
Interaction of Cholesterol with Perfringolysin O: What Have We Learned from Functional Analysis?


Abstract
:1. Introduction
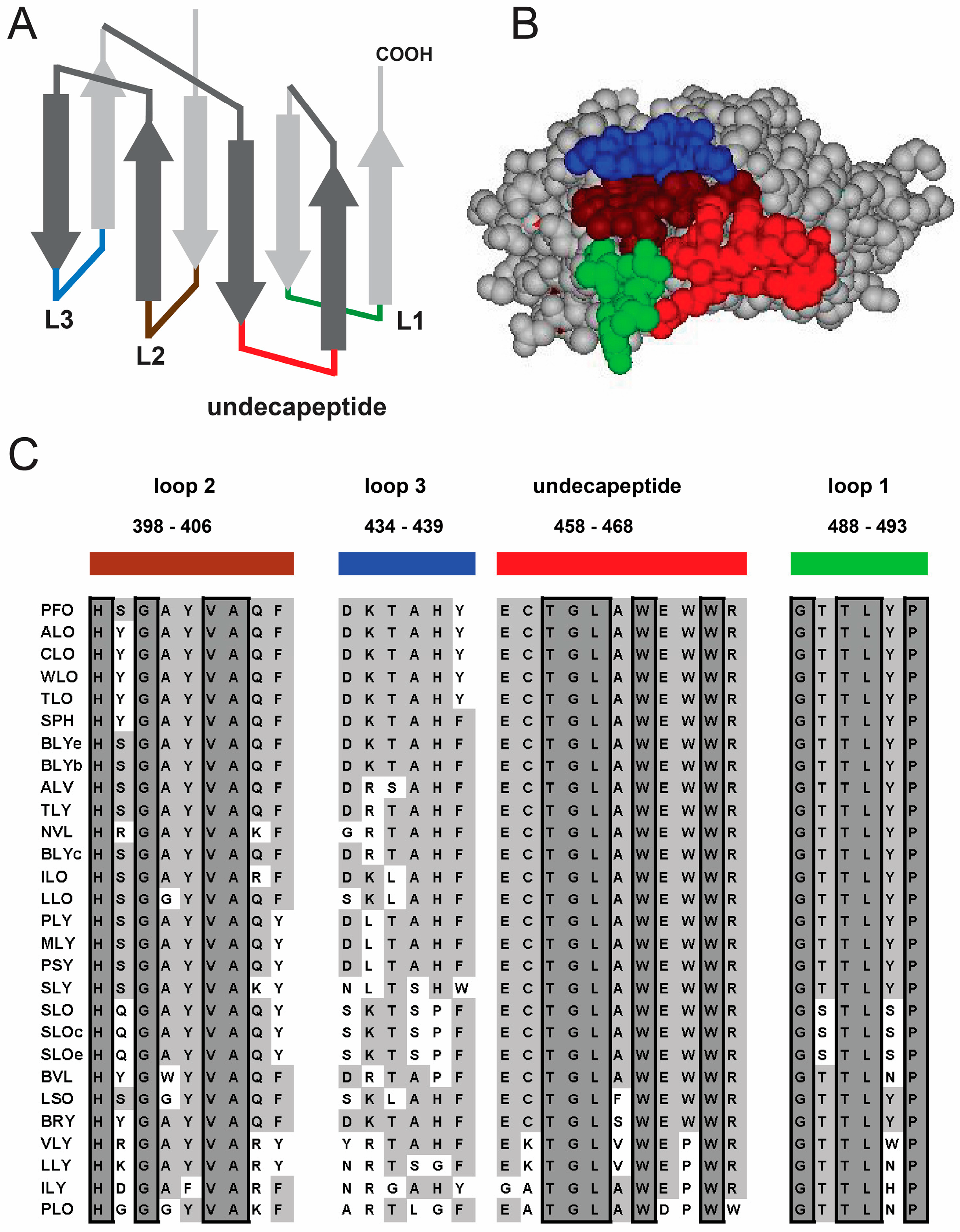
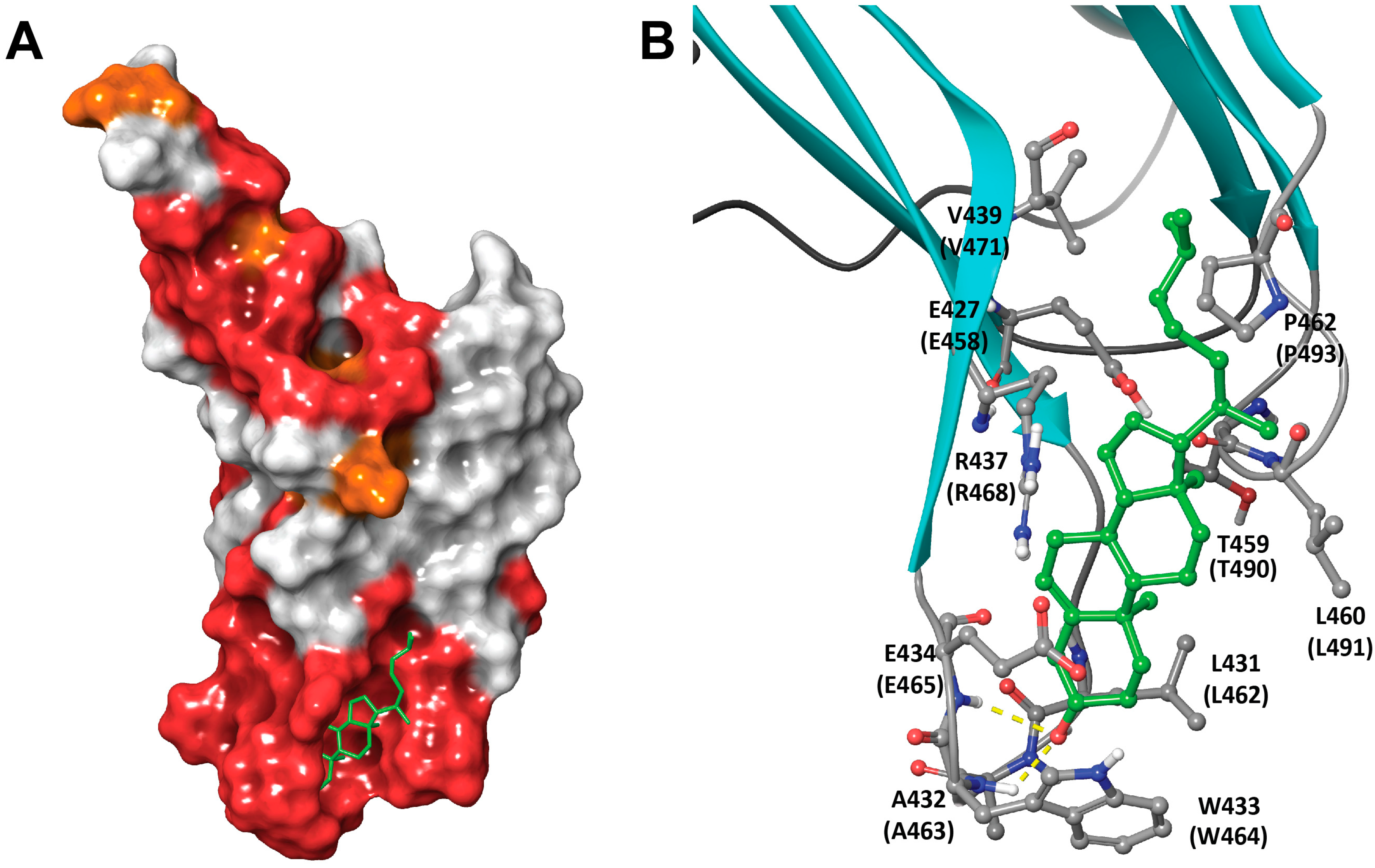
1.1. Structural Elements of Domain 4 Involved in Cholesterol Recognition
1.2. The Effects of Membrane Lipids on the Cholesterol Threshold Required for CDC Binding
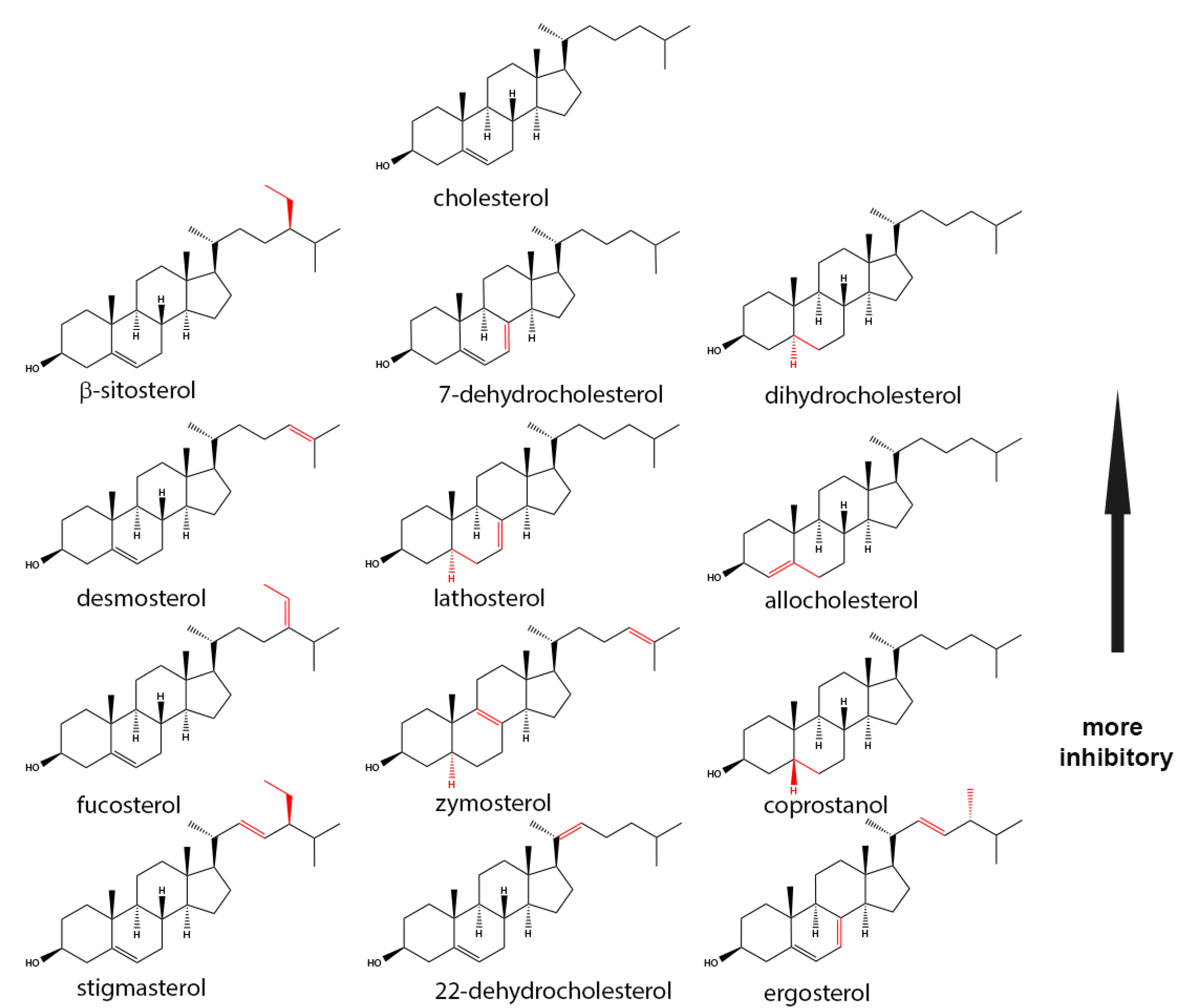
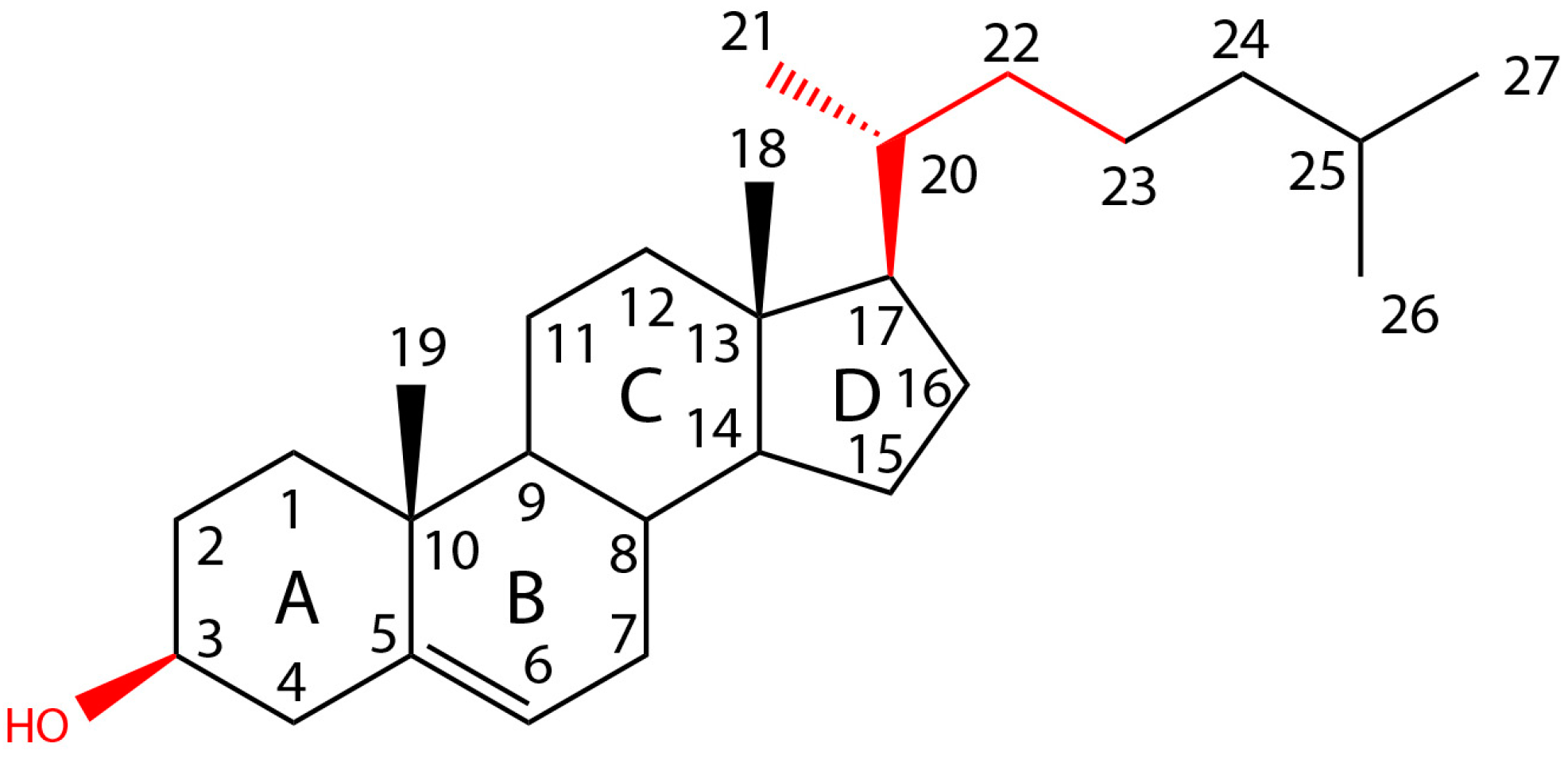
1.3. Structure Elements of Cholesterol that Influence CDC Activity
1.3.1. The Presence of a Lateral Aliphatic Side Chain of Suitable Size at Carbon 17 Is Required
1.3.2. The Presence of a 3 β-Hydroxy Group on Ring A Is Required
1.3.3. An Intact Ring B Is Required
2. Results
2.1. Selective Solubilization of Sterol Aggregates by Methyl-β-cyclodextrin
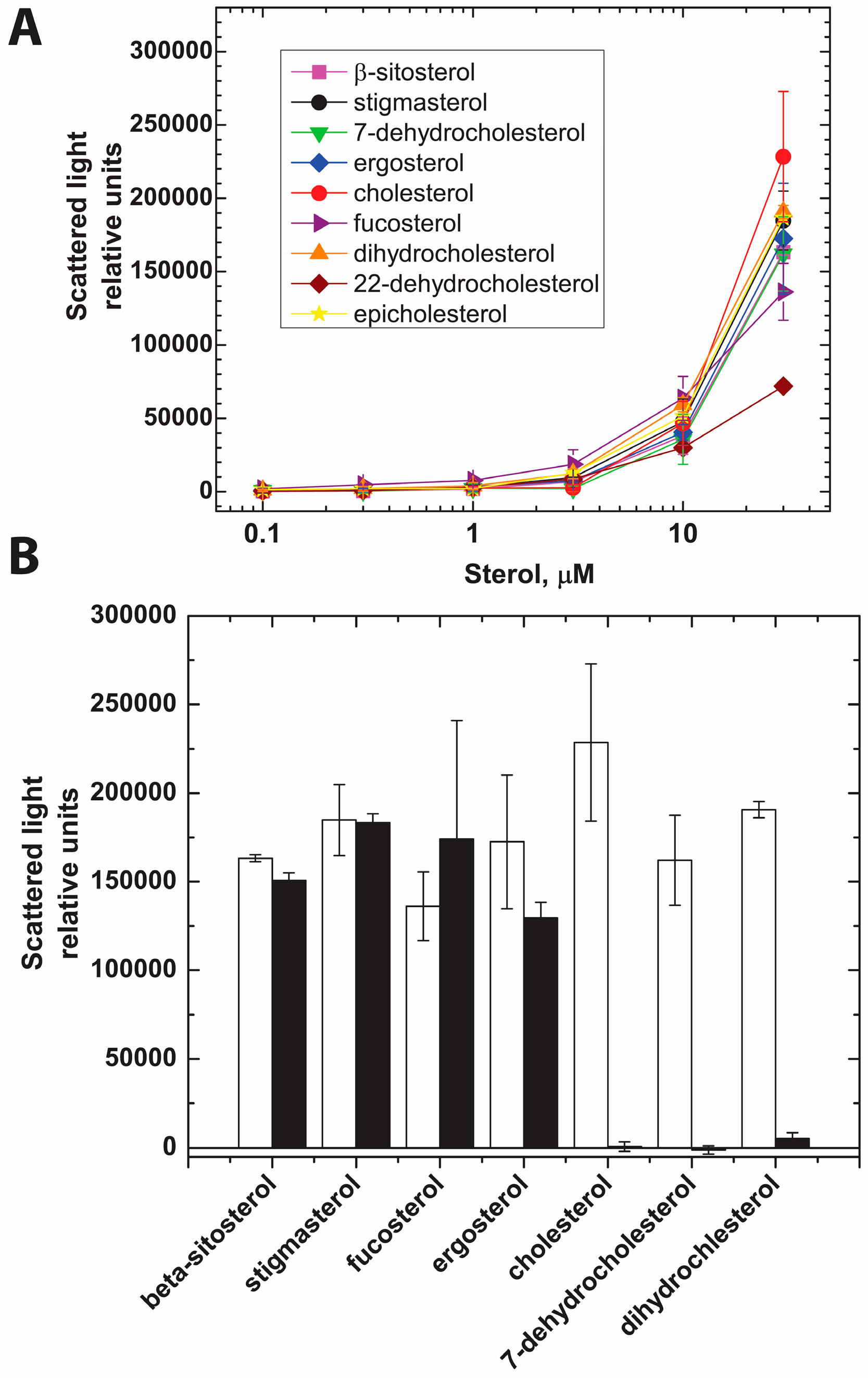
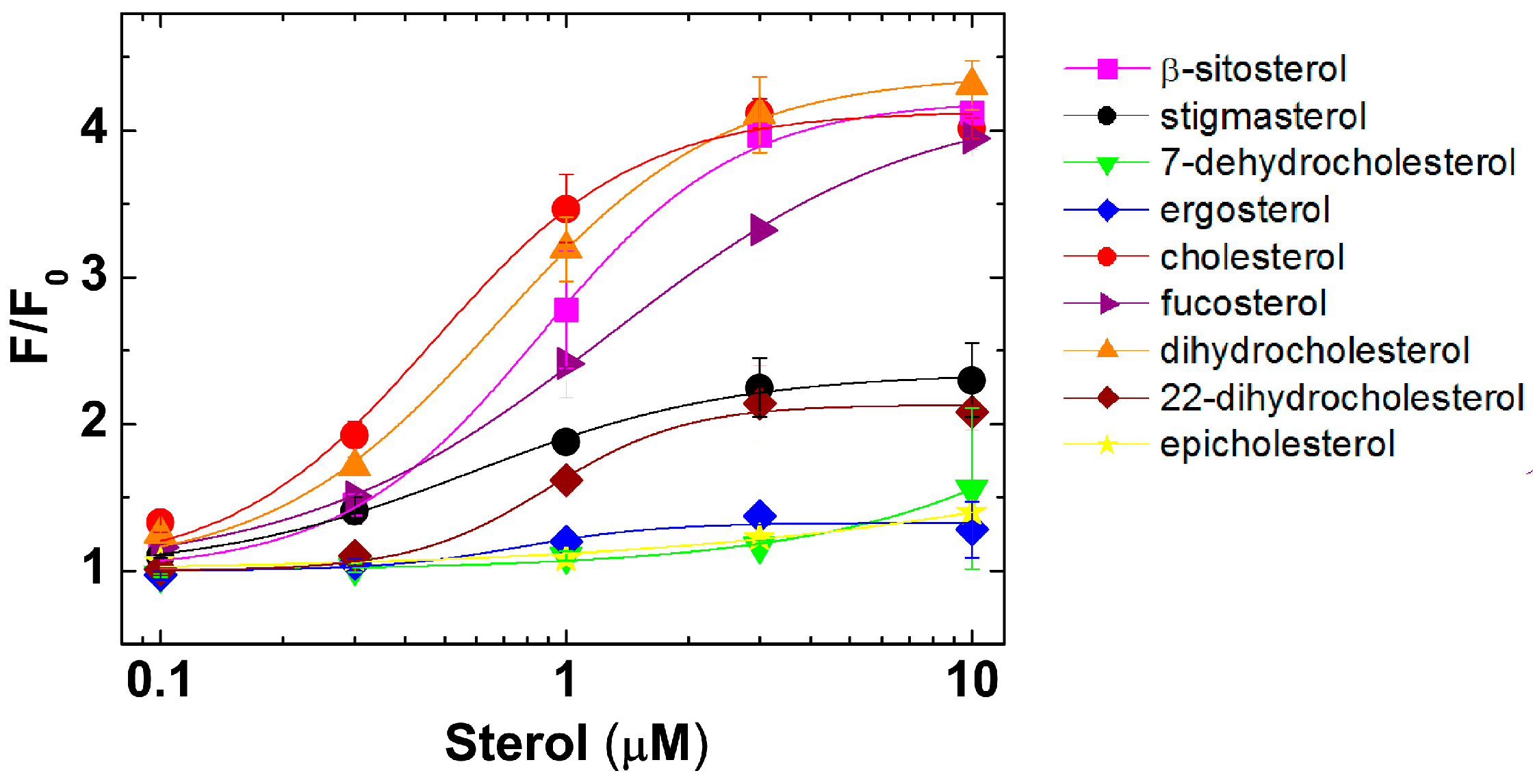
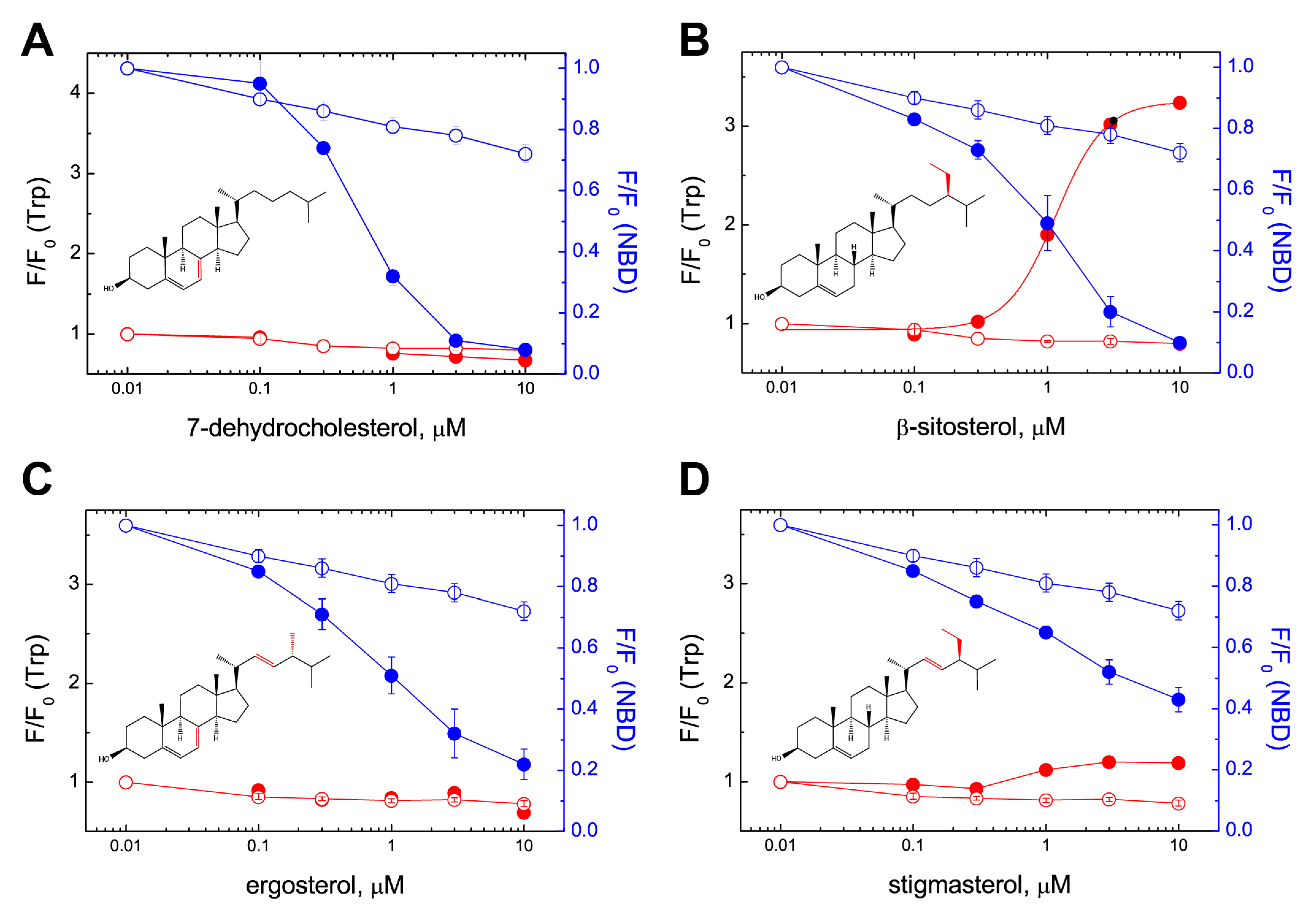
2.2. PFO Interaction with Free Sterols
2.3. Molecular Modeling Rationales for the Observed Cholesterol Structure–Activity Relationship and Mutagenesis Data
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Incubation with Sterol Dispersions in Aqueous Solutions
4.3. Steady-State Fluorescence Spectroscopy
4.4. Molecular Modeling
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tweten, R.K. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 2005, 73, 6199–6209. [Google Scholar] [CrossRef] [PubMed]
- Heuck, A.P.; Moe, P.C.; Johnson, B.B. The cholesterol-dependent cytolysins family of Gram-positive bacterial toxins. In Cholesterol Binding Proteins and Cholesterol Transport; Harris, J.R., Ed.; Springer: Dordrecht, The Netherlands, 2010; Volume 51, pp. 551–577. [Google Scholar]
- Johnson, B.; Heuck, A. Perfringolysin O structure and mechanism of pore formation as a paradigm for cholesterol-dependent cytolysins. In Macpf/cdc Proteins—Agents of Defence, Attack and Invasion; Anderluh, G., Gilbert, R., Eds.; Springer: Dordrecht, The Netherlands, 2014; Volume 80, pp. 63–81. [Google Scholar]
- Giddings, K.S.; Zhao, J.; Sims, P.J.; Tweten, R.K. Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat. Struct. Mol. Biol. 2004, 11, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Giddings, K.S.; Johnson, A.E.; Tweten, R.K. Redefining cholesterol’s role in the mechanism of the cholesterol-dependent cytolysins. Proc. Natl. Acad. Sci. USA 2003, 100, 11315–11320. [Google Scholar] [CrossRef] [PubMed]
- Alving, C.R.; Habig, W.H.; Urban, K.A.; Hardegree, M.C. Cholesterol-dependent tetanolysin damage to liposomes. Biochim. Biophys. Acta 1979, 551, 224–228. [Google Scholar] [CrossRef]
- Rosenqvist, E.; Michaelsen, T.E.; Vistnes, A.I. Effect of streptolysin O and digitonin on egg lecithin/cholesterol vesicles. Biochim. Biophys. Acta 1980, 600, 91–102. [Google Scholar] [CrossRef]
- Ohno-Iwashita, Y.; Iwamoto, M.; Ando, S.; Iwashita, S. Effect of lipidic factors on membrane cholesterol topology—Mode of binding of θ-toxin to cholesterol in liposomes. Biochim. Biophys. Acta 1992, 1109, 81–90. [Google Scholar] [CrossRef]
- Nelson, L.D.; Johnson, A.E.; London, E. How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low ph: Insights into the origin of perfringolysin O-lipid raft interaction. J. Biol. Chem. 2008, 283, 4632–4642. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.J.; Tweten, R.K.; Johnson, A.E.; Heuck, A.P. Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 2009, 48, 3977–3987. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Heuck, A.P.; Tweten, R.K.; Johnson, A.E. Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat. Struct. Mol. Biol. 2002, 9, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Soltani, C.E.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 20226–20231. [Google Scholar] [CrossRef] [PubMed]
- Farrand, A.J.; LaChapelle, S.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc. Natl. Acad. Sci. USA 2010, 107, 4341–4346. [Google Scholar] [CrossRef] [PubMed]
- Polekhina, G.; Giddings, K.S.; Tweten, R.K.; Parker, M.W. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc. Natl. Acad. Sci. USA 2005, 102, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Moe, P.C.; Heuck, A.P. Phospholipid hydrolysis caused by Clostridium perfringens α-toxin facilitates the targeting of perfringolysin o to membrane bilayers. Biochemistry 2010, 49, 9498–9507. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.B.; Moe, P.C.; Wang, D.; Rossi, K.; Trigatti, B.L.; Heuck, A.P. Modifications in perfringolysin O domain 4 alter the cholesterol concentration threshold required for binding. Biochemistry 2012, 51, 3373–3382. [Google Scholar] [CrossRef] [PubMed]
- Lally, C.C.M.; Bauer, B.; Selent, J.; Sommer, M.E. C-edge loops of arrestin function as a membrane anchor. Nat. Commun. 2017, 8, 14258. [Google Scholar] [CrossRef] [PubMed]
- Saunders, F.K.; Mitchell, T.J.; Walker, J.A.; Andrew, P.W.; Boulnois, G.J. Pneumolysin, the thiol-activated toxin of Streptococcus pneumoniae, does not require a thiol group for in vitro activity. Infect. Immun. 1989, 57, 2547–2552. [Google Scholar] [PubMed]
- Pinkney, M.; Beachey, E.; Kehoe, M. The thiol-activated toxin streptolysin O does not require a thiol group for cytolytic activity. Infect. Immun. 1989, 57, 2553–2558. [Google Scholar] [PubMed]
- Michel, E.; Reich, K.A.; Favier, R.; Berche, P.; Cossart, P. Attenuated mutants of the intracellular bacterium Listeria monocytogenes obtained by single amino acid substitutions in listeriolysin O. Mol. Microbiol. 1990, 4, 2167–2178. [Google Scholar] [CrossRef] [PubMed]
- Sekino-Suzuki, N.; Nakamura, M.; Mitsui, K.-I.; Ohno-Iwashita, Y. Contribution of individual tryptophan residues to the structure and activity of θ-toxin (perfringolysin O), a cholesterol-binding cytolysin. Eur. J. Biochem. 1996, 241, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Korchev, Y.E.; Bashford, C.L.; Pederzolli, C.; Pasternak, C.A.; Morgan, P.J.; Andrew, P.W.; Mitchell, T.J. A conserved tryptophan in pneumolysin is a determinant of the characteristics of channels formed pneumolysin in cells and planar lipid bilayers. Biochem. J. 1998, 329, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Billington, S.J.; Songer, J.G.; Jost, B.H. The variant undecapeptide sequence of the Arcanobacterium pyogenes haemolysin, pyolysin, is required for full cytolytic activity. Microbiology 2002, 148, 3947–3954. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.J.; Tweten, R.K. The cholesterol-dependent cytolysin signature motif: A critical element in the allosteric pathway that couples membrane binding to pore assembly. PLoS Pathog. 2012, 8, e1002787. [Google Scholar] [CrossRef]
- Johnson, B.B.; Breña, M.; Anguita, J.; Heuck, A.P. Mechanistic insights into the cholesterol-dependent binding of perfringolysin O-based probes and cell membranes. Sci. Rep. 2017, 7, 13793. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Park, Y.S.; Bong, S.M.; Lee, K.S. Structure-based functional studies for the cellular recognition and cytolytic mechanism of pneumolysin from Streptococcus pneumoniae. J. Struct. Biol. 2016, 193, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Heuck, A.P.; Hotze, E.M.; Tweten, R.K.; Johnson, A.E. Mechanism of membrane insertion of a multimeric β-barrel protein: Perfringolysin O creates a pore using ordered and coupled conformational changes. Mol. Cell 2000, 6, 1233–1242. [Google Scholar] [CrossRef]
- Bavdek, A.; Gekara, N.O.; Priselac, D.; Gutierrez Aguirre, I.; Darji, A.; Chakraborty, T.; MacÌŒek, P.; Lakey, J.H.; Weiss, S.; Anderluh, G. Sterol and pH interdependence in the binding, oligomerization, and pore formation of listeriolysin O. Biochemistry 2007, 46, 4425–4437. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.; Shimada, Y.; Heijnen, H.F.G.; Nakamura, M.; Inomata, M.; Hayashi, M.; Iwashita, S.; Slot, J.W.; Ohno-Iwashita, Y. Selective binding of perfringolysin O derivative to cholesterol-rich membrane microdomains (rafts). Proc. Natl. Acad. Sci. USA 2001, 98, 4926–4931. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Iwashita, Y.; Shimada, Y.; Waheed, A.; Hayashi, M.; Inomata, M.; Nakamura, M.; Maruya, M.; Iwashita, M. Perfringolysin O, a cholesterol-binding cytolysin, as a probe for lipid rafts. Anaerobe 2004, 10, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, A.; Radhakrishnan, A. Accessibility of cholesterol in endoplasmic reticulum membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. J. Biol. Chem. 2010, 285, 29480–29490. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.N.; Bielska, A.A.; Lee, T.; Daily, M.D.; Covey, D.F.; Schlesinger, P.H.; Baker, N.A.; Ory, D.S. The structural basis of cholesterol accessibility in membranes. Biophys. J. 2013, 105, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Heuck, A.P.; Savva, C.G.; Holzenburg, A.; Johnson, A.E. Conformational changes that effect oligomerization and initiate pore formation are triggered throughout perfringolysin O upon binding to cholesterol. J. Biol. Chem. 2007, 282, 22629–22637. [Google Scholar] [CrossRef] [PubMed]
- Prigent, D.; Alouf, J.E. Interaction of streptolysin o with sterols. Biochim. Biophys. Acta 1976, 443, 288–300. [Google Scholar] [CrossRef]
- Kenneth, C.; Watson, K.C.; Kerr, E.J. Sterol structural requirements for inhibition of streptolysin O activity. Biochem. J. 1974, 140, 95–98. [Google Scholar]
- Hase, J.; Mitsui, K.; Shonaka, E. Clostridium perfringens exotoxins. Iv. Inhibition of the theta-toxin induced hemolysis by steroids and related compounds. Jpn. J. Exp. Med. 1976, 46, 45–50. [Google Scholar] [PubMed]
- Alouf, J.E. Streptococcal toxins (streptolysin O, streptolysin S, erythrogenic toxin). Pharmacol. Ther. 1980, 11, 661–717. [Google Scholar] [CrossRef]
- Megha; Bakht, O.; London, E. Cholesterol precursors stabilize ordinary and ceramide-rich ordered lipid domains (lipid rafts) to different degrees: Implications for the bloch hypothesis and sterol biosynthesis disorders. J. Biol. Chem. 2006, 281, 21903–21913. [Google Scholar]
- Haberland, M.E.; Reynolds, J.A. Self-association of cholesterol in aqueous solution. Proc. Natl. Acad. Sci. USA 1973, 70, 2313–2316. [Google Scholar] [CrossRef] [PubMed]
- Gimpl, G.; Burger, K.; Fahrenholz, F. Cholesterol as modulator of receptor function. Biochemistry 1997, 36, 10959–10974. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.D.; Chiantia, S.; London, E. Perfringolysin O association with ordered lipid domains: Implications for transmembrane protein raft affinity. Biophys. J. 2010, 99, 3255–3263. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Tweten, R.K.; Johnson, A.E. Membrane-dependent conformational changes initiate cholesterol-dependent cytolysin oligomerization and intersubunit beta-strand alignment. Nat. Struct. Mol. Biol. 2004, 11, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.C.; Ascher, D.B.; Kuiper, M.J.; Tweten, R.K.; Parker, M.W. Structural studies of Streptococcus pyogenes streptolysin O provide insights into the early steps of membrane penetration. J. Mol. Biol. 2014, 426, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R. Structural features of cholesterol dependent cytolysins and comparison to other MACPF-domain containing proteins. In MACPF/CDC Proteins—Agents of Defence, Attack and Invasion; Anderluh, G., Gilbert, R., Eds.; Springer: Dordrecht, The Netherlands, 2014; Volume 80, pp. 47–62. [Google Scholar]
- Chou, P.Y.; Fasman, G.D. Empirical predictions of protein conformation. Ann. Rev. Biochem. 1978, 47, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Kulma, M.; Kacprzyk-Stokowiec, A.; Kwiatkowska, K.; Traczyk, G.; Sobota, A.; Dadlez, M. R468A mutation in perfringolysin O destabilizes toxin structure and induces membrane fusion. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Farrand, A.J.; Hotze, E.M.; Sato, T.K.; Wade, K.R.; Wimley, W.C.; Johnson, A.E.; Tweten, R.K. The cholesterol-dependent cytolysin membrane-binding interface discriminates lipid environments of cholesterol to support β-barrel pore insertion. J. Biol. Chem. 2015, 290, 17733–17744. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.; Stahelin, R.V. Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 119–151. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Benoff, B.; Liou, H.-L.; Lobel, P.; Stock, A.M. Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick typeC2 disease. J. Biol. Chem. 2007, 282, 23525–23531. [Google Scholar] [CrossRef] [PubMed]
- Rezai, T.; Bock, J.E.; Zhou, M.V.; Kalyanaraman, C.; Lokey, R.S.; Jacobson, M.P. Conformational flexibility, internal hydrogen bonding, and passive membrane permeability: Successful in silico prediction of the relative permeabilities of cyclic peptides. J. Am. Chem. Soc. 2006, 128, 14073–14080. [Google Scholar] [CrossRef] [PubMed]
- Shepard, L.A.; Heuck, A.P.; Hamman, B.D.; Rossjohn, J.; Parker, M.W.; Ryan, K.R.; Johnson, A.E.; Tweten, R.K. Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: An α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry 1998, 37, 14563–14574. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hemolysis Inhibition | Sterol in Aqueous Buffer | Sterol in Liposomes | ||||
|---|---|---|---|---|---|---|
| SLO | PFO | PFO | PFO | PFO | PFO | |
| Trp D4 | NBD D3 | Trp D4 | Oligo SDS | |||
| cholesterol | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | +++ |
| 7-dehydrocholesterol | (1.7) | 0.67 | quenched | 0.89 | quenched | |
| dihydrocholesterol | 0.50 | 0.46 | 0.83 | 1.1 | +++ | |
| β-sitosterol | 0.50 | 0.61 | 0.59 | 0.80 | 0.94 | +++ |
| lathosterol | 0.50 | 0.85 | +++ | |||
| allocholesterol | 0.40 | 0.68 | ++ | |||
| desmosterol | 0.18 | 1.2 | +++ | |||
| coprostanol | 0.71 | 0.11 | 0.65 | + | ||
| zymosterol | 0.61 | + | ||||
| ergosterol | 0.10 | 0.13 | quenched | 0.44 | quenched | |
| fucosterol | 0.42 | |||||
| stigmasterol | 0.33 | 0.037 | low | <0.08 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savinov, S.N.; Heuck, A.P. Interaction of Cholesterol with Perfringolysin O: What Have We Learned from Functional Analysis? Toxins 2017, 9, 381. https://doi.org/10.3390/toxins9120381
Savinov SN, Heuck AP. Interaction of Cholesterol with Perfringolysin O: What Have We Learned from Functional Analysis? Toxins. 2017; 9(12):381. https://doi.org/10.3390/toxins9120381
Chicago/Turabian StyleSavinov, Sergey N., and Alejandro P. Heuck. 2017. "Interaction of Cholesterol with Perfringolysin O: What Have We Learned from Functional Analysis?" Toxins 9, no. 12: 381. https://doi.org/10.3390/toxins9120381
APA StyleSavinov, S. N., & Heuck, A. P. (2017). Interaction of Cholesterol with Perfringolysin O: What Have We Learned from Functional Analysis? Toxins, 9(12), 381. https://doi.org/10.3390/toxins9120381