik, I.; Linhartova, I.; Link, M.; Cerny, O.; Stulik, J.; Sebo, P. Phosphoproteomics of cAMP signaling of Bordetella adenylate cyclase toxin in mouse dendritic cells. Sci. Rep. under revision.
- Yong Kim, S.; Jeong, S.; Chah, K.H.; Jung, E.; Baek, K.H.; Kim, S.T.; Shim, J.H.; Chun, E.; Lee, K.Y. Salt-inducible kinases 1 and 3 negatively regulate Toll-like receptor 4-mediated signal. Mol. Endocrinol. 2013, 27, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; MacKenzie, K.F.; Petkevicius, K.; Kristariyanto, Y.; Zhang, J.; Choi, H.G.; Peggie, M.; Plater, L.; Pedrioli, P.G.; McIver, E.; et al. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, 16986–16991. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, K.F.; Clark, K.; Naqvi, S.; McGuire, V.A.; Nöehren, G.; Kristariyanto, Y.; van den Bosch, M.; Mudaliar, M.; McCarthy, P.C.; Pattison, M.J.; et al. PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J. Immunol. 2013, 190, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Khelef, N.; Zychlinsky, A.; Guiso, N. Bordetella pertussis induces apoptosis in macrophages: Role of adenylate cyclase-hemolysin. Infect. Immun. 1993, 61, 4064–4071. [Google Scholar] [PubMed]
- Moujalled, D.; Weston, R.; Anderton, H.; Ninnis, R.; Goel, P.; Coley, A.; Huang, D.C.; Wu, L.; Strasser, A.; Puthalakath, H. Cyclic-AMP-dependent protein kinase A regulates apoptosis by stabilizing the BH3-only protein Bim. EMBO Rep. 2011, 12, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Palen, D.I.; Belmadani, S.; Lucchesi, P.A.; Matrougui, K. Role of SHP-1, Kv.1.2, and cGMP in nitric oxide-induced ERK1/2 map kinase dephosphorylation in rat vascular smooth muscle cells. Cardiovasc. Res. 2005, 68, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.; Ewings, K.E.; Hadfield, K.; Howes, E.; Balmanno, K.; Cook, S.J. Extracellular signal-regulated kinases 1/2 are serum-stimulated “Bim(EL) kinases” that bind to the BH3-only protein Bim(EL) causing its phosphorylation and turnover. J. Biol. Chem. 2004, 279, 8837–8847. [Google Scholar] [CrossRef] [PubMed]
- Van Haarst, J.M.; de Wit, H.J.; Drexhage, H.A.; Hoogsteden, H.C. Distribution and immunophenotype of mononuclear phagocytes and dendritic cells in the human lung. Am. J. Respir. Cell Mol. Biol. 1994, 10, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Baharom, F.; Rankin, G.; Blomberg, A.; Smed-Sorensen, A. Human lung mononuclear phagocytes in health and disease. Front. Immunol. 2017, 8, 499. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Lambrecht, B.N.; Hammad, H. Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal Immunol. 2013, 6, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.; Heit, A.; Dreher, S.; Eisenächer, K.; Mages, J.; Haas, T.; Krug, A.; Janssen, K.P.; Kirschning, C.J.; Wagner, H. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur. J. Immunol. 2008, 38, 2981–2992. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Costantino, G.; Poglitsch, M.; Rosner, M.; Zeyda, M.; Stuhlmeier, K.M.; Kolbe, T.; Stulnig, T.M.; Hörl, W.H.; Hengstschläger, M.; et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 2008, 29, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.J.; Gray, M.C.; Hewlett, E.L.; Vogel, S.N. Bordetella pertussis adenylate cyclase toxin (ACT) induces cyclooxygenase-2 (COX-2) in murine macrophages and is facilitated by act interaction with CD11b/CD18 (Mac-1). Mol. Microbiol. 2007, 66, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Kreisel, D.; Nava, R.G.; Li, W.; Zinselmeyer, B.H.; Wang, B.; Lai, J.; Pless, R.; Gelman, A.E.; Krupnick, A.S.; Miller, M.J. In Vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 18073–18078. [Google Scholar] [CrossRef] [PubMed]
- Pechous, R.D. With friends like these: The complex role of neutrophils in the progression of severe pneumonia. Front. Cell. Infect. Microbiol. 2017, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Eby, J.C.; Hoffman, C.L.; Gonyar, L.A.; Hewlett, E.L. Review of the neutrophil response to Bordetella pertussis infection. Pathog. Dis. 2015, 73, ftv081. [Google Scholar] [CrossRef] [PubMed]
- Weingart, C.L.; Mobberley-Schuman, P.S.; Hewlett, E.L.; Gray, M.C.; Weiss, A.A. Neutralizing antibodies to adenylate cyclase toxin promote phagocytosis of Bordetella pertussis by human neutrophils. Infect. Immun. 2000, 68, 7152–7155. [Google Scholar] [CrossRef] [PubMed]
- Weingart, C.L.; Weiss, A.A. Bordetella pertussis virulence factors affect phagocytosis by human neutrophils. Infect. Immun. 2000, 68, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Mobberley-Schuman, P.S.; Connelly, B.; Weiss, A.A. Phagocytosis of Bordetella pertussis incubated with convalescent serum. J. Infect. Dis. 2003, 187, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, M.S.; Weiss, A.A. Adenylate cyclase toxin is critical for colonization and pertussis toxin is critical for lethal infection by Bordetella pertussis in infant mice. Infect. Immun. 1990, 58, 3445–3447. [Google Scholar] [PubMed]
- Gross, M.K.; Au, D.C.; Smith, A.L.; Storm, D.R. Targeted mutations that ablate either the adenylate cyclase or hemolysin function of the bifunctional cyaa toxin of Bordetella pertussis abolish virulence. Proc. Natl. Acad. Sci. USA 1992, 89, 4898–4902. [Google Scholar] [CrossRef] [PubMed]
- Guiso, N.; Rocancourt, M.; Szatanik, M.; Alonso, J.M. Bordetella adenylate cyclase is a virulence associated factor and an immunoprotective antigen. Microb. Pathog. 1989, 7, 373–380. [Google Scholar] [CrossRef]
- Guiso, N.; Szatanik, M.; Rocancourt, M. Protective activity of Bordetella adenylate cyclase-hemolysin against bacterial colonization. Microb. Pathog. 1991, 11, 423–431. [Google Scholar] [CrossRef]
- Weiss, A.A.; Hewlett, E.L.; Myers, G.A.; Falkow, S. Tn5-induced mutations affecting virulence factors of Bordetella pertussis. Infect. Immun. 1983, 42, 33–41. [Google Scholar] [PubMed]
- Chenal-Francisque, V.; Caro, V.; Boursaux-Eude, C.; Guiso, N. Genomic analysis of the adenylate cyclase-hemolysin C-terminal region of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Res. Microbiol. 2009, 160, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Bart, M.J.; Harris, S.R.; Advani, A.; Arakawa, Y.; Bottero, D.; Bouchez, V.; Cassiday, P.K.; Chiang, C.S.; Dalby, T.; Fry, N.K.; et al. Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. mBio 2014, 5, e01074. [Google Scholar] [CrossRef] [PubMed]
- Khelef, N.; Bachelet, C.M.; Vargaftig, B.B.; Guiso, N. Characterization of murine lung inflammation after infection with parental Bordetella pertussis and mutants deficient in adhesins or toxins. Infect. Immun. 1994, 62, 2893–2900. [Google Scholar] [PubMed]
- Khelef, N.; Sakamoto, H.; Guiso, N. Both adenylate cyclase and hemolytic activities are required by Bordetella pertussis to initiate infection. Microb. Pathog. 1992, 12, 227–235. [Google Scholar] [CrossRef]
- Andreasen, C.; Carbonetti, N.H. Role of neutrophils in response to Bordetella pertussis infection in mice. Infect. Immun. 2009, 77, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Gorgojo, J.; Scharrig, E.; Gomez, R.M.; Harvill, E.T.; Rodriguez, M.E. Bordetella parapertussis circumvents neutrophil extracellular bactericidal mechanisms. PLoS ONE 2017, 12, e0169936. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novak, J.; Cerny, O.; Osickova, A.; Linhartova, I.; Masin, J.; Bumba, L.; Sebo, P.; Osicka, R. Structure–Function Relationships Underlying the Capacity of Bordetella Adenylate Cyclase Toxin to Disarm Host Phagocytes. Toxins 2017, 9, 300. https://doi.org/10.3390/toxins9100300
Novak J, Cerny O, Osickova A, Linhartova I, Masin J, Bumba L, Sebo P, Osicka R. Structure–Function Relationships Underlying the Capacity of Bordetella Adenylate Cyclase Toxin to Disarm Host Phagocytes. Toxins. 2017; 9(10):300. https://doi.org/10.3390/toxins9100300
Chicago/Turabian StyleNovak, Jakub, Ondrej Cerny, Adriana Osickova, Irena Linhartova, Jiri Masin, Ladislav Bumba, Peter Sebo, and Radim Osicka. 2017. "Structure–Function Relationships Underlying the Capacity of Bordetella Adenylate Cyclase Toxin to Disarm Host Phagocytes" Toxins 9, no. 10: 300. https://doi.org/10.3390/toxins9100300
APA StyleNovak, J., Cerny, O., Osickova, A., Linhartova, I., Masin, J., Bumba, L., Sebo, P., & Osicka, R. (2017). Structure–Function Relationships Underlying the Capacity of Bordetella Adenylate Cyclase Toxin to Disarm Host Phagocytes. Toxins, 9(10), 300. https://doi.org/10.3390/toxins9100300